
Abstract
RTP801 expression is induced by cellular stress and has a pro-apoptotic function in non-proliferating differentiated cells such as neurons. In several neurodegenerative disorders, including Parkinson’s disease and Alzheimer’s disease, elevated levels of RTP801 have been observed, which suggests a role for RTP801 in neuronal death. Neuronal death is also a pathological hallmark in Huntington’s disease (HD), an inherited neurodegenerative disorder caused by a CAG repeat expansion in the huntingtin gene. Currently, the exact mechanisms underlying mutant huntingtin (mhtt)-induced toxicity are still unclear. Here, we investigated whether RTP801 is involved in (mhtt)-induced cell death. Ectopic exon-1 mhtt elevated RTP801 mRNA and protein levels in nerve growth factor (NGF)-differentiated PC12 cells and in rat primary cortical neurons. In neuronal PC12 cells, mhtt also contributed to RTP801 protein elevation by reducing its proteasomal degradation rate, in addition to promoting RTP801 gene expression. Interestingly, silencing RTP801 expression with short hairpin RNAs (shRNAs) blocked mhtt-induced cell death in NGF-differentiated PC12 cells. However, RTP801 protein levels were not altered in the striatum of HdhQ7/Q111 and R6/1 mice, two HD models that display motor deficits but not neuronal death. Importantly, RTP801 protein levels were elevated in both neural telencephalic progenitors differentiated from HD patient-derived induced pluripotent stem cells and in the putamen and cerebellum of human HD postmortem brains. Taken together, our results suggest that RTP801 is a novel downstream effector of mhtt-induced toxicity and that it may be relevant to the human disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
RTP801/REDD1 protein, encoded by the DDIT4 gene, was first identified on the basis of its induction by hypoxia [1] and DNA damage [2]. Other stressors induced its expression such as dexamethasone, thapsigargin, tunicamycin, heat shock in murine T cell lymphoma cells [3], or cigarette smoke in lung cells [4].
In the central nervous system, RTP801 expression is increased in response to ischemia, β-amyloid peptide [5, 6], 6-hydroxydopamine (6-OHDA) [7, 8], and 1-methy-4-phenyl-1,2,3,6-tetrahydropyridine [8]. Importantly, RTP801 not only accumulates in cellular and animal toxic models but also in samples from patients suffering from neurodegenerative diseases. RTP801 protein levels are elevated in dopaminergic neurons from the substantia nigra in sporadic and mutant parkin Parkinson’s disease (PD) patients [8, 9] and in lymphocytes from Alzheimer’s disease (AD) patients [10].
RTP801 is sufficient to induce cell death in nerve growth factor (NGF)-differentiated PC12 cells [1, 8] and sympathetic neurons [8]. RTP801 inactivates sequentially mechanistic target of rapamycin (mTOR) and Akt survival kinases via Tuberous Sclerosis Complex proteins 1 and 2 [8, 11]. As a consequence, the neuronal survival kinase Akt, which is also a substrate of mTOR, cannot be phosphorylated at residue Ser473, and so, it is unable to enhance pro-survival signals thereby triggering neuronal death [12].
The activity of mTOR has been studied in cellular and mouse models of neurodegenerative diseases, as its regulation also controls autophagy and, as a consequence, the clearance of unfolded proteins and protein aggregates [13, 14]. Huntington’s disease (HD) is one of the neurodegenerative diseases in which inhibition of mTOR with rapamycin or rapalogs has been suggested to be beneficial [15, 16]. HD is caused by a dominantly inherited expansion of a CAG repeat (≥37 repeats) in the huntingtin (htt) gene that generates an aberrant protein [17]. In spite of the ubiquitous expression pattern of htt, the most vulnerable brain regions are the striatum [18] and the motor cortex [19]. However, mutant htt (mhtt) toxicity also extends to other brain structures such as the hippocampus [20] and the cerebellum where atrophy and cell death have been shown recently [21]. Neural dysfunction in these brain areas leads to motor, cognitive, and psychiatric symptoms [22].
Here, we investigated whether RTP801 mediates mhtt-induced toxicity. Ectopic exon-1 mhtt upregulated RTP801 protein levels, both in NGF-differentiated PC12 cells and in primary cortical neurons, by increasing RTP801 gene expression and by decreasing its degradation. RTP801 accumulation was also observed in brain samples and HD-induced pluripotent stem cells (iPSC)-derived telencephalic progenitors. Interestingly, RTP801 knockdown prevents mhtt-induced cell death. Therefore, blockade of RTP801 emerges as a new possible therapeutic target to counteract cell death in HD.
Materials and Methods
Antibodies, Plasmids, and Materials
Rabbit polyclonal anti-RTP801 antibody was purchased from Proteintech Group Inc., Chicago, IL, USA. We used different lot numbers of this antibody with one of them detecting an unspecific band below RTP801 band in the Western blots. Whenever this band appears, the RTP801-specific band is always indicated with an arrow in the figures. The mouse monoclonal antibody against green fluorescent protein (GFP) was obtained from Santa Cruz Biotechnology. Anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was purchased from Merck Millipore. Anti-α-actin antibody was purchased from MP Biomedicals. Goat anti-mouse and anti-rabbit secondary antibodies conjugated to horseradish peroxidase were obtained from Pierce Thermo Scientific. Goat anti-mouse and anti-rabbit secondary antibodies conjugated with Alexa 488 or Alexa 568 were purchased from Life Technologies.
The shRTP801 1 and 4 and scrambled shCT constructs were generated as previously described [8, 23]. The specificity and the effectiveness of shRTP801 1 and 4 were previously proved in cellular models [8, 9, 12] and in vivo, in utero rat brain electroporations [23]. The constructs Q25, Q72, and Q103 were a kind gift of Dr. G.M. Lawless (Cure HD Initiative, Reagent Resource Bank of the Hereditary Disease Foundation, New York, NY). All constructs were verified by DNA sequencing. Actinomycin D was purchased from Gibco, cycloheximide was purchased from Merck Millipore, D-2-Amino-5-phosphonovaleric acid (APV) was obtained from Sigma-Aldrich, and CNQX and NGF were from Alomone Labs.
Cell Culture and Transfection
PC12 cells were cultured and differentiated with NGF as described previously [24]. For NGF treatment, cells were grown in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) and supplemented with 1 % heat-inactivated horse serum (Sigma-Aldrich, St. Louis, MO, USA), penicillin/streptomycin (Gibco Life Technologies, Grand Island, NY, USA), and 50 ng/ml recombinant human β-NGF (Alomone Labs, Jerusalem, Israel) for 7–8 days, in a 7.5 % CO2 atmosphere at 37 °C. Medium was changed every other day and before transfection. Neuronal PC12 cells were transfected at day 5 of NGF exposure, with Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. Media were replaced 4 h later with serum-supplemented RPMI media.
Rat primary cortical cultures were prepared as previously described [25]. Briefly, neurons from embryonic (E18) Sprague–Dawley rat cortex were dissociated in 0.05 % trypsin and plated at a density of 250 cells/mm2 on poly-l-lysine-coated coverslips and maintained in neurobasal medium with B27 and 2 mM GlutaMAX (all from Gibco). After 19 days in vitro (DIV 19), cultured neurons in coverslips were transfected with Lipofectamine 2000 in neurobasal medium supplemented with B27, 2 mM GlutaMAX, 50 μM APV (Sigma-Aldrich) and 10 μM CNQX (Alomone Labs). Sixty minutes later, the coverslips were transferred back into the original neurobasal medium for further 2 days.
HD Mouse Models
For this study, we used male R6/1 transgenic mice (B6CBA background) expressing exon-1 of mhtt containing 145 repeats [26, 27] and their corresponding WT littermates and heterozygous mutant HdhQ7/Q111 and wild-type HdhQ7/Q7 knock-in mice (C57BL/6 background) [28]. Mouse genotype and CAG repeat length were determined as described elsewhere [28, 29]. Mice were housed together in numerical birth order in groups of mixed genotypes, and data were recorded for analysis by microchip mouse number. The animals were housed with access to food and water ad libitum in a colony room kept at 19–22 °C and 40–60 % humidity, under a 12:12-h light/dark cycle. Experiments were carried out according to European regulation (2010/63/UE) for the care and use of laboratory animals.
Ventral Telencephalic Differentiation of Human-Induced Pluripotent Stem Cells
Two non-integrating human iPSC lines CS83iCTR33n1 and CS21iHD60n5 obtained from iPSCs-Core from CEDARS-Sinai (Los Angeles, CA, USA) were differentiated until 12 DIV. hiPSCs were cultured on Matrigel-coated plates (BD Biosciences) in mTeSR1 medium following the manufacturers’ protocols (Stem Cell Technologies). For neural induction, hPSCs grown to 70 % confluency were washed three times with phosphate-buffered saline (PBS) and cultured in SLI neural induction medium (advanced DMEM/F12, 2 mM l-glutamine, 1 % penicillin/streptomycin (Life Technologies), 10 μM SB431542 (Abcam), 1 μM LDN 193189 (Stemgent), 1.5 μM IWR1 (Tocris), 2 % NeuroBrew-21 without RA (Miltenyi Biotec)). On day 4, confluent cultures were treated with 10 μM Y-27632 (Abcam) for 1 h prior to passaging with Accutase (Life Technologies) onto fresh Matrigel-coated plates, with a split ratio of 1:2. On day 8, cultures were passaged 1:2 and cultured in LI medium (SLI without SB431542). At day 12, human iPSC-derived telencephalic progenitors’ total protein was extracted from the cultures using TRI Reagent (Sigma-Aldrich) as described elsewhere.
Post-Mortem Brain Tissue
Samples of putamen, motor cortex, hippocampus, and cerebellum from control subjects and HD patients were obtained from the Neurological Tissue Bank of the Biobanc-Hospital Clinic-IDIBAPS, following the guidelines of the local ethics committees (for details, see Table 1). Post-mortem tissue was homogenized as described previously [30].
Immunocytochemistry
Cells were fixed in 4 % paraformaldehyde for 15 min at room temperature. After washing with PBS, cells were incubated for 1 h at room temperature with the blocking solution Superblock-PBS (Life Technologies, Carlsbad, CA, USA) plus 0.3 % Triton X-100 and then incubated overnight with the primary antibody diluted in PBS. The following primary antibodies were used: rabbit polyclonal anti-RTP801 antibody (1:80) and mouse monoclonal anti-GFP (1:1,000; Santa Cruz Biotechnology, Dallas, TX, USA). The cells were then washed with PBS and incubated for 2 h with the corresponding secondary antibody (1:500 for goat anti-rabbit conjugated with Alexa 568 and 1:1,000 for goat anti-mouse conjugated with Alexa 488; Life Technologies), diluted in PBS, and co-stained with Hoechst 33342 (1:5,000; Invitrogen Life Technologies) for nuclear staining. After washing in PBS, cells were mounted with mounting medium Prolong Gold Antifade Mountant (Invitrogen Life Technologies). In survival assays, eGFP-positive viable cells were scored by strip-counting, as described previously [8]. Highly stained cells for RTP801 were scored with the help of ImageJ software by setting a common color threshold in each image. Only the brightest cells that overcame this threshold were counted as positive.
Western Blot
Whole cell extracts were collected and processed as described previously [8]. Animals were anesthetized and killed by decapitation at different ages. Brains were removed quickly, and the striata were dissected out and homogenized in lysis buffer. Protein extraction and Western blot analyses were performed as described elsewhere [8, 31]. The following primary antibodies were used: anti-RTP801 (1:1,000, Proteintech Group Inc.) and rabbit polyclonal anti-living colors (1:1,000; Clontech Laboratories Inc., Mountain View, CA, USA). Loading control was obtained by incubation with anti- α-actin (1:20000; MP Biomedicals, Santa Ana, CA, USA) or anti-GAPDH (1:1000; Merck Millipore; Darmstadt, Germany) antibodies. Goat anti-mouse or anti-rabbit secondary antibodies conjugated to horseradish peroxidase were obtained from Pierce Thermo Fisher Scientific (Rockford, IL, USA). Chemiluminescent images were acquired using a LAS-3000 imager (Fuji) and quantified by computer-assisted densitometric analysis (ImageJ).
Quantitative Reverse Transcription-PCR
Total RNA was isolated from NGF-differentiated PC12 cells using the High Pure RNA Isolation Kit (Roche Diagnostics Corporation, Indianapolis, IN, USA). Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics Corporation) was used to reverse transcribe cDNA from total RNA. Specific primers for quantitative PCR in amplification were used as follows: RTP801 forward primer, 5′-GCTCTGGACCCCAGTCTAGT-3′; RTP801 reverse primer, 5′-GGGACAGTCCTTCAGTCCTT-3′; α-actin forward primer, 5′-GGGTATGGGTCAGAAGGACT-3′; and α-actin reverse primer, 5′-GAGGCATACAGGGACAACAC-3′. Total RNA extraction from 12-week-old wild-type and R6/1 mice striatal samples and cDNA synthesis were performed as described elsewhere [31]. Specific mouse RTP801 primers were used as follows: forward primer, 5′-ACCTGTGTGCCAACCTGAT-3′; reverse primer, 5′-TAACAGCCCCTGGATCTTG-3′. Quantitative PCR was performed with a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using equal amounts of cDNA template, normalized by α-actin. The RT-PCR data were analyzed and quantified using the comparative quantification.
RTP801 Protein Half-Life
Neuronal PC12 cells were transfected with eGFP, Q25, Q72, or Q103 constructs. Twenty-four hours later, cell cultures were treated with 1 μM of cycloheximide (Calbiochem Merck Millipore, Darmstadt, Germany) for 10 or 60 min. Subsequently, cells were harvested and subjected to Western blot. Membranes were probed for RTP801 or eGFP and reprobed for α-actin as a loading control. The half-life of RTP801 was calculated by fitting the curve to a one-phase decay type exponential equation (GraphPad Prism).
RTP801 mRNA Half-Life
NGF-differentiated PC12 cells were transfected with eGFP, Q25, Q72, or Q103 constructs. Twenty-four hours later, cell cultures were treated with 3 μg/μl of actinomycin D (Gibco) for 10 or 60 min. Subsequently, total RNA was isolated and reverse transcribed to cDNA, and quantitative PCR was performed as described above. The half-life of the mRNA was calculated by fitting the curve to a one-phase decay type exponential equation (GraphPad Prism).
Statistics
All experiments were performed at least in triplicate, and results are reported as the mean ± SEM. Statistical analyses were performed by using the unpaired Student’s t test (95 % confidence) or one-way ANOVA with Dunnett’s multiple comparison test as a post hoc for the comparison of multiple groups, as appropriate and indicated in the figure legends. Values of P < 0.05 were considered as statistically significant.
Results
Ectopic mhtt Increases RTP801 Protein Levels in Neural Cells and Induces Cell Death
RTP801 is upregulated in neural cells as a response to different types of toxic stimuli [5, 6, 8, 32]. Thus, we asked whether mhtt could also upregulate RTP801 levels. To this end, NGF-differentiated PC12 cells were transfected with eGFP alone (empty vector) or with exon-1-encoded N-terminal htt with 25 (Q25, non-toxic form), 72, or 103 (Q72 and Q103, toxic forms) CAG repeats fused to eGFP. Twenty-four hours after transfection, RTP801 and ectopic htt protein levels were analyzed by Western blot (WB). We detected the protein product of all three forms of htt by WB and, as expected, Q72 and Q103 were found in both the soluble and insoluble fractions. We also observed insoluble Q72 and Q103 retained in the stacking gel (Fig. 1a). We observed that RTP801 protein levels were increased by about 70 % in cells overexpressing Q72 or Q103 in comparison to those cells transfected with the control eGFP- or Q25-expressing plasmids (Fig. 1a). By immunofluorescence, we confirmed that Q72- and Q103-overexpressing cells showed higher levels of RTP801 (Fig. 1b1, b2), and that in many of these cells, RTP801 formed aggregates that often co-localized with mhtt aggregates (Fig. 1b1). Interestingly, around 35 % of mhtt-transfected cells that were highly stained for RTP801 also displayed pyknotic nuclei (Fig. 1b3). Similar results were obtained in rat primary cortical neurons that were transfected with mhtt (Fig. 1c1–c3).

RTP801 protein levels are increased in cells overexpressing mhtt. (a) Extracts from transfected neuronal PC12 cells with eGFP, Q25, Q72, or Q103 constructs for 24 h were subjected to Western immnunoblotting. Membranes of the soluble and insoluble fractions were probed with antibodies against eGFP and RTP801 and then re-probed with an anti-α-actin antibody as a loading control. Representative immunoblots are shown along with densitometry analysis for RTP801 signals from at least three independent experiments. Immunostaining of neuronal PC12 cells (transfected cells, marked with white arrows) (b1) or cortical neurons (c1) reveals that RTP801 (in red) is increased in cells transfected with Q72 or Q103 fused to eGFP (in green). Nuclei were stained with Hoechst 33342 (in blue). Scale bar, 5 μm. Proportions of transfected neuronal PC12 cells (eGFP+) (b2) or cortical neurons (eGFP+) (c2) highly stained for RTP801 were scored under fluorescence microscopy for each condition. (b3 and c3) Proportions of transfected cells (eGFP+) highly positive for RTP801 and with pyknotic nuclei were also scored for each condition. Values represent mean ± SEM for at least three independent experiments performed in triplicate. Data was analyzed by one-way ANOVA with Dunnett’s multiple comparison test (**P < 0.01 and ***P < 0.001 vs eGFP; # P < 0.05, ## P < 0.01, and ### P < 0.001 vs Q25)
Both RTP801 mRNA Levels and Protein Degradation Are Altered in Cells Overexpressing mhtt
Next, we explored the mechanisms by which mhtt induces RTP801 protein accumulation. We first analyzed RTP801 mRNA levels in NGF-differentiated PC12 cells transfected with eGFP, Q25, Q72, or Q103 mhtt constructs to assess whether mhtt induces RTP801 expression at the transcriptional level. Pathogenic Q72- or Q103-overexpressing cells displayed a threefold increase in RTP801 mRNA levels compared to cells transfected with eGFP- or Q25-expressing plasmids (Fig. 2a).

mhtt alters both RTP801 mRNA levels and protein degradation rate. a mhtt regulates RTP801 transcriptionally. NGF-differentiated PC12 cells were transfected with eGFP, Q25, Q72, or Q103 constructs. RNA was extracted 24 h post-transfection, and samples were analyzed by reverse transcription-qPCR to quantify RTP801 mRNA under the indicated conditions. Values represent mean ± SEM of at least three independent experiments. b mhtt does not alter RTP801 mRNA half-life. NGF-differentiated PC12 cells were transfected with eGFP, Q25, Q72, or Q103 constructs. Actinomycin D was added to the media 24 h post-transfection for 10 or 60 min. RNA was extracted, and reverse transcription-qPCR was performed to quantify RTP801 mRNA under the indicated conditions. RTP801 mRNA half-life (min) was calculated and expressed as the mean ± SEM of three independent experiments performed in triplicate (n.s., not significant). c mhtt alters RTP801 protein half-life. NGF-differentiated PC12 cells transfected with eGFP, Q25, Q72, or Q103 constructs for 24 h were treated with cycloheximide (CHX) for 10 or 60 min. Cell extracts were harvested, and insoluble and soluble protein fractions were subjected to Western blot. Membranes were probed with antibodies against eGFP and RTP801 and with anti- α-actin antibody as a loading control. Representative immunoblots are shown. Note that the RTP801 specific band in the WB is denoted with an arrow. RTP801 half-life (min) was calculated and expressed as the mean ± SEM of three independent experiments performed in triplicate. Data (a, b, and c) were analyzed by one-way ANOVA with Dunnett’s multiple comparison test (*P < 0.05, **P < 0.01 vs eGFP; # P < 0.05, ## P < 0.01 vs Q25)
To investigate whether the increase in RTP801 mRNA levels could result from an upregulation of gene expression or a reduction of the mRNA degradation rate, we analyzed the mRNA half-life in NGF-differentiated PC12 cells transfected with eGFP, Q25, Q72, or Q103 constructs. Twenty-four hours after transfection, actinomycin D, an inhibitor of RNA synthesis, was added to the culture medium for 10 or 60 min. The cells were subsequently harvested and the RNA was isolated. RTP801 transcripts were analyzed by reverse transcriptase-qPCR. As shown in Fig. 2b, mhtt did not alter the half-life of RTP801 mRNA.
RTP801 degradation mostly depends on the proteasome, as shown by its short half-life of 5–7 min [33, 34]. Thus, we next assessed whether mhtt, apart from inducing RTP801 gene expression, could impair RTP801 protein degradation. To this end, NGF-differentiated PC12 cells were transfected with eGFP-, Q25-, Q72-, or Q103-expressing plasmids. Twenty-four hours later, cultures were treated with cycloheximide, a protein synthesis inhibitor, for 10 or 60 min. Relative RTP801 protein levels were assessed by WB, and the data were used to calculate the protein degradation rate. We observed that ectopic mhtt (Q72 or Q103) increased the RTP801 protein half-life by 4 and 10 min, respectively (Fig. 2c). Taken together, our results show that mhtt elevates RTP801 protein levels by increasing RTP801 mRNA levels and impairing RTP801 proteasomal degradation.
RTP801 Upregulation Mediates Death in Neuronal PC12 Cells Overexpressing mhtt
RTP801 is sufficient and necessary to cause cell death in cellular models of PD [1, 8]. Hence, we next explored whether RTP801 is involved in mhtt-induced cell death. To explore whether RTP801 elevation mediated mhtt-induced toxicity, we co-transfected NGF-differentiated PC12 cells with control Q25 htt or pathogenic mhtt Q72 and shRNAs to knock down RTP801 expression. We tested two different nucleotide sequences (shRTP801 1 and shRTP801 4) to discard off-target effects. Forty-eight hours after transfection, we analyzed RTP801 protein levels by Western blot. RTP801 knock down was about 20–30 % in cells transfected with RTP801 shRNAs compared to those transfected with the scrambled shRNA (a 3A). Interestingly, when mhtt-induced RTP801 accumulation was abrogated with the shRNAs, mhtt-induced cell death was significantly prevented (Fig. 3b). These results thus highlight an important contribution of RTP801 to mhtt-induced toxicity.

RTP801 mediates mhtt-induced toxicity. a Specific shRNAs against RTP801 abrogated mhtt-induced RTP801 expression. NGF-differentiated PC12 cells were co-transfected with eGFP, Q25, or Q72 and either pCMS eGFP-shG (scrambled shRNA as a control), pCMS eGFP-shRTP801_1, or pCMS eGFP-shRTP801_4. Two days later, RTP801 protein levels were analyzed by Western blot. Representative immunoblots for RTP801 and α-actin (loading control) are shown in the lower panel. The upper panel shows a graph with the values of densitometry analysis as the mean ± SEM of three independent experiments. Note that the RTP801-specific band in the WB is denoted with an arrow. b Under the same transfection conditions, cell survival (eGFP+ cells) was scored using fluorescence microscopy. The graph shows the number of surviving cells in each condition. Values represent the mean ± SEM of three independent experiments. Data were analyzed using one-way ANOVA with Dunnett’s multiple comparison test (*P < 0.05, **P < 0.01 vs eGFP-shG; # P < 0.05, ## P < 0.01 vs Q25 shG; +P < 0.05, ++ P < 0.01, and +++ P < 0.001 vs Q72 shG)
RTP801 Protein Levels Are Not Altered in the Striatum of HD Mouse Models
Next, we analyzed whether RTP801 levels were altered in the striatum of two different HD mouse models: the R6/1 mouse that overexpresses human exon-1 mhtt and the knock-in model HdhQ7/Q111 that expresses wild-type and full-length mhtt. We observed that RTP801 mRNA levels did not vary in 12-week-old R6/1 mice in comparison to their wild-type littermates (WT = 1.000 ± 0.0977, n = 7; R6/1 = 1.002 ± 0.1619, n = 7; P = 0.9917, Student’s t test). RTP801 protein levels were analyzed by WB at different stages of the disease. Interestingly, we did not detect changes in RTP801 protein levels in the striatum of either R6/1 or HdhQ7/Q111 mice at any stage when compared with the corresponding wild-type littermates (Fig. 4a, b).

RTP801 does not accumulate in the striatum of HD mouse models. a Striata from WT and R6/1 mice at different stages of disease progression (from 8 to 30 weeks (w) of age) were subjected to SDS-PAGE and Western blot. Membranes were probed with antibodies against RTP801 and α-actin as a loading control. The left panel shows densitometric analysis with the values represented as the mean ± SEM of six animals per condition. b Striata from 7- and 13-month-old Hdh Q7/Q7 (WT) and HdhQ7/Q111 knock-in mice were subjected to SDS-PAGE and Western blot. Membranes were probed with antibodies against RTP801 and α-actin as a loading control. The left panel shows densitometric analysis with the values represented as the mean ± SEM of five animals per condition. The specific RTP801 band is denoted with an arrow. Data were analyzed using Student’s t test
RTP801 Accumulates in Differentiated HD-iPS Cells and in Human Post-Mortem HD Brains
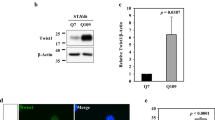
We next examined whether RTP801 protein levels were also increased in human HD-iPSCs and brain tissue. To achieve this, we analyzed RTP801 levels by WB in iPSCs derived from a non-affected individual (expressing htt containing 33 CAG repeats, referred as control; Ctr33) and from an HD patient (expressing mhtt containing 60 CAG repeats, referred as HD60) that were differentiated according to a neuronal differentiation protocol that generates striatal medium-sized spiny neurons. Cells were harvested 12 days after starting the differentiation process, when they display a medial telencephalic identity. As shown in Fig. 5a, mhtt-expressing cells displayed a 37 % increase in RTP801 protein levels compared to control cells.

RTP801 is increased human HD models. a iPSCs expressing control (Ctr33) or mutant (HD60) htt were subjected to Western blot after 12 days of differentiation towards medium spiny neurons. Membranes were probed with antibodies against RTP801 and GAPDH, as a loading control. The graph displays values obtained by densitometric analysis, shown as the mean ± SEM of three independent iPSC differentiations. Student’s t test (*P < 0.05 vs Ctr33). b–e RTP801 protein levels were analyzed by Western blot in protein extracts obtained from b putamen, c cerebellum, d cortex, and e hippocampus of control individuals and HD patients. The graphs display values obtained by densitometric analysis of Western blot data. Note that the specific RTP801 band is indicated with an arrow. Results are shown as the mean ± SEM. Student’s t test. *P < 0.05 and **P < 0.01 vs control
Our previous studies showed that RTP801 protein is highly expressed in degenerating nigral neurons from PD patients [8, 9, 12]. Thus, we examined whether this protein was also increased in the brain tissue of HD patients. We analyzed RTP801 protein levels by WB in protein extracts obtained from the putamen, frontal cortex, hippocampus, and cerebellum of seven control and six HD donors (Table 1). We observed increased levels of RTP801 in the putamen and cerebellum (Fig. 5b, c), whereas no changes were detected in frontal cortex and hippocampus (Fig. 5d, e) when compared with controls. Thus, our results show for the first time that RTP801 protein levels are increased in telencephalic progenitors derived from human HD-induced pluripotent stem cells (iPSCs) and in the striatum and cerebellum of HD patients.
Discussion
This study shows that mhtt regulates the expression of RTP801 in several cellular models of HD. The mechanism by which mhtt elevates RTP801 levels involves increasing gene expression as well as lengthening the half-life of RTP801 protein. Moreover, RTP801 acts as a downstream effector of mhtt toxicity since silencing of RTP801 expression prevents mutant htt-induced cell death. In accordance with these results, we observed increased levels of RTP801 in the striatum and cerebellum of human HD postmortem samples and also in differentiated HD-iPSC cells. However, no alterations of RTP801 protein levels or neuronal death were detected in the striatum of HD mouse models.
Here we show that mhtt induces an increase in RTP801 mRNA and protein levels in cultured cells as has been shown previously to occur in neuronal cells in response to other stressors [5, 6, 8, 32]. This effect was similar in different cell types such as rat NGF-differentiated PC12 cells and rat primary cortical neurons overexpressing exon-1 mhtt with 72 or 103 CAG repeats. Moreover, our results indicate that changes in RTP801 protein and mRNA levels in cellular HD models seem independent of the number of CAG repeats, as similar results were obtained by overexpression of exon-1 mhtt with 72 or 103 CAG repeats.
Many transcription factors regulate RTP801 mRNA levels depending on the stimulus. For example, RTP801 expression is induced by ATF4 via eIF2ɑ in response to endoplasmic reticulum stress, by p53 in response to DNA damage [2], and by HIF-1 and Sp1 in response to hypoxia [35]. Interestingly, activation of endoplasmic reticulum stress [36, 37] and increased p53 and Sp1 protein levels [38–42] have been shown in cellular models of HD and thus could explain the induction of RTP801 mRNA that we have observed. In addition to alterations in gene expression, our results also show that mhtt elevates RTP801 protein levels by impairing its proteasomal degradation in NGF-differentiated PC12 cells, which is in accordance with the impairment of the UPS system detected in HD cellular models [43].
Our results show that overexpression of exon-1 mhtt in NGF-differentiated PC12 cells and rat cortical neurons results in htt aggregate formation and induces cell death, in accordance with previous reports [44–49]. One unexpected observation was that mhtt also induced the appearance of RTP801 aggregates in both neuronal PC12 cells and cortical neurons. Interestingly, RTP801 aggregates did not always co-localize with mhtt aggregates. This mhtt effect could also interfere with RTP801 degradation thereby contributing to RTP801 elevation.
Several mechanisms have been proposed to mediate mhtt-induced cell death including mitochondrial dysfunction, excitotoxicity, and lack of trophic support [50, 51]. Here, we identified RTP801 protein as a new player in mediating mhtt toxicity as demonstrated by the prevention of mhtt-induced cell death by silencing RTP801 expression. Similarly, knockdown of RTP801 expression confers neuroprotection in cellular models of AD [5] or PD [8], and its inhibition protects the brain from ischemic injury [32]. Upregulation of RTP801 also occurs in non-neuronal cells where it has been shown to be an essential mediator of cigarette smoke-induced pulmonary injury and emphysema [4] and to participate in several ocular disorders [52–54]. Interestingly, the administration of a siRNA targeting RTP801 is being used for the treatment of diabetic macular edema with promising results [55]. In addition, RTP801 has been identified recently as a novel negative regulator of Schwann cell myelination [56]. Taken together, these results identify RTP801 as an important mediator of cellular damage.
Importantly, increased levels of RTP801 are not only detected in cellular models of neurodegenerative diseases but also in cell types such as lymphocytes from AD patients [10] and dopaminergic neurons from the substantia nigra of PD patients [8, 9], thus suggesting a role in the pathophysiology of these diseases. Here, we extended this observation to telencephalic progenitors differentiated from HD-iPSCs, which display many of the biological properties found in the human HD brain, although the lack of the brain architecture and interaction with other non-neuronal cells limit the model. However, since these cells are still proliferative, RTP801 does not induce cell death as it occurs in non-dividing mature neurons [1, 8].
We also studied brain regions affected in HD, and we detected elevated levels of RTP801 in the putamen and cerebellum of HD patients. Consistent with a role of RTP801 in mhtt-induced cell death, massive cell death occurs in the putamen of HD patients [19]. Furthermore, the cerebellum displays considerable atrophy, as well as a consistent loss of Purkinje cells and nerve cells of cerebellar nuclei [21]. Degeneration seen in putamen is much higher than in the cerebellum. This could be explained by differential sensitivity to mhtt observed in each brain region that is still not well understood. Interestingly, we did not detect changes in RTP801 protein levels in the striatum of two mouse models of HD, the R6/1 and the HdhQ7/Q111 mice, at any of the ages analyzed. These results are in accordance with a role of RTP801 as a mediator of mhtt-induced cell death since these mice have almost no neuronal death in the striatum [57–62]. Supporting this observation, R6/1 and R6/2 mice displayed less neuronal damage following intrastriatal injection of 6-OHDA [63], a parkinsonian toxin that induces RTP801 and neuronal death [1, 7, 8]. These results suggest that striatal neurons from these models would activate mechanisms to counteract oxidative stress involving the blockade of stress-induced RTP801 protein elevation. In accordance with our results, RTP801/DDIT4 mRNA expression has been shown to be increased in the striatum of HD patients, not altered in R6/1 mice, and decreased in other HD murine models [64]. Therefore, RTP801 protein levels in HD striatum could be the product of both gene regulation and altered degradation.
In summary, mhtt elevates RTP801 by inducing its gene expression and impairing its proteasomal degradation in cellular models of HD. Blockade of RTP801 expression prevents mhtt-induced cell death in cellular models of HD. In addition, vulnerable brain regions that degenerate in HD pathogenesis present increased levels of RTP801. Hence, RTP801 is a novel downstream effector of mhtt that is involved in mediating its toxicity.
References
Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S (2002) Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol 22:2283–2293
Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, Oliner JD, McKeon F et al (2002) REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell 10:995–1005
Wang Z, Malone MH, Thomenius MJ, Zhong F, Xu F, Distelhorst CW (2003) Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. J Biol Chem 278:27053–27058
Yoshida T, Mett I, Bhunia AK, Bowman J, Perez M, Zhang L, Gandjeva A, Zhen L et al (2010) Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nat Med 16:767–773
Kim JR, Lee SR, Chung HJ, Kim S, Baek SH, Kim JH, Kim YS (2003) Identification of amyloid beta-peptide responsive genes by cDNA microarray technology: involvement of RTP801 in amyloid beta-peptide toxicity. Exp Mol Med 35:403–411
Morel M, Couturier J, Pontcharraud R, Gil R, Fauconneau B, Paccalin M, Page G (2009) Evidence of molecular links between PKR and mTOR signalling pathways in Abeta neurotoxicity: role of p53, Redd1 and TSC2. Neurobiol Dis 36:151–161
Ryu EJ, Angelastro JM, Greene LA (2005) Analysis of gene expression changes in a cellular model of Parkinson disease. Neurobiol Dis 18:54–74
Malagelada C, Ryu EJ, Biswas SC, Jackson-Lewis V, Greene LA (2006) RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson's disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci 26:9996–10005
Romani-Aumedes J, Canal M, Martin-Flores N, Sun X, Perez-Fernandez V, Wewering S, Fernandez-Santiago R, Ezquerra M et al (2014) Parkin loss of function contributes to RTP801 elevation and neurodegeneration in Parkinson's disease. Cell Death Dis 5, e1364
Damjanac M, Page G, Ragot S, Laborie G, Gil R, Hugon J, Paccalin M (2009) PKR, a cognitive decline biomarker, can regulate translation via two consecutive molecular targets p53 and Redd1 in lymphocytes of AD patients. J Cell Mol Med 13:1823–1832
Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW et al (2004) Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18:2893–2904
Malagelada C, Jin ZH, Greene LA (2008) RTP801 is induced in Parkinson's disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci 28:14363–14371
Sarkar S (2013) Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans 41:1103–1130
Maiese K, Chong ZZ, Shang YC, Wang S (2013) mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med 19:51–60
Ravikumar B, Rubinsztein DC (2006) Role of autophagy in the clearance of mutant huntingtin: a step towards therapy? Mol Aspects Med 27:520–527
Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC (2009) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 16:46–56
The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44:559–577
Mann DM, Oliver R, Snowden JS (1993) The topographic distribution of brain atrophy in Huntington's disease and progressive supranuclear palsy. Acta Neuropathol 85:553–559
Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Caplan K, Marek K et al (2003) Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology 60:1615–1620
Rub U, Hoche F, Brunt ER, Heinsen H, Seidel K, Del Turco D, Paulson HL, Bohl J et al (2013) Degeneration of the cerebellum in Huntington's disease (HD): possible relevance for the clinical picture and potential gateway to pathological mechanisms of the disease process. Brain Pathol 23:165–177
Martin JB, Gusella JF (1986) Huntington's disease. Pathogenesis and management. N Engl J Med 315:1267–1276
Malagelada C, Lopez-Toledano MA, Willett RT, Jin ZH, Shelanski ML, Greene LA (2011) RTP801/REDD1 regulates the timing of cortical neurogenesis and neuron migration. J Neurosci 31:3186–3196
Greene LA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A 73:2424–2428
Leal-Ortiz S, Waites CL, Terry-Lorenzo R, Zamorano P, Gundelfinger ED, Garner CC (2008) Piccolo modulation of Synapsin1a dynamics regulates synaptic vesicle exocytosis. J Cell Biol 181:831–846
Giralt A, Rodrigo T, Martin ED, Gonzalez JR, Mila M, Cena V, Dierssen M, Canals JM et al (2009) Brain-derived neurotrophic factor modulates the severity of cognitive alterations induced by mutant huntingtin: involvement of phospholipaseCgamma activity and glutamate receptor expression. Neuroscience 158:1234–1250
Rue L, Alcala-Vida R, Lopez-Soop G, Creus-Muncunill J, Alberch J, Perez-Navarro E (2014) Early down-regulation of PKCdelta as a pro-survival mechanism in Huntington's disease. Neuromol Med 16:25–37
Wheeler VC, Auerbach W, White JK, Srinidhi J, Auerbach A, Ryan A, Duyao MP, Vrbanac V et al (1999) Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum Mol Genet 8:115–122
Giralt A, Saavedra A, Carreton O, Xifro X, Alberch J, Perez-Navarro E (2011) Increased PKA signaling disrupts recognition memory and spatial memory: role in Huntington's disease. Hum Mol Genet 20:4232–4247
Saavedra A, Giralt A, Arumi H, Alberch J, Perez-Navarro E (2013) Regulation of hippocampal cGMP levels as a candidate to treat cognitive deficits in Huntington's disease. PLoS One 8, e73664
Saavedra A, Garcia-Martinez JM, Xifro X, Giralt A, Torres-Peraza JF, Canals JM, Diaz-Hernandez M, Lucas JJ et al (2010) PH domain leucine-rich repeat protein phosphatase 1 contributes to maintain the activation of the PI3K/Akt pro-survival pathway in Huntington's disease striatum. Cell Death Differ 17:324–335
Wu XM, Qian ZM, Zhu L, Du F, Yung WH, Gong Q, Ke Y (2011) Neuroprotective effect of ligustilide against ischaemia-reperfusion injury via up-regulation of erythropoietin and down-regulation of RTP801. Br J Pharmacol 164:332–343
Kimball SR, Do AN, Kutzler L, Cavener DR, Jefferson LS (2008) Rapid turnover of the mTOR complex 1 (mTORC1) repressor REDD1 and activation of mTORC1 signaling following inhibition of protein synthesis. J Biol Chem 283:3465–3475
Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA (2010) Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci 30:1166–1175
Jin HO, An S, Lee HC, Woo SH, Seo SK, Choe TB, Yoo DH, Lee SB et al (2007) Hypoxic condition- and high cell density-induced expression of Redd1 is regulated by activation of hypoxia-inducible factor-1alpha and Sp1 through the phosphatidylinositol 3-kinase/Akt signaling pathway. Cell Signal 19:1393–1403
Duennwald ML, Lindquist S (2008) Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev 22:3308–3319
Reijonen S, Putkonen N, Norremolle A, Lindholm D, Korhonen L (2008) Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp Cell Res 314:950–960
Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME (2000) Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 9:2799–2809
Qiu Z, Norflus F, Singh B, Swindell MK, Buzescu R, Bejarano M, Chopra R, Zucker B et al (2006) Sp1 is up-regulated in cellular and transgenic models of Huntington disease, and its reduction is neuroprotective. J Biol Chem 281:16672–16680
Illuzzi J, Yerkes S, Parekh-Olmedo H, Kmiec EB (2009) DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J Neurosci Res 87:733–747
Chae JI, Kim DW, Lee N, Jeon YJ, Jeon I, Kwon J, Kim J, Soh Y et al (2012) Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington's disease patient. Biochem J 446:359–371
HD iPSC Consortium. (2012) Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 11:264–278
Li XJ, Li S (2011) Proteasomal dysfunction in aging and Huntington disease. Neurobiol Dis 43:4–8
Igarashi S, Morita H, Bennett KM, Tanaka Y, Engelender S, Peters MF, Cooper JK, Wood JD et al (2003) Inducible PC12 cell model of Huntington's disease shows toxicity and decreased histone acetylation. Neuroreport 14:565–568
Poirier MA, Jiang H, Ross CA (2005) A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: evidence for a compact beta-sheet structure. Hum Mol Genet 14:765–774
Tagawa K, Marubuchi S, Qi ML, Enokido Y, Tamura T, Inagaki R, Murata M, Kanazawa I et al (2007) The induction levels of heat shock protein 70 differentiate the vulnerabilities to mutant huntingtin among neuronal subtypes. J Neurosci 27:868–880
Bertoni A, Giuliano P, Galgani M, Rotoli D, Ulianich L, Adornetto A, Santillo MR, Porcellini A et al (2011) Early and late events induced by polyQ-expanded proteins: identification of a common pathogenic property of polYQ-expanded proteins. J Biol Chem 286:4727–4741
Scotter EL, Goodfellow CE, Graham ES, Dragunow M, Glass M (2010) Neuroprotective potential of CB1 receptor agonists in an in vitro model of Huntington's disease. Br J Pharmacol 160:747–761
Sontag EM, Lotz GP, Agrawal N, Tran A, Aron R, Yang G, Necula M, Lau A et al (2012) Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington's disease models. J Neurosci 32:11109–11119
Perez-Navarro E, Canals JM, Gines S, Alberch J (2006) Cellular and molecular mechanisms involved in the selective vulnerability of striatal projection neurons in Huntington's disease. Histol Histopathol 21:1217–1232
Cisbani G, Cicchetti F (2012) An in vitro perspective on the molecular mechanisms underlying mutant huntingtin protein toxicity. Cell Death Dis 3, e382
Brafman A, Mett I, Shafir M, Gottlieb H, Damari G, Gozlan-Kelner S, Vishnevskia-Dai V, Skaliter R et al (2004) Inhibition of oxygen-induced retinopathy in RTP801-deficient mice. Invest Ophthalmol Vis Sci 45:3796–3805
del Olmo-Aguado S, Nunez-Alvarez C, Ji D, Manso AG, Osborne NN (2013) RTP801 immunoreactivity in retinal ganglion cells and its down-regulation in cultured cells protect them from light and cobalt chloride. Brain Res Bull 98:132–144
Rittenhouse KD, Johnson TR, Vicini P, Hirakawa B, Kalabat D, Yang AH, Huang W, Basile AS (2014) RTP801 gene expression is differentially upregulated in retinopathy and is silenced by PF-04523655, a 19-Mer siRNA directed against RTP801. Invest Ophthalmol Vis Sci 55:1232–1240
Nguyen QD, Schachar RA, Nduaka CI, Sperling M, Basile AS, Klamerus KJ, Chi-Burris K, Yan E et al (2012) Dose-ranging evaluation of intravitreal siRNA PF-04523655 for diabetic macular edema (the DEGAS study). Invest Ophthalmol Vis Sci 53:7666–7674
Noseda R, Belin S, Piguet F, Vaccari I, Scarlino S, Brambilla P, Martinelli Boneschi F, Feltri ML et al (2013) DDIT4/REDD1/RTP801 is a novel negative regulator of Schwann cell myelination. J Neurosci 33:15295–15305
Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y et al (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87:493–506
Martin-Aparicio E, Yamamoto A, Hernandez F, Hen R, Avila J, Lucas JJ (2001) Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington's disease. J Neurosci 21:8772–8781
Wheeler VC, Gutekunst CA, Vrbanac V, Lebel LA, Schilling G, Hersch S, Friedlander RM, Gusella JF et al (2002) Early phenotypes that presage late-onset neurodegenerative disease allow testing of modifiers in Hdh CAG knock-in mice. Hum Mol Genet 11:633–640
Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martin-Ibanez R, Munoz MT, Mengod G, Ernfors P et al (2004) Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington's disease. J Neurosci 24:7727–7739
Diaz-Hernandez M, Torres-Peraza J, Salvatori-Abarca A, Moran MA, Gomez-Ramos P, Alberch J, Lucas JJ (2005) Full motor recovery despite striatal neuron loss and formation of irreversible amyloid-like inclusions in a conditional mouse model of Huntington's disease. J Neurosci 25:9773–9781
Garcia-Martinez JM, Perez-Navarro E, Xifro X, Canals JM, Diaz-Hernandez M, Trioulier Y, Brouillet E, Lucas JJ et al (2007) BH3-only proteins Bid and Bim(EL) are differentially involved in neuronal dysfunction in mouse models of Huntington's disease. J Neurosci Res 85:2756–2769
Petersen A, Hansson O, Puschban Z, Sapp E, Romero N, Castilho RF, Sulzer D, Rice M et al (2001) Mice transgenic for exon 1 of the Huntington's disease gene display reduced striatal sensitivity to neurotoxicity induced by dopamine and 6-hydroxydopamine. Eur J Neurosci 14:1425–1435
Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA et al (2007) Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet 16:1845–1861
Acknowledgments
The authors thank Dr. M. MacDonald (Massachusetts General Hospital, Boston, Massachusetts, USA) for the HdhQ7/Q111 mice, Neurological Tissue Bank of the Biobanc-Hospital Clinic-IDIBAPS (Barcelona, Spain), and Institute of Neuropathology (Hospital de Bellvitge, L’Hospitalet de Llobregat, Barcelona, Spain) for human tissue samples, Dr. G.M. Lawless (Cure HD Initiative, Reagent Resource Bank of the Hereditary Disease Foundation, New York, NY) for exon-1-mhtt-expressing plasmids and Dr. C. Svendsen (Regenerative Medicine Institute, Cedars-Sinai Medical Center, Los Angeles, CA, USA) for the iPS cells. iPSCs were obtained and characterized in the context of the HD-iPSC Consortium supported by the NINDS and CHDI Foundation, USA. M. MacDonald, C.S., PS, MS, NA, and JMC are members of the HD iPSC consortium. We also thank Ana López, Maria Teresa Muñoz, and Georgina Bombau for technical assistance and Dr. Teresa Rodrigo and the staff of the animal care facility (Facultat de Psicologia, Universitat de Barcelona) for their help. We thank Dr. Sílvia Ginés for helpful discussion. Financial support was obtained from the Ministerio de Economia y Competitividad (grants SAF2010-21058 and SAF2013-45888R to C.M., SAF2012-37417 to J.M.C., and SAF2011-29507 to J.A.), projects integrated in the Plan Nacional de I + D + I y cofinanciado por el ISCIII-Subdirección General de Evaluación y el Fondo Europeo de Desarrollo Regional (FEDER; grants PI13/01250 to E.P.-N. and RETICS RD12/0019/0002 to J.M.C.), the European Commission with a Marie Curie International Reintegration Grant (PIRG08-GA-2010-276957), Spain, CHDI Foundation, USA (grants A-4528 to N.A. and A-7332 to J.M.C.), and funds obtained via the crowdfunding platform Goteo.org, sponsored by “Mememtum: early detection of neurological disorders” and Portal d’Avall SL. to C.M.
Conflict of Interest
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Martín-Flores, N., Romaní-Aumedes, J., Rué, L. et al. RTP801 Is Involved in Mutant Huntingtin-Induced Cell Death. Mol Neurobiol 53, 2857–2868 (2016). https://doi.org/10.1007/s12035-015-9166-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9166-6