
Abstract
SIRT1 induces cell survival and has shown neuroprotection against amyloid and tau pathologies in Alzheimer’s disease (AD). However, protective effects against memory loss or the enhancement of cognitive functions have not yet been proven. We aimed to investigate the benefits induced by SIRT1 overexpression in the hippocampus of the AD mouse model 3xTg-AD and in control non-transgenic mice. A lentiviral vector encoding mouse SIRT1 or GFP, selectively transducing neurons, was injected into the dorsal CA1 hippocampal area of 4-month-old mice. Six-month overexpression of SIRT1 fully preserved learning and memory in 10-month-old 3xTg-AD mice. Remarkably, SIRT1 also induced cognitive enhancement in healthy non-transgenic mice. Neuron cultures of 3xTg-AD mice, which show traits of AD-like pathology, and neuron cultures from non-transgenic mice were also transduced with lentiviral vectors to analyze beneficial SIRT1 mechanisms. We uncovered novel pathways of SIRT1 neuroprotection through enhancement of cell proteostatic mechanisms and activation of neurotrophic factors not previously reported such as GDNF, present in both AD-like and healthy neurons. Therefore, SIRT1 may increase neuron function and resilience against AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The progressive gain of human life expectancy has led to an increase of the incidence of age-related neurodegenerative diseases [1]. In 2015, 46.8 million people worldwide were living with dementia and this number is estimated to increase to 131.5 million by 2050 [2]. Alzheimer’s disease (AD) is the most common cause of dementia in older adults, becoming an obstacle to healthy aging.
AD is characterized by brain depositions of amyloid-β (Aβ) and tangles of hyperphosphorylated tau (p-tau), leading to synapse dysfunction, dementia, and death [3]. The amyloid precursor protein (APP) is cleaved by α-secretase to yield the neurotrophic and -protective APPα fragment, while the accumulation of amyloid deposits results from the sequential APP cleavage by β- and γ-secretases [4]. Hyperphosphorylation of tau reduces the binding to the microtubules, which become unstable, disintegrate, and form neurofibrillary tangles [5].
Many efforts are being invested in finding an effective therapy against AD [6]. Recent research has identified the anti-aging pathway mediated by the NAD+-dependent deacetylase sirtuin 1 (SIRT1) [7], a member of the sirtuin family of epigenetic regulators [8]. SIRT1 has been proposed to induce protective effects against AD brain pathology through regulating the acetylation homeostasis of key proteins [9–11]. Specifically, SIRT1 deacetylates the retinoic acid receptor β (RARβ), a transcriptional activator of the α-secretase enzyme disintegrin and metalloproteinase 10 (ADAM10), resulting in the reduction of Aβ production [12, 13]. SIRT1 can also prevent tau tangle formation, reducing tau acetylation, thereby allowing its degradation [14]. SIRT1 is highly expressed in neurons of the hippocampus, an important region for learning and memory that is vulnerable to AD [15]. The deficiency of SIRT1 causes cognitive impairment, as shown in brain-specific SIRT1 knockout mice [16]. SIRT1 expression is decreased in the affected brain regions of patients with AD [17, 18]. Decreased expression [19] and lower protein levels of SIRT1 [20–22] have also been found in the hippocampus of the 3xTg-AD mouse model of AD.
Both AD models and old animals can respond to therapies by upregulating SIRT1 expression in the hippocampus, as shown by a neuroprotective treatment of physical exercise in 3xTg-AD mice [21] and calorie restriction in aged rats [23]. Furthermore, injection of SIRT1 lentivirus in the hippocampus of p25 transgenic mouse model of AD and tauopathies conferred significant neuroprotection [24]. SIRT1 overexpression in primary neuron cultures from Tg2576 AD mouse induced a reduction of Aβ secretion [25]. Regarding anti-aging effects, SIRT1 overexpression in mice improves healthy aging [26] and survival [27].
There is a functional decline of protein clearance mechanisms with aging, which contributes to the accumulation of aberrant proteins in age-related diseases [28]. SIRT1 is involved in maintaining protein quality control [29], although an effect of SIRT1 on UPS activation has not been reported to date. Clearance of aberrant proteins, including both Aβ and p-tau, is performed by the ubiquitin-proteasome system (UPS) [30], although a proteasome-independent proteolysis of Aβ through insulin-degrading enzyme (IDE) has also been described [31]. The contribution of further enzymes catabolizing Aβ and its relevance in AD is not fully clarified [32].
Furthermore, SIRT1 overexpression in cultured neurons enhances neurite outgrowth, dendritic arbor complexity, dendritic spine density, and survival [33–35]. Plasticity effects of SIRT1 may be mediated through the neurotrophic factor BDNF. Two mechanisms have been proposed for SIRT1-mediated BDNF enhancement: either deacetylation of CREB-regulated transcription coactivator 1 (TORC1) [36] or inhibition of miR-134 [37].
The previously referred studies pointed to SIRT1 as a promising new tool in the fight against AD. It remained to be proven if the SIRT1 beneficial effects against AD pathology were able to rescue from cognitive loss in AD mouse models. Common anti-aging and -AD mechanisms of SIRT1 may include enhancement of cell proteostasis and neurotrophic pathways. In this study, we aimed to analyze the pro-cognitive effects of SIRT1 in the hippocampus of 3xTg-AD mice and the underlying neuroprotective mechanisms with emphasis in proteolysis and neurotrophism-mediated plasticity. For this purpose, we transduced the hippocampal neurons in vivo of 3xTg-AD mice and their control non-transgenic (NoTg) strain with lentiviral vectors bearing the mouse SIRT1 gene. After 6 months of gene overexpression, animals were fully analyzed for cognitive and neuronal changes. Neuron cultures of both mouse strains were transduced and analyzed in parallel. Our results demonstrated that overexpression of SIRT1 in the hippocampus of 3xTg-AD mice induced complete protection against memory loss and brain pathology. Novel pathways of neuroprotection through proteostasis and neurotrophism were explored, adding more effector mechanisms to this pleiotropic molecule. Furthermore, we showed for the first time that SIRT1 induced memory enhancement in healthy non-transgenic mice.
Material and Methods
Lentiviral Vectors
Recombinant lentiviral vectors encoding mouse SIRT1 protein were constructed for the neuron transduction. For in vivo injection into the hippocampus, the combination of the VSV-G (vesicular stomatitis virus G glycoprotein) pseudotype with the human PGK (phosphoglycerate kinase 1 promoter) allowed neuron specificity and efficient transgene expression. Vectors used for neuronal transduction in vitro were pseudotyped with VSV-G, and its gene expression was under control of the human CMV (cytomegalovirus immediate early promoter). Vectors encoding the humanized recombinant green fluorescent protein II (GFP) instead of SIRT1 were utilized as control vectors. Details of the obtaining and handling of the viral stocks are described elsewhere [38]. The viral titers were obtained by ELISA of the p24 capsid protein (HIV-1 p24 Antigen ELISA; Zeptometrix) and also by employing real-time quantitative PCR (qPCR)-based method previously described [39].
Animals
Male 3xTg-AD mice and control NoTg mice with the same genetic background hybrid 129 × C57BL/6 were used in this study. This AD mouse model expresses familial mutations of the amyloid precursor protein (APPSwe) and Presenilin 1 (PS1M146V), and a tau gene mutation (TauP301L) [40]. 3xTg-AD mice mimic many of the critical hallmarks of AD including Aβ and tau pathologies, impaired learning and memory, presence of behavioral and psychological symptoms of dementia (BPSD)-like, and oxidative stress [41, 42]. Genotypes were confirmed by PCR analysis of DNA obtained from tail biopsies. Mice were bred at the University of Barcelona Animal House (UB, Barcelona, Spain). At 4 months of age, mice were assigned to SIRT1 treatment by gene overexpression or control treatment by GFP gene expression. The experimental groups were as follows: NoTg-GFP (n = 8), NoTg-SIRT1 (n = 9), 3xTg-GFP (n = 9), and 3xTg-SIRT1 (n = 10). Animals were individually housed in cages with filters, complying with level 2 safety requirements and under standard laboratory conditions of food and water ad libitum, 22 ± 2 °C, and 12 h:12 h light-dark cycle. Animal handling and procedures were approved by the local Animal Ethics Committee (Ref: DAAM 5981, CEEA, UB), in accordance with the Spanish legislation and the European Union Directive 2010/63/EU for animal experiments.
Hippocampal Surgery and Gene Transduction
Mice were anesthetized with 10 mg/kg xylazine (Rompun 2 %, Bayer) intraperitoneally (i.p.) and 80 mg/kg ketamine (Ketolar 50 mg/mL, Pfizer) i.p. and placed in a stereotactic apparatus (David Kopf Instruments). Bilateral infusions of lentiviral vectors were performed into the dorsal CA1 hippocampal area, a brain region critically involved in cognitive processes. Stereotactic injections were performed at a rate of 0.5 μL/min at relative Bregma coordinates of −2.0-mm anteroposterior, ±1.2-mm mediolateral, and −1.5-mm dorsoventral. Coordinates were selected in preliminary studies with Fast Green colorant injection. One microliter per side of SIRT1 vector solution, containing 3.2 × 108 genomes/μL, was delivered to the application point with a 25-gauge stainless steel cannula (Small Parts, Inc.) connected to a Hamilton syringe through a Teflon tube. Control animals transduced with GFP received 1-μL injections of 6.6 × 108 genomes/μL solution. The syringe was attached to a micro-infusion pump (Bioanalytical Systems, Inc.). The cannula was left in position for 10 min after delivery to prevent the solution from backward surging. The incision was sutured, and the mice were allowed to recover from anesthesia on a thermal pad and placed back into their cages. Long-term gene expression was monitored at termination by qPCR, and specific neuron transduction was monitored by immunohistochemistry, as described later.
Behavioral and Cognitive Tests
Animals were tested for behavior and cognitive improvement at 6 months of the hippocampal injection, at 10 months of age. Selected BPSD-like symptoms were analyzed as previously described [42]. Briefly, the Corner test was utilized to assess neophobia to a new home cage during 30 s. The Open field test was used to evaluate vertical and horizontal locomotor activity and general behavior in a white chamber during 5 min. The Novel object recognition (NOR) test was employed to evaluate recognition memory and based on the spontaneous tendency of rodents to spend more time exploring a novel object than a familiar one. Animals were placed in the middle of a black maze with two arms angled 90°, each measuring 25 cm × 5 cm. The 15-cm high walls could be lifted off for easy cleaning. The objects to be discriminated were made of wood (objects A, B, and C were 6.2, 4.75, and 5 cm high, respectively, and distinctly colored). After 2 previous days of habituation, the animals were submitted to 10-min acquisition trial in the presence of two identical novel objects (A + A′) placed at the end of each arm. A 10-min retention trial occurred 2 h later, replacing the object A by object B. Another 10-min retention trial occurred 24 h later, replacing object A by object C. Object exploration was defined as the orientation of the nose to the object at a distance of less than 2 cm. Exploration times were recorded and used to calculate a discrimination index [novel (t) − familiar (t)] / [total time (t) at novel + familiar]. The Morris water maze (MWM) test was employed to evaluate the spatial learning and memory, consisting of 1 day of cue learning, 6 days of acquisition of learning for spatial reference memory, and 1 last day of memory retrieval through the removal of the platform. Mice were trained to locate a hidden platform (10 cm diameter, located 20 cm from the wall, and 0.5 cm below the water surface) in a circular tank (100 cm diameter, 40 cm height, 25 °C opaque water, surrounded by black curtains) by relying on distinctive landmarks as visual cues (four trial sessions of 60 s per day). On day 7, after one trial of place learning, the platform was removed and the mice performed a probe trial of 60 s to test the retention of learning. A computerized tracking system (SMART, Panlab S.A., Barcelona, Spain) was used to measure the distances and quadrants covered.
Brain Samples and Histology
After completion of the behavioral and cognitive tests, three animals per group were processed for brain histologic analysis. The remaining animals (5–7 mice per group) were decapitated and the hippocampus were dissected and stored at −80 °C for further analysis. For brain histology, mice were anesthetized as described previously for hippocampal injection of vectors. Animals were transcardially perfused through a ventricular catheter with 100 mM phosphate buffer pH 7.4, containing 0.1 mg/mL heparin (Mayne Pharma) followed by 4 % paraformaldehyde in phosphate buffer. Brains were removed, post-fixed overnight in cold paraformaldehyde solution, and then rinsed with cold phosphate buffer. Brains were cryopreserved in successive 20 and 30 % sucrose solutions and frozen on dry ice. Fixed mouse brains were processed for immunofluorescence by standard procedures. Coronal slices of 30 μm were stained with the following primary antibodies: SIRT1, glial fibrillary acidic protein (GFAP), neuronal specific nuclear protein (NeuN), Aβ clone 4G8, and p-tau clone AT8. Secondary antibodies used were Alexa Fluor 488 and 546 species-specific conjugated (1:1000) (Life Technologies). Primary antibody source and dilution are listed in Supplementary Table 1. Cell nuclei were counterstained with TO-PRO-3 iodide (1:1000) from Molecular Probes. Images were obtained in a Leica TCS SP2 confocal microscope.
Real-Time Quantitative PCR
Total RNA was isolated from the mouse hippocampus using the mirVana™ RNA Isolation Kit (Applied Biosystems) following the manufacturer’s instructions. RNA yield, purity, and quality were determined using a NanoDrop™ ND1000 spectrophotometer. RNA with 260/280 ratio of >1.9 were selected. Random-primed cDNA synthesis was performed using the High-Capacity cDNA Archive kit (Applied Biosystems). Gene expression was measured in an ABI Prism 7900HT qPCR system using TaqMan FAM-labeled specific probes (Applied Biosystems). Gene expression of ADAM10, brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), SIRT1, tropomyosin receptor kinase B (TrkB), and vascular endothelial growth factor A (VEGF-A) were analyzed in the mouse hippocampus. Data were normalized to TATA-binding protein (TBP) gene expression. A list of primers utilized is presented in Supplementary Table 2.
Validation of 3xTg-AD In Vitro Model
There is controversy about the usefulness of neuron cultures of transgenic AD mice for reproducing AD-like pathology. In the 3xTg-AD mice, the human transgenes are expressed under the transcriptional control of the Thy1.2 expression cassette [40]. Expression and activity of Thy1.2-driven transgenes is very low until postnatal days 6–12 [43]. In this regard, 3xTg-AD cortical brain tissue from embryonic day 16 (E16) has shown a threefold increase of both APP and tau expression [44] as compared to a sixfold expression in adult mice [40]. However, cortical and hippocampal neuron cultures of 3xTg-AD embryo mice have been shown useful for studying a range of AD-related alterations such as those in calcium signaling [44], neuron excitability [45], mitochondrial function [46], and zinc ion homeostasis [47]. Furthermore, Vale et al. [48] reported specific culture conditions leading to an accumulation of Aβ and p-tau in cortical neuron cultures of 3xTg-AD embryos, as early as 8 days in vitro (DIV). These authors proposed that primary 3xTg-AD neuron cultures constitute an in vitro model that mimics AD-like pathology and can be useful for studies of neuroprotection [49–51]. In the present study, we used the cell culture protocol of the later authors and confirmed the accumulation of Aβ and p-tau and signs of neurodegeneration at 11 DIV.
Neuron Cultures and Gene Transduction
Primary cortical neurons were prepared from E16–17 NoTg and 3xTg-AD mice. Cerebral hemispheres including cortices and hippocampi were dissected out in Krebs buffer, cut into small dices, trypsinized, and mechanically triturated into a single cell suspension. The cell suspension was centrifuged and resuspended in Dulbecco’s modified Eagle’s medium (Biochrom AG, Berlin, Germany) supplemented with 26.2 mM NaHCO3, 0.2 mM glutamine, 100 mU/L insulin B, 7 μM p-aminobenzoic acid, 25 mM KCl, and 10 % fetal bovine serum (Gibco-BRL, Invitrogen, Paisley, UK). Plates were precoated with 25 mg/L poly-D-lysine. Cells were seeded for immunocytochemistry analysis at a density of 3 × 105 cells/cm2 in 24-well plates onto glass coverslips. A density of 4 × 105 cells/cm2 was used in 12-well plates for Western blot analysis. Cultures were maintained at 37 °C in a humidified incubator with 5 % CO2. Two days after plating, 2 μM of cytosine arabinoside was added to the culture medium to prevent the proliferation of non-neuronal cells. At 4 DIV, vector solution containing 1.3 × 107 SIRT1 genomes/μL or 2.5 × 109 GFP genomes/μL were added directly to the culture medium. Neuron cultures were analyzed at 11 DIV, when amyloid and p-tau accumulation was manifested. Neuron transduction was monitored by immunohistochemistry and Western blotting, as described below.
Immunocytochemistry
Neuron cultures grown onto glass coverslips were fixed with 4 % paraformaldehyde after 11 DIV. Cells were processed by immunofluorescence with the following primary antibodies: Aβ clone 4G8, C-terminal fragment of APP (APP-CTF), microtubule-associated protein 2 (MAP2), NeuN, postsynaptic density protein 95 (PSD95), SIRT1, p-tau clone AT8, and tau clone HT7. Secondary antibodies used were Alexa Fluor 488 and 546 species-specific conjugated (1:1000) (Life Technologies). Primary antibody source and dilution are listed in Supplementary Table 1.
Western Blotting
Mature cultured neurons were homogenized in ice-cold RIPA buffer supplemented with protease and phosphatase inhibitors. Twenty micrograms of protein were analyzed for Western blot analysis by standard procedures [21]. The following antibodies were employed for immunodetection: ADAM10, APP-CTF, BDNF, GDNF, heat shock protein 70 (Hsp70), IDE, MAP2, acetylated p53 (ac-p53), proteasome 20S core subunits, PSD95, SIRT1, acetylated tau (ac-tau), p-tau clone AT8, and ubiquitin. Secondary antibodies were peroxidase-conjugated (1:2000) (GE Healthcare). Details of primary antibodies used are presented in Supplementary Table 1. The immunoreactive bands were detected by a chemiluminiscence method and digitized. Optical density of the studied proteins was normalized to that of actin, unless otherwise stated. Protein levels were expressed relative to the amount in the NoTg-GFP mouse group.
Statistical Analysis
Results are expressed as mean ± SEM. Data were analyzed with ANOVA procedures. Two-way ANOVA was followed by Bonferroni’s post hoc test for comparison of groups where interaction between variables was present. One-way ANOVA was followed by Bonferroni’s post hoc test for group comparison. Statistical analyses were performed using IBM SPSS Statistics v22.
Results
Expression of SIRT1 and GFP in CA1 Hippocampal Area
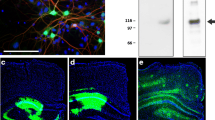
The injection of lentiviral vectors into the dorsal CA1 hippocampal area yielded a selective expression of the transduced proteins in the pyramidal neurons of NoTg and 3xTg-AD mice. Neurons transduced with lenti-SIRT1 showed intense immunostaining as compared with faint staining of the endogenous SIRT1 levels (Fig. 1a). Recombinant SIRT1 was expressed in neuronal nuclei (Fig. 1b) whereas GFP was expressed in the whole neuron cell body (Fig. 1c). The neural nature of the transduced cells was confirmed by the specific neuronal marker NeuN (Fig. 1c) and the astrocyte GFAP staining (Fig. 1b). Protein overexpression was restricted to the CA1 injected area and it was not detected outside of the hippocampus. Analysis of the whole hippocampus by RNA quantification revealed an increase of SIRT1 gene expression in the 3xTg-SIRT1 group, as expected (Fig. 1d) [ANOVA, F(2,14) = 28.62, p < 0.0001].

Expression of SIRT1 and GFP in the hippocampus and neuron cultures. a–c Confocal images of NoTg mouse hippocampus transduced with lentiviral vectors. a Endogenous (arrow) and transgene (arrowhead) SIRT1 expression in the CA1 hippocampal area. b SIRT1 expression in the nucleus of CA1 pyramidal. c GFP expression in the whole neuronal body demonstrated by the overlapping with the neuronal nuclei NeuN. d Gene expression measured by RNA levels of SIRT1 in the hippocampus tissue obtained by qPCR. e Confocal images of NoTg-SIRT1 neuron cultures stained with SIRT1 and NeuN. GFAP and NeuN were shown by red fluorescence; GFP and SIRT1 were labeled with green fluorescence. f–g Western blot analysis of SIRT1 and ac-p53 protein levels in neuron cultures of NoTg and 3xTg-AD mice. Optical density of ac-p53 was normalized to that of total p53. Values are mean ± SEM (n = 5–7). Scale bars: a = 200 μm; b, c, e = 50 μm. Statistical analysis: d One-way ANOVA followed by Bonferroni’s, *p < 0.05, **p < 0.01, and ***p < 0.001 compared to indicated group; f–g Two-way ANOVA followed by Bonferroni’s, ***p < 0.001 compared to NoTg mice, ###p < 0.001 compared to GFP treatment
Expression of SIRT1 and GFP in Neuron Cultures
In order to further analyze the mechanisms involved in neuroprotection, 3xTg-AD neuron cultures were also transduced to overexpress SIRT1. Lenti-SIRT1 transduction confirmed the expression of SIRT1 in the nuclei of the neurons in the same manner as in vivo. The neuronal specificity of lenti-SIRT1 vector was demonstrated by the overlapping of SIRT1 and neuronal marker NeuN (Fig. 1e). Western blot analysis showed that SIRT1 protein is reduced in 3xTg-GFP compared with NoTg-GFP neuron cultures and was greatly enhanced by SIRT1 lentiviral vector in NoTg-SIRT1 and 3xTg-SIRT1 neuron cultures (Fig. 1f) [genotype, F(1,16) = 11.25, p = 0.0040; treatment, F(1,16) = 219.6, p < 0.0001, and interaction treatment × genotype F(1,16) = 6.776, p = 0.0192]. SIRT1 deacetylase activity was confirmed through the decrease of ac-p53 levels in 3xTg-SIRT1 (Fig. 1g). Interestingly, although SIRT1 was similarly increased in the neuron cultures of both strains, ac-p53 was only reduced in 3xTg-AD cultures, where it was abnormally enhanced [genotype, F(1,20) = 22,72, p = 0.0001; treatment, F(1,20) = 16.22, p = 0.0007, and interaction treatment × genotype F(1,20) = 11.28, p = 0.0031].
Beneficial Effects of SIRT1 Overexpression on BPSD-Like Behavior
Six-month chronic SIRT1 overexpression induced a significant protective effect against the AD-like pathology underlying BPSD-like behavioral alterations (Fig. 2a–d). Behavior was normalized in 10-month-old 3xTg-SIRT1 mice, and there was also an enhancement of exploratory ambulation in both NoTg-SIRT1 and 3xTg-SIRT1 mice, whereas no unwanted effects were detected in either of the two strains. In the Corner test, the latency of initial vertical activity (rearing) was higher (Fig. 2a) and the total number of rearings was lower (Fig. 2b) in 3xTg-GFP compared with that in NoTg-GFP mice [latency of rearing: genotype, F(1,23) = 11.53, p = 0.0025; treatment, F(1,23) = 15.42, p = 0.0007, and interaction treatment × genotype F(1,23) = 9.935, p = 0.0045; and total number of rearings: interaction treatment × genotype F(1,24) = 4.425, p = 0.0461]. Therefore, lenti-SIRT1 recovered the neophobia responses in 3xTg-SIRT1 mice to NoTg mouse levels. In the Open field test, the latency of initial movement (freezing time) was higher (Fig. 2c) in 3xTg-GFP compared with that in NoTg-GFP mice [interaction treatment × genotype F(1,28) = 4.201, p = 0.0499]. Therefore, lenti-SIRT1 reduced freezing time behavior in 3xTg-SIRT1 mice to NoTg mouse levels. Lenti-SIRT1 also increased total distance covered (Fig. 2d) in both NoTg-SIRT1 and 3xTg-SIRT1 mice [treatment, F(1,22) = 5.221, p = 0.0323].

SIRT1 overexpression protects against cognitive loss in 3xTg-AD and induces cognitive enhancement in NoTg mice. Latency of vertical activity (a) and number of rearings (b) in the Corner test. Freezing time (c) and total distance covered (d) in the Open field test. NOR test at time 0 h (e), 2 h (f), and 24 h (g). MWM test with the distances covered to reach the platform (h) and the distance covered in the platform quadrant after the platform removal (i). A dotted line in the removal test graph indicates chance performance. Values are mean ± SEM (n = 6–9). Statistical analysis: a–c Two-way ANOVA followed by Bonferroni’s, *p < 0.05 and ***p < 0.001 compared to NoTg mice; #p < 0.05 and ###p < 0.001 compared to GFP treatment; d–g, i Two-way ANOVA, effect of genotype &p < 0.05 and &&p < 0.01, and treatment $p < 0.05 and $$p < 0.01; h One-way ANOVA followed by Bonferroni’s, *p < 0.05 and ***p < 0.001 P1 as compared with P6
Beneficial Effects of SIRT1 Overexpression on Cognitive Behavior
Six-month chronic SIRT1 overexpression induced a significant protective effect against AD-like pathology involved in learning and memory capacities (Fig. 2e–i). Cognition was preserved in 10-month-old 3xTg-SIRT1 mice. Additionally, cognitive enhancement effects were also induced in NoTg-SIRT1 mice. In the NOR test, 3xTg-GFP mice exhibited a deficit of recognition memory, while 3xTg-SIRT1 mice increased their capacity to remember familial objects at 2 h (Fig. 2f) and at 24 h (Fig. 2g). Two-way ANOVA demonstrated a significant effect of SIRT1 overexpression at 2 and 24 h [F(1,21) = 6.072, p = 0.0224 and F(1,21) = 6.542, p = 0.0183, respectively] and an effect of genotype at 24 h [F(1,21) = 8.574, p = 0.0080]. In the MWM test, the distances covered to locate the platform decreased over days of learning in NoTg-GFP and in NoTg-SIRT1 mice [F(5,24) = 2.933, p = 0.0332 and F(5,30) = 3.099, p = 0.0226, respectively], but the learning acquisition curves did not decrease significantly in 3xTg-GFP mice (Fig. 2h). Lenti-SIRT1 treatment reduced distances over learning days in 3xTg-SIRT1 mice [F(5,36) = 6.909, p = 0.0001]. In learning retrieval (Fig. 2i), both NoTg groups and the 3xTg-SIRT1 group covered a distance in the platform quadrant significantly greater than the distance expected at random [one-sample t test column statistics, at least p < 0.05], indicating good memory response. This effect was not observed in 3xTg-GFP mice, which swam at random in the pool, unaware of the former position of the escape platform. ANOVA showed a general effect of SIRT1 overexpression suggesting a better memory response for both mouse strains [genotype, F(1,22) = 6.023, p = 0.0225, and treatment, F(1,22) = 8.861, p = 0.0070]. No differences in average swimming speed and total distance were detected among groups [F(1,22) = 0.7141, p = 0.4072 and F(1,22) = 2.936, p = 0.1007, respectively].
SIRT1-Induced Neuroprotective Effects Against Amyloid-β and Tau Pathology
We confirmed the protective effects of SIRT1 against AD-like pathology in vivo. The presence of Aβ and p-tau was highly reduced in the 3xTg-AD hippocampal pyramidal neurons after SIRT1 overexpression, as demonstrated by immunostaining (Supplementary Fig. 1a, b). Increased expression of SIRT1 in the hippocampus was paralleled by an increased gene expression of α-secretase ADAM10 (Supplementary Fig. 1c) [F(2,14) = 18.97, p = 0,0001]. Neuron cultures obtained from 3xTg-AD mice reproduced amyloid and tau pathologies in vitro. We showed full neuroprotection against these pathologies in SIRT1-transduced neurons.
Immunostaining of neuron cultures with both Aβ (Fig. 3a) and APP-CTF (Fig. 3b) antibodies demonstrated a decrease in Aβ load within the 3xTg-SIRT1 as compared with 3xTg-GFP group. Furthermore, analysis of immunoblotting from neuron culture protein extracts confirmed the reduction of full length APP (Fig. 3c) [treatment, F(1,16) = 6.674, p = 0.0200, and interaction treatment × genotype F(1,16) = 6.907, p = 0.0183] and APP-CTF (Fig. 3d) [genotype, F(1,16) = 8.363, p = 0.0106; treatment, F(1,16) = 7.675, p = 0.0136, and interaction treatment × genotype F(1,16) = 11.20, p = 0.0041] in 3xTg-SIRT1 compared with 3xTg-GFP neuron cultures.

SIRT1 overexpression reduces amyloid-β and tau pathology in neuron cultures of 3xTg-AD mice. Representative confocal images of Aβ (a), APP-CTF (b), p-tau (e), and total tau (f) load in neuron cultures of 3xTg-AD mice. Amyloid and tau are shown by red fluorescence; GFP and SIRT1 were labeled with green fluorescence. Western blot analysis of full length APP (c), APP-CTF (d), p-tau (g), and ac-tau (h) protein levels in NoTg and 3xTg-AD neuron cultures. Scale bars: a, b, e, f = 50 μm. Values are mean ± SEM (n = 5–10). Statistical analysis: Two-way ANOVA followed by Bonferroni’s, **p < 0.01 and ***p < 0.001 compared to NoTg mice; ##p < 0.01, ###p < 0.001 compared to GFP treatment
Immunostaining of neuron cultures incubated with tau antibodies demonstrated a decrease in p-tau (Fig. 3e) and total tau (Fig. 3f) within the 3xTg-SIRT1 as compared with 3xTg-GFP group. Analysis of immunoblotting from neuron cultures also confirmed the reduction of p-tau (Fig. 3g) [genotype, F(1,12) = 13.74, p = 0.0030; treatment, F(1,12) = 19.27, p = 0.0009, and interaction treatment × genotype F(1,12) = 13.26, p = 0.0034] and ac-tau (Fig. 3h) [genotype, F(1,28) = 22.49, p < 0.0001; treatment, F(1,28) = 8.451, p = 0.0071, and interaction treatment × genotype F(1,28) = 5.061, p = 0.0325] in 3xTg-SIRT1 compared with 3xTg-GFP neuron cultures.
SIRT1-Induced Neuroprotective Effects Against Proteostatic Dysfunction
The functionality of UPS, the main cell system for aberrant protein disposal, was analyzed by determining the levels of selected key proteins in the protein extract of neuron cultures.
Immunoblotting analysis showed reduced expression of ubiquitinated proteins and free ubiquitin (Fig. 4a), and proteasome 20S core subunits (Fig. 4b) in 3xTg-GFP as compared with those in NoTg-GFP neuron cultures. SIRT1 overexpression induced total recovery of the levels of UPS proteins analyzed in 3xTg-SIRT1 cultures [F(2,24) = 8.527, p = 0.0016; F(2,24) = 6.287, p = 0.0064, and F(2,24) = 9.528, p = 0.0009, respectively]. A different pattern was observed for the Hsp70 heat shock protein, which showed higher expression in 3xTg-GFP than in NoTg-GFP and was further increased in 3xTg-SIRT1 neurons (Fig. 4c) [F(2,22) = 13.40, p = 0.0002]. SIRT1 reversed the deficit of UPS of 3xTg-AD neurons.

SIRT1 overexpression protects against UPS dysfunction in 3xTg-AD neuron cultures and increases non-amyloidogenic pathways. Western blot analysis of ubiquitinated proteins and monomeric ubiquitin (a), three bands attributable to the proteasome 20S core subunits (b), Hsp70 (c), IDE (d), and ADAM10 (e) protein levels in NoTg and 3xTg-AD neuron cultures. Values are mean ± SEM (n = 5–9). Statistical analysis: a–c One-way ANOVA followed by Bonferroni’s, *p < 0.05, **p < 0.01, and ***p < 0.001; d, e Two-way ANOVA, effect of treatment $$$p < 0.001
Furthermore, we analyzed protein levels of the amyloid-metabolizing enzyme IDE and the α-secretase ADAM10. Lenti-SIRT1 treatment induced higher expression of enzymes IDE (Fig. 4d) and ADAM10 (Fig. 4e) in both NoTg-SIRT1 and 3xTg-SIRT1 neuron cultures [treatment, F(1,28) = 36.30, p < 0.0001 and F(1,36) = 23.02, p < 0.0001, respectively].
SIRT1-Induced Increase of Neurotrophism
SIRT1 overexpression increased neurotrophic pathways in hippocampal tissue and neuron cultures. The analysis of neurotrophic factors in the hippocampus of SIRT1-transduced 3xTg-AD mice showed that the increase of GDNF (Fig. 5a) [F(2,12) = 17.79, p = 0.0003] and VEGF-A (Fig. 5b) [F(2,13) = 40.29, p < 0.0001] gene expression was highly significant. Protein levels of GDNF (Fig. 5c) were also highly increased in both NoTg-SIRT1 and 3xTg-SIRT1 neuron cultures [F(1,36) = 21.15, p < 0.0001].

SIRT1 overexpression increases neurotrophism in 3xTg-AD mice hippocampus and in 3xTg-AD and NoTg neuron cultures. Gene expression measured by RNA levels of the trophic factors GDNF (a) and VEGF-A (b) in the hippocampus tissue. Western blot analysis of GDNF (c) and BDNF (d) protein levels in NoTg and 3xTg-AD neuron cultures. Values are mean ± SEM (n = 5–10). Statistical analysis: a, b One-way ANOVA followed by Bonferroni’s, **p < 0.01 and ***p < 0.001 compared to the indicated group; c, d Two-way ANOVA, effect of treatment $p < 0.05 and $$$p < 0.001
Furthermore, we confirmed the enhancement of the neurotrophic factor BDNF by SIRT1 in vivo and in vitro. BDNF gene expression (Supplementary Fig. 1d) exhibited an increase in the hippocampus of 3xTg-SIRT1 mice [F(2,13) = 3.764, p = 0.0513], whereas the expression levels of the TrkB neurotrophin receptor were unchanged (Supplementary Fig. 1e). Protein levels of BDNF (Fig. 5d) were increased in both NoTg-SIRT1 and 3xTg-SIRT1 neuron cultures [treatment, F(1,28) = 5.084, p = 0.0322].
SIRT1-Induced Increase of Neuron Plasticity
To evaluate the effects of SIRT1 on neuroplasticity, synaptic structures and neurite growth were analyzed in neuron cultures by immunodetection of PSD95 and MAP2 antibodies, respectively.
Lenti-SIRT1 increased synaptic marker density (Fig. 6a, c) and neurite arborization (Fig. 6b, d) in both NoTg-SIRT1 and 3xTg-SIRT1 neuron cultures in comparison with the corresponding GFP-transduced cultures, as shown by immunostaining images.

SIRT1 overexpression increases plasticity markers in 3xTg-AD and NoTg neuron cultures. Representative confocal images of PSD95 (a, c) and MAP2 (b, d) labeled with red fluorescence. GFP and SIRT1 were labeled with green fluorescence. Western blot analysis of PSD95 (e) and MAP2 (f) protein levels in NoTg and 3xTg-AD neuron cultures. Scale bars: a, b, c, d = 50 μm. Values are mean ± SEM (n = 8–9). Statistical analysis: Two-way ANOVA, effect of genotype &&p < 0.01, and treatment $$$p < 0.001
Immunoblotting showed that PSD95 protein levels were lower in 3xTg-AD as compared with those in NoTg neuron cultures (Fig. 6e) [genotype, F(1,28) = 10.06, p = 0.0037], and SIRT1 overexpression induced an increase of protein expression of the PSD95, and also of MAP2 (Fig. 6f) in both NoTg and 3xTg-AD neurons [treatment, F(1,28) = 21.19, p < 0.0001 and F(1,32) = 28.59, p < 0.0001, respectively].
Discussion
SIRT1 overexpression demonstrated an outstanding neuroprotective effect against cognitive loss in 3xTg-AD mice and also cognitive enhancement in NoTg mice. Studies in hippocampus and in neuron cultures of these mice showed activation of proteolytic mechanisms (UPS and IDE) and enhancement of neurotrophic factors (GDNF and VEGF-A) not previously reported. Furthermore, the expected increase of ADAM10 and BDNF expression and decrease of ac-tau by SIRT1 were also reproduced in vivo and/or in vitro.
Locally transduced hippocampal neurons in 3xTg-AD mice maintained SIRT1 overexpression throughout 6 months of chronic treatment and until termination at an age of advanced AD-like pathology. The hippocampus is one area selectively affected by AD neurodegeneration [15], and the impairment of hippocampus circuitry greatly contributes to the devastating effects of memory loss in the disease. 3xTg-AD mice exhibited deficient learning and impaired retention in an MWM spatial task, as previously reported [41, 42, 52]. This task, which is primarily dependent on the dorsal hippocampus [53], was totally protected by SIRT1 overexpression. Furthermore, 3xTg-AD mice presented impairment of recognition memory evaluated by the NOR test [54], a task involving the hippocampus and other undefined brain regions [55]. Recognition memory was also preserved by SIRT1 overexpression in 3xTg-AD mice. Benefits of SIRT1 beyond those in the hippocampus were also proven by the reversal of the abnormal behaviors included to the BPSD phenotype linked with AD. 3xTg-AD mice experience BPSD-like behaviors [41] including neophobia and freezing behavior, which were totally abolished by SIRT1. Remarkably, the improved responses in the MWM and NOR tasks and in open field exploratory activity were generally detected in both strains, proving SIRT1 cognitive enhancement properties. This cognitive enhancement of the reference strain of NoTg mice has not been obtained in the NeSTO (Nestin-Cre × SIRT1 STOP) transgenic mouse expressing SIRT1 in nerve brain cells [56] in which no changes in long-term potentiation, associative learning, or spatial memory were found [16]. These authors also reported increased baseline synaptic activity. These SIRT1 transgenic mice possess broader distribution of SIRT1 gene expression in brain than that obtained with the present lentiviral injection in the CA1 hippocampus. Besides, age or unknown factors related to the genetic background may have caused the differences obtained.
In the AD brain, the origin of the excess of Aβ is not known although both increased generation and unbalanced degradation are assumed [57]. SIRT1 gene overexpression reduced APP and Aβ peptides in mouse brain and in cultured neurons of 3xTg-AD. Mice deficient in ADAM10 [58] and IDE [59] enzymatic activities show increased cerebral accumulation of endogenous Aβ. However, no changes in the levels of these enzymes were present in untreated 3xTg-AD, in agreement with previous reports [60, 61]. We found enhancement of ADAM10 by SIRT1 overexpression, thus indicating a shift to the non-amyloidogenic pathway of APP processing. A parallel increase in the expression of Aβ metabolizing enzyme IDE also contributed to the anti-amyloidogenic effect of SIRT1. Remarkably, the expression of both ADAM10 and IDE enzymes was generally enhanced by SIRT1 overexpression and would induce neuroprotection in NoTg mice against any potential age-related increase of Aβ accumulation.
Tau pathology is proposed to be triggered by amyloid pathology in the AD brain [62]. In the mouse model utilized here, there is a transgene encoding a tauopathy mutation [40], therefore facilitating the appearance of p-tau. Similar to the practical abolition of Aβ accumulation by SIRT1 overexpression, p-tau levels in 3xTg-AD neurons were reduced to those of NoTg neurons by SIRT1 gene overexpression. We found that the increase of p-tau levels in the 3xTg-AD neuron cultures was paralleled by an increase in ac-tau. Acetylation of lysine residues has been reported as a novel modification in the brain tissue of patients with AD and familial tauopathies [63, 14]. These authors proved that SIRT1 directly deacetylates tau and that mice with SIRT1 deletion undergo accumulation of ac-tau in the brain. Accordingly, ac-tau levels in 3xTg-AD neuron cultures increased concomitantly with the decrease of SIRT1. Interestingly, SIRT1 gene overexpression decreased the levels of both ac-tau and p-tau. Lysine deacetylation allows polyubiquitination in these residues of tau proteins and their subsequent degradation by proteasome [64, 14]. Therefore, p-tau degradation and clearance would be facilitated by its deacetylation through SIRT1 enzymatic activity. We analyzed tau acetylation at Lys 280 (K280); this and other Lys residue acetylation are increasingly considered as critical deleterious changes in AD [65]. Moreover, the sirtuin inhibitor nicotinamide has been reported to decrease tau pathology in 3xTg-AD mice through selective reduction of a specific p-tau species and increase of microtubule stability [66], showing the involvement of alternative neuroprotective pathways.
Functional proteasome degrades ubiquitin-tagged misfolded or aggregated proteins. Neurons of 3xTg-AD mice showed severely decreased levels of monomeric ubiquitin and consequential decreased levels of ubiquitinated proteins. There was an increase of Hsp70 levels in these neurons, probably as a compensatory mechanism. We found that SIRT1 induced a further increase of Hsp70, in agreement with a SIRT1 regulation of Hsp70 through maintaining the deacetylation state of heat shock factor 1 [67]. Chaperone Hsp70 recruits the carboxyl-terminus of Hsp70-interacting protein (CHIP) and enhances ligation between monomeric ubiquitin and misfolded proteins including Aβ and p-tau [68, 69]. Furthermore, SIRT1 overexpression induced an increase of monomeric ubiquitin in 3xTg-AD neurons up to the levels of NoTg neurons. SIRT1 overexpression normalized ubiquitin conjugation and in addition normalized ubiquitinated protein levels. Proteasome 20S core subunit levels were decreased in 3xTg-AD neurons, indicating impairment of the proteasome function. This is in agreement with the previous results of decreased proteasomal enzymatic activities in AD brains [70] and in hippocampal homogenates of 3xTg-AD mice [71]. The latter authors proved that Aβ oligomers inhibit proteasomal activity and that proteasome inhibition leads to the accumulation of Aβ and tau. SIRT1 normalized the levels of proteasome 20S core subunits and ubiquitinated proteins in transduced 3xTg-AD neurons, suggesting a recovery of UPS functionality. Benefits in proteostasis regulation induced by SIRT1 enhancement of UPS have recently been suggested in synucleinopathies [72].
SIRT1 knockout mice have shown defects in synaptic plasticity [16]. We found that SIRT1 overexpression induced gene expression of the neurotrophic factors BDNF, GDNF, and VEGF-A in the hippocampus tissue. BDNF is highly expressed in the hippocampus and comprises a crucial regulator of the synaptic plasticity mechanisms involved in learning and memory [73]. BDNF levels are reduced in the AD brain [74], and BDNF overexpression has induced neuroprotection in experimental models of AD [75]. GDNF has been proposed as a therapeutic agent against Parkinson’s disease [76]. Additionally, we previously reported its potent neuroprotective effect against hippocampus-related cognitive loss in 3xTg-AD mice [77]. VEGF-A upregulation induces angiogenesis, a process intimately linked with brain plasticity mechanisms. Furthermore, VEGF-A was reported to protect against endothelial damage by Aβ and memory loss in a mouse model of AD [78]. Analysis of BDNF and GDNF protein levels in SIRT1-transduced neurons showed marked enhancement in cultures from both strains. Therefore, these neurotrophic factors may contribute to the increase in neurite outgrowth and synaptic structures demonstrated by MAP2 and PSD95 immunodetection. These changes suggest an increase of synaptic plasticity induced by SIRT1. SIRT1 regulation of GDNF is a novel finding in which the mechanisms are not known. SIRT1 can increase VEGF-A expression by deacetylation of peroxisome proliferator-activated receptor-γ 1α (PGC1α), as described in muscle cells after exercise [79].
SIRT1 may also activate other neuroprotective pathways in addition to those analyzed here involving proteostatic and neurotrophic mechanisms. For instance, SIRT1 is a negative regulator of p53, since acetylation is an essential step in p53 tumor-suppression functions [80]. p53 was hyperacetylated in 3xTg-AD. Remarkably, SIRT1 overexpression normalized the levels of ac-p53 in 3xTg-AD neurons but did not lowered ac-p53 in control neurons. Therefore, SIRT1 overexpression would promote p53-induced genomic stability in AD-like neurons without increasing tumor risk in control neurons. Among other neuroprotective pathways modulated by SIRT1 deacetylation of specific substrates, inflammatory and oxidative processes are inhibited by deacetylation of nuclear factor κB, mitochondrial biogenesis is activated by deacetylation of PGC1α, and regulation of insulin signaling and cell survival are activated by deacetylation of forkhead box O proteins [9–11]. Furthermore, the regulation of cell fate of neural stem cells through SIRT1-mediated mechanisms [81] may also contribute to neuroprotection against AD. Finally, SIRT1 mediates metabolic adaptation to nutrient availability, and therefore SIRT1 activation may be crucial against metabolic alterations developed in neurodegenerative disorders [82].
SIRT1 is a valuable target for neuroprotection because it may be modulated by diet and lifestyles. For instance, calorie restriction [23, 83], physical exercise [21, 84], and resveratrol [85, 86] activate SIRT1 pathways that contribute to halt and/or reverse cellular aging and neurodegeneration. Although the cognitive effects of SIRT1 activators in healthy humans have not been clarified, the neurotrophic effects and the cognitive enhancement induced by SIRT1 overexpression in mouse neurons suggest that they may induce a general strengthening of learning and memory processes. In this regard, some studies reported cognitive improvement by physical exercise [87, 88], calorie restriction [89], and resveratrol supplementation [90] in humans. Furthermore, small-molecule SIRT1 activators, which may be the basis for emerging pharmacological therapies, are being actively developed [91, 92]. The results obtained here on novel proteostatic and neurotrophic pathways induced by SIRT1 warrant further investigation to better unveil the anti-AD and pro-cognitive potential of SIRT1 activators.
Conclusions
SIRT1 overexpression led to complete protection against memory loss in this transgenic mouse model of AD and to cognitive enhancement in normal mice. Analysis of SIRT1-transduced hippocampal neurons and neurons cultured from AD and normal mice leads to uncover novel SIRT1 mechanisms: (i) activation of IDE that will reduce amyloid load, (ii) enhancement of UPS that will lead to a reduction of aberrant amyloid and tau proteins, and (iii) upregulation of trophic factors GDNF and VEGF-A that will contribute to increased plasticity and neuroprotection. Additionally, we confirmed previously proposed neuroprotective mechanisms of SIRT1: (i) activation of ADAM10 that leads to an increase of the non-amyloidogenic pathway of APP processing, (ii) tau deacetylation that increases p-tau degradation, (iii) activation of stress response pathways such as Hsp70, and (iv) increase of the neurotrophic factor BDNF and neuron plasticity. A schematic representation of the proposed mechanisms activated by SIRT1 overexpression in this study is depicted in Fig. 7. Therefore, the dual effect of neural enhancement and protection against AD-like pathology converts the SIRT1 pathway into a promising target for developing preventive or therapeutic treatments against frailty and dementia.

Proposed pathways involved in the neuroprotective and cognitive enhancement effects of SIRT1 overexpression. Schematic representation of the mechanisms regulated by SIRT1, whether those already known (black arrows) or those uncovered here (dotted black arrows). See text for discussion of mechanisms. SIRT1 overexpression in mouse neurons led to a reduction in AD-like pathology and strengthened neuronal function. Ac acetylated, Aβ amyloid-β, AD Alzheimer’s disease, ADAM10 a disintegrin and metalloproteinase 10, APP amyloid precursor protein, BDNF brain-derived neurotrophic factor, CHIP carboxyl-terminus of Hsp70 interacting protein, GDNF glial cell line-derived neurotrophic factor, Hsp70 heat shock protein 70, IDE insulin-degrading enzyme, MAP2 microtubule-associated protein 2, ac-tau acetylated tau, p-tau hyperphosphorylated tau, RARβ retinoic acid receptor β, TORC1 CREB-regulated transcription coactivator 1, Ub ubiquitin, VEGF-A vascular endothelial growth factor A
References
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153(6):1194–1217. doi:10.1016/j.cell.2013.05.039
Alzheimer’s Disease International (2015) World Alzheimer Report 2015. Alzheimer’s Disease International (ADI) London
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356. doi:10.1126/science.1072994
Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120(4):545–555. doi:10.1016/j.cell.2005.02.008
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81(2):741–766
Parsons CG, Danysz W, Dekundy A, Pulte I (2013) Memantine and cholinesterase inhibitors: complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox Res 24(3):358–369. doi:10.1007/s12640-013-9398-z
Haigis MC, Sinclair DA (2010) Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5:253–295. doi:10.1146/annurev.pathol.4.110807.092250
Bosch-Presegue L, Vaquero A (2015) Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J 282(9):1745–1767. doi:10.1111/febs.13053
Herskovits AZ, Guarente L (2014) SIRT1 in neurodevelopment and brain senescence. Neuron 81(3):471–483. doi:10.1016/j.neuron.2014.01.028
Paraiso AF, Mendes KL, Santos SH (2013) Brain activation of SIRT1: role in neuropathology. Mol Neurobiol 48(3):681–689. doi:10.1007/s12035-013-8459-x
Donmez G (2012) The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol Sci 33(9):494–501. doi:10.1016/j.tips.2012.05.007
Saftig P, Lichtenthaler SF (2015) The alpha secretase ADAM10: a metalloprotease with multiple functions in the brain. Prog Neurobiol 135:1–20. doi:10.1016/j.pneurobio.2015.10.003
Lee HR, Shin HK, Park SY, Kim HY, Lee WS, Rhim BY, Hong KW, Kim CD (2014) Cilostazol suppresses beta-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-beta. J Neurosci Res 92(11):1581–1590. doi:10.1002/jnr.23421
Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67(6):953–966. doi:10.1016/j.neuron.2010.08.044
Mu Y, Gage FH (2011) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6:85. doi:10.1186/1750-1326-6-85
Michan S, Li Y, Chou MM, Parrella E, Ge H, Long JM, Allard JS, Lewis K, Miller M, Xu W, Mervis RF, Chen J, Guerin KI, Smith LE, McBurney MW, Sinclair DA, Baudry M, de Cabo R, Longo VD (2010) SIRT1 is essential for normal cognitive function and synaptic plasticity. J Neurosci 30(29):9695–9707. doi:10.1523/JNEUROSCI.0027-10.2010
Julien C, Tremblay C, Emond V, Lebbadi M, Salem N Jr, Bennett DA, Calon F (2009) Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol 68(1):48–58. doi:10.1097/NEN.0b013e3181922348
Lutz MI, Milenkovic I, Regelsberger G, Kovacs GG (2014) Distinct patterns of sirtuin expression during progression of Alzheimer’s disease. Neruomol Med 16(2):405–414. doi:10.1007/s12017-014-8288-8
Marques SC, Lemos R, Ferreiro E, Martins M, de Mendonca A, Santana I, Outeiro TF, Pereira CM (2012) Epigenetic regulation of BACE1 in Alzheimer’s disease patients and in transgenic mice. Neuroscience 220:256–266. doi:10.1016/j.neuroscience.2012.06.029
Torres-Lista V, Parrado-Fernandez C, Alvarez-Monton I, Frontinan-Rubio J, Duran-Prado M, Peinado JR, Johansson B, Alcain FJ, Gimenez-Llort L (2014) Neophobia, NQO1 and SIRT1 as premorbid and prodromal indicators of AD in 3xTg-AD mice. Behav Brain Res 271:140–146. doi:10.1016/j.bbr.2014.04.055
Revilla S, Sunol C, Garcia-Mesa Y, Gimenez-Llort L, Sanfeliu C, Cristofol R (2014) Physical exercise improves synaptic dysfunction and recovers the loss of survival factors in 3xTg-AD mouse brain. Neuropharmacology 81:55–63. doi:10.1016/j.neuropharm.2014.01.037
Rodriguez-Ortiz CJ, Baglietto-Vargas D, Martinez-Coria H, LaFerla FM, Kitazawa M (2014) Upregulation of miR-181 decreases c-Fos and SIRT-1 in the hippocampus of 3xTg-AD mice. J Alzheimers Dis 42(4):1229–1238. doi:10.3233/JAD-140204
Quintas A, de Solis AJ, Diez-Guerra FJ, Carrascosa JM, Bogonez E (2012) Age-associated decrease of SIRT1 expression in rat hippocampus: prevention by late onset caloric restriction. Exp Gerontol 47(2):198–201. doi:10.1016/j.exger.2011.11.010
Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J 26(13):3169–3179. doi:10.1038/sj.emboj.7601758
Qin W, Zhao W, Ho L, Wang J, Walsh K, Gandy S, Pasinetti GM (2008) Regulation of forkhead transcription factor FoxO3a contributes to calorie restriction-induced prevention of Alzheimer’s disease-type amyloid neuropathology and spatial memory deterioration. Ann N Y Acad Sci 1147:335–347. doi:10.1196/annals.1427.024
Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, Serrano M (2010) Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun 1:3. doi:10.1038/ncomms1001
Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, Yamada KA, Imai S (2013) Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab 18(3):416–430. doi:10.1016/j.cmet.2013.07.013
Vilchez D, Saez I, Dillin A (2014) The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun 5:5659. doi:10.1038/ncomms6659
Tomita T, Hamazaki J, Hirayama S, McBurney MW, Yashiroda H, Murata S (2015) Sirt1-deficiency causes defective protein quality control. Sci Rep 5:12613. doi:10.1038/srep12613
Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47:e147. doi:10.1038/emm.2014.117
Himeno E, Ohyagi Y, Ma L, Nakamura N, Miyoshi K, Sakae N, Motomura K, Soejima N, Yamasaki R, Hashimoto T, Tabira T, LaFerla FM, Kira J (2011) Apomorphine treatment in Alzheimer mice promoting amyloid-beta degradation. Ann Neurol 69(2):248–256. doi:10.1002/ana.22319
Wang DS, Dickson DW, Malter JS (2006) Beta-amyloid degradation and Alzheimer’s disease. J Biomed Biotechnol 2006(3):58406. doi:10.1155/JBB/2006/58406
Godoy JA, Zolezzi JM, Braidy N, Inestrosa NC (2014) Role of Sirt1 during the ageing process: relevance to protection of synapses in the brain. Mol Neurobiol 50(3):744–756. doi:10.1007/s12035-014-8645-5
Codocedo JF, Allard C, Godoy JA, Varela-Nallar L, Inestrosa NC (2012) SIRT1 regulates dendritic development in hippocampal neurons. PLoS One 7(10):e47073. doi:10.1371/journal.pone.0047073
Guo W, Qian L, Zhang J, Zhang W, Morrison A, Hayes P, Wilson S, Chen T, Zhao J (2011) Sirt1 overexpression in neurons promotes neurite outgrowth and cell survival through inhibition of the mTOR signaling. J Neurosci Res 89(11):1723–1736. doi:10.1002/jnr.22725
Jeong H, Cohen DE, Cui L, Supinski A, Savas JN, Mazzulli JR, Yates JR 3rd, Bordone L, Guarente L, Krainc D (2012) Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med 18(1):159–165. doi:10.1038/nm.2559
Gao J, Wang WY, Mao YW, Graff J, Guan JS, Pan L, Mak G, Kim D, Su SC, Tsai LH (2010) A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466(7310):1105–1109. doi:10.1038/nature09271
Pertusa M, Garcia-Matas S, Mammeri H, Adell A, Rodrigo T, Mallet J, Cristofol R, Sarkis C, Sanfeliu C (2008) Expression of GDNF transgene in astrocytes improves cognitive deficits in aged rats. Neurobiol Aging 29(9):1366–1379. doi:10.1016/j.neurobiolaging.2007.02.026
Kutner RH, Zhang XY, Reiser J (2009) Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 4(4):495–505. doi:10.1038/nprot.2009.22
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39(3):409–421
Garcia-Mesa Y, Colie S, Corpas R, Cristofol R, Comellas F, Nebreda AR, Gimenez-Llort L, Sanfeliu C (2015) Oxidative stress is a central target for physical exercise neuroprotection against pathological brain aging. J Gerontol A Biol Sci Med Sci. doi:10.1093/gerona/glv005
Garcia-Mesa Y, Lopez-Ramos JC, Gimenez-Llort L, Revilla S, Guerra R, Gruart A, Laferla FM, Cristofol R, Delgado-Garcia JM, Sanfeliu C (2011) Physical exercise protects against Alzheimer’s disease in 3xTg-AD mice. J Alzheimers Dis 24(3):421–454. doi:10.3233/JAD-2011-101635
Caroni P (1997) Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods 71(1):3–9
Smith IF, Hitt B, Green KN, Oddo S, LaFerla FM (2005) Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. J Neurochem 94(6):1711–1718. doi:10.1111/j.1471-4159.2005.03332.x
Frazzini V, Guarnieri S, Bomba M, Navarra R, Morabito C, Mariggio MA, Sensi SL (2016) Altered Kv2.1 functioning promotes increased excitability in hippocampal neurons of an Alzheimer’s disease mouse model. Cell Death Dis 7:e2100. doi:10.1038/cddis.2016.18
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106(34):14670–14675. doi:10.1073/pnas.0903563106
Sensi SL, Rapposelli IG, Frazzini V, Mascetra N (2008) Altered oxidant-mediated intraneuronal zinc mobilization in a triple transgenic mouse model of Alzheimer’s disease. Exp Gerontol 43(5):488–492. doi:10.1016/j.exger.2007.10.018
Vale C, Alonso E, Rubiolo JA, Vieytes MR, LaFerla FM, Gimenez-Llort L, Botana LM (2010) Profile for amyloid-beta and tau expression in primary cortical cultures from 3xTg-AD mice. Cell Mol Neurobiol 30(4):577–590. doi:10.1007/s10571-009-9482-3
Alonso E, Vale C, Vieytes MR, Botana LM (2013) Translocation of PKC by yessotoxin in an in vitro model of Alzheimer’s disease with improvement of tau and beta-amyloid pathology. ACS Chem Neurosci 4(7):1062–1070. doi:10.1021/cn400018y
Alonso E, Vale C, Vieytes MR, Laferla FM, Gimenez-Llort L, Botana LM (2011) 13-Desmethyl spirolide-C is neuroprotective and reduces intracellular Abeta and hyperphosphorylated tau in vitro. Neurochem Int 59(7):1056–1065. doi:10.1016/j.neuint.2011.08.013
Alonso E, Vale C, Vieytes MR, Laferla FM, Gimenez-Llort L, Botana LM (2011) The cholinergic antagonist gymnodimine improves Abeta and tau neuropathology in an in vitro model of Alzheimer disease. Cell Physiol Biochem 27(6):783–794. doi:10.1159/000330086
Gimenez-Llort L, Blazquez G, Canete T, Johansson B, Oddo S, Tobena A, LaFerla FM, Fernandez-Teruel A (2007) Modeling behavioral and neuronal symptoms of Alzheimer’s disease in mice: a role for intraneuronal amyloid. Neurosci Biobehav Rev 31(1):125–147. doi:10.1016/j.neubiorev.2006.07.007
D’Hooge R, De Deyn PP (2001) Applications of the Morris water maze in the study of learning and memory. Brain Res Brain Res Rev 36(1):60–90
Slevin M, Matou S, Zeinolabediny Y, Corpas R, Weston R, Liu D, Boras E, Di Napoli M, Petcu E, Sarroca S, Popa-Wagner A, Love S, Font MA, Potempa LA, Al-Baradie R, Sanfeliu C, Revilla S, Badimon L, Krupinski J (2015) Monomeric C-reactive protein—a key molecule driving development of Alzheimer’s disease associated with brain ischaemia? Sci Rep 5:13281. doi:10.1038/srep13281
Broadbent NJ, Gaskin S, Squire LR, Clark RE (2010) Object recognition memory and the rodent hippocampus. Learn Mem 17(1):5–11. doi:10.1101/lm.1650110
Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch P, Wright SM, Mills KD, Bonni A, Yankner BA, Scully R, Prolla TA, Alt FW, Sinclair DA (2008) SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135(5):907–918. doi:10.1016/j.cell.2008.10.025
Agostinho P, Pliassova A, Oliveira CR, Cunha RA (2015) Localization and trafficking of amyloid-beta protein precursor and secretases: impact on Alzheimer’s disease. J Alzheimers Dis 45(2):329–347. doi:10.3233/JAD-142730
Epis R, Marcello E, Gardoni F, Vastagh C, Malinverno M, Balducci C, Colombo A, Borroni B, Vara H, Dell’Agli M, Cattabeni F, Giustetto M, Borsello T, Forloni G, Padovani A, Di Luca M (2010) Blocking ADAM10 synaptic trafficking generates a model of sporadic Alzheimer’s disease. Brain 133(11):3323–3335. doi:10.1093/brain/awq217
Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 100(7):4162–4167. doi:10.1073/pnas.0230450100
Chu J, Giannopoulos PF, Ceballos-Diaz C, Golde TE, Pratico D (2012) 5-Lipoxygenase gene transfer worsens memory, amyloid, and tau brain pathologies in a mouse model of Alzheimer disease. Ann Neurol 72(3):442–454. doi:10.1002/ana.23642
Stargardt A, Gillis J, Kamphuis W, Wiemhoefer A, Kooijman L, Raspe M, Benckhuijsen W, Drijfhout JW, Hol EM, Reits E (2013) Reduced amyloid-beta degradation in early Alzheimer’s disease but not in the APPswePS1dE9 and 3xTg-AD mouse models. Aging Cell 12(3):499–507. doi:10.1111/acel.12074
Lloret A, Fuchsberger T, Giraldo E, Vina J (2015) Molecular mechanisms linking amyloid beta toxicity and tau hyperphosphorylation in Alzheimers disease. Free Radic Biol Med 83:186–191. doi:10.1016/j.freeradbiomed.2015.02.028
Irwin DJ, Cohen TJ, Grossman M, Arnold SE, Xie SX, Lee VM, Trojanowski JQ (2012) Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 135(Pt 3):807–818. doi:10.1093/brain/aws013
Morris M, Knudsen GM, Maeda S, Trinidad JC, Ioanoviciu A, Burlingame AL, Mucke L (2015) Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci 18(8):1183–1189. doi:10.1038/nn.4067
Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, Sohn PD, Schilling B, Cong X, Ellerby L, Gibson BW, Johnson J, Krogan N, Shamloo M, Gestwicki J, Masliah E, Verdin E, Gan L (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21(10):1154–1162. doi:10.1038/nm.3951
Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, Thompson LM, LaFerla FM (2008) Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J Neurosci 28(45):11500–11510. doi:10.1523/JNEUROSCI.3203-08.2008
Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI (2009) Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323(5917):1063–1066. doi:10.1126/science.1165946
Magrane J, Smith RC, Walsh K, Querfurth HW (2004) Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci 24(7):1700–1706. doi:10.1523/JNEUROSCI.4330-03.2004
Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M (2004) CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet 13(7):703–714. doi:10.1093/hmg/ddh083
Keller JN, Hanni KB, Markesbery WR (2000) Impaired proteasome function in Alzheimer’s disease. J Neurochem 75(1):436–439
Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM (2008) Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging 29(11):1607–1618. doi:10.1016/j.neurobiolaging.2007.04.014
Sampaio-Marques B, Ludovico P (2015) Sirtuins and proteolytic systems: implications for pathogenesis of synucleinopathies. Biomolecules 5(2):735–757. doi:10.3390/biom5020735
Cunha C, Brambilla R, Thomas KL (2010) A simple role for BDNF in learning and memory? Front Mol Neurosci 3:1. doi:10.3389/neuro.02.001.2010
Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M (1997) Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Brain Res Mol Brain Res 49(1–2):71–81
Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15(3):331–337. doi:10.1038/nm.1912
d’Anglemont de Tassigny X, Pascual A, Lopez-Barneo J (2015) GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson’s disease. Front Neuroanat 9:10. doi:10.3389/fnana.2015.00010
Revilla S, Ursulet S, Alvarez-Lopez MJ, Castro-Freire M, Perpina U, Garcia-Mesa Y, Bortolozzi A, Gimenez-Llort L, Kaliman P, Cristofol R, Sarkis C, Sanfeliu C (2014) Lenti-GDNF gene therapy protects against Alzheimer’s disease-like neuropathology in 3xTg-AD mice and MC65 cells. CNS Neurosci Ther 20(11):961–972. doi:10.1111/cns.12312
Religa P, Cao R, Religa D, Xue Y, Bogdanovic N, Westaway D, Marti HH, Winblad B, Cao Y (2013) VEGF significantly restores impaired memory behavior in Alzheimer’s mice by improvement of vascular survival. Sci Rep 3:2053. doi:10.1038/srep02053
Silvennoinen M, Ahtiainen JP, Hulmi JJ, Pekkala S, Taipale RS, Nindl BC, Laine T, Hakkinen K, Selanne H, Kyrolainen H, Kainulainen H (2015) PGC-1 isoforms and their target genes are expressed differently in human skeletal muscle following resistance and endurance exercise. Physiol Rep 3(10). doi:10.14814/phy2.12563
Reed SM, Quelle DE (2014) p53 acetylation: regulation and consequences. Cancers (Basel) 7(1):30–69. doi:10.3390/cancers7010030
Cai Y, Xu L, Xu H, Fan X (2016) SIRT1 and neural cell fate determination. Mol Neurobiol 53(5):2815–2825. doi:10.1007/s12035-015-9158-6
Lima LC, Saliba SW, Andrade JM, Cunha ML, Cassini-Vieira P, Feltenberger JD, Barcelos LS, Guimaraes AL, de-Paula AM, de Oliveira AC, Santos SH (2016) Neurodegeneration alters metabolic profile and Sirt 1 signaling in high-fat-induced obese mice. Mol Neurobiol. doi:10.1007/s12035-016-9927-x
Michan S (2014) Calorie restriction and NAD(+)/sirtuin counteract the hallmarks of aging. Front Biosci (Landmark Ed) 19:1300–1319
Suwa M, Sakuma K (2013) The potential role of sirtuins regarding the effects of exercise on aging- related diseases. Curr Aging Sci 6(2):178–188
Porquet D, Grinan-Ferre C, Ferrer I, Camins A, Sanfeliu C, Del Valle J, Pallas M (2014) Neuroprotective role of trans-resveratrol in a murine model of familial Alzheimer’s disease. J Alzheimers Dis 42(4):1209–1220. doi:10.3233/JAD-140444
Pallas M, Porquet D, Vicente A, Sanfeliu C (2013) Resveratrol: new avenues for a natural compound in neuroprotection. Curr Pharm Des 19(38):6726–6731
Cox EP, O’Dwyer N, Cook R, Vetter M, Cheng HL, Rooney K, O’Connor H (2016) Relationship between physical activity and cognitive function in apparently healthy young to middle-aged adults: a systematic review. J Sci Med Sport 19(8):616–628. doi:10.1016/j.jsams.2015.09.003
Griffin EW, Mullally S, Foley C, Warmington SA, O’Mara SM, Kelly AM (2011) Aerobic exercise improves hippocampal function and increases BDNF in the serum of young adult males. Physiol Behav 104(5):934–941. doi:10.1016/j.physbeh.2011.06.005
Witte AV, Fobker M, Gellner R, Knecht S, Floel A (2009) Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A 106(4):1255–1260. doi:10.1073/pnas.0808587106
Witte AV, Kerti L, Margulies DS, Floel A (2014) Effects of resveratrol on memory performance, hippocampal functional connectivity, and glucose metabolism in healthy older adults. J Neurosci 34(23):7862–7870. doi:10.1523/JNEUROSCI.0385-14.2014
Hubbard BP, Sinclair DA (2014) Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci 35(3):146–154. doi:10.1016/j.tips.2013.12.004
Mercken EM, Mitchell SJ, Martin-Montalvo A, Minor RK, Almeida M, Gomes AP, Scheibye-Knudsen M, Palacios HH, Licata JJ, Zhang Y, Becker KG, Khraiwesh H, Gonzalez-Reyes JA, Villalba JM, Baur JA, Elliott P, Westphal C, Vlasuk GP, Ellis JL, Sinclair DA, Bernier M, de Cabo R (2014) SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell 13(5):787–796. doi:10.1111/acel.12220
Acknowledgments
This study was supported by Grants CSD2010-00045 and SAF2012-39852 from the Spanish Ministry of Economy and Competitiveness (MINECO) and the European Regional Development Fund (ERDF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Corpas, R., Revilla, S., Ursulet, S. et al. SIRT1 Overexpression in Mouse Hippocampus Induces Cognitive Enhancement Through Proteostatic and Neurotrophic Mechanisms. Mol Neurobiol 54, 5604–5619 (2017). https://doi.org/10.1007/s12035-016-0087-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0087-9