
Abstract
First principles calculations of the structural, electronic and optical properties of GaSb, InSb and their ternary \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys (x = 0.25, 0.50 and 0.75) have been performed using the full-potential linear muffin-tin orbital (FP-LAPW) method within density functional theory (DFT). The generalized gradient approximation and modified Becke and Janson functional with local density approximation (mBJ–LDA) are utilized for the treatment of exchange and correlation potentials. The results show the calculated lattice constants increase linearity with increasing the In concentration. The electronic band structure indicates that these alloys are direct band gap semiconductors for all the values of x and the band gap decreases as x increases from x = 0 to x = 1. The results also show that the there is a non-linear dependence of the band gap on composition x in \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys. Regarding optical properties, the real and imaginary parts of the dielectric function have been calculated. The results show that the static dielectric constant increases with increase in concentration of In, consistent with reduction in energy band gap. The onset point and major peaks in the imaginary parts of the dielectric function spectra are identified for the \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys and shown that they are related to the corresponding band gap values. The results also show that for energy values higher than \(\cong 4\) eV, the electromagnetic wave transition through \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys is nearly zero.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The properties of semiconductor materials can be tuned through alloying. Using III–V group compounds and alloys, optoelectronic devices working at different wavelengths can be fabricated. Thus, exploring new materials provide opportunities to improve existing devices or to demonstrate new devices designed for new applications. Like other III–V semiconductor materials, GaSb and InSb have also shown their potential for many applications including electronic and optoelectronic devices due to their direct band gap nature and other fundamental properties. Since the electronic and optical properties of alloys can be changed in a wide range between the corresponding properties of the starting compounds, thus, GaSb and InSb compounds having different physical properties can be combined to obtain new materials, \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) ternary alloys with intermediate properties [1].
\({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloy crystals are widely used as substrate materials to make various optoelectronic devices, such as lasers [2], detectors [3,4], satellite navigations [5] and so on, due to both band gap and lattice parameters can be regulated by the x values in \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys. Thus, Studies of basic properties of these alloys are very important for the understanding of devices characteristics and the improvement of their performance. Motivated by the technological importance of these alloys, in the recent years, some works on \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) crystals for different x values have been published [6,7,8,9]. These previous theoretical and experimental studies limited to growth of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys and notwithstanding the potential device applications of these alloys, to the best of our knowledge, no experimental or theoretical investigations on the electronic and optical properties of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys have been appeared in the literatures. In fact, experimental and theoretical results on the optical and electronic properties were mainly restricted to just InSb and GaSb and not for \({{\text{Ga}}}_{1-{\text{x}}}{{\text{In}}}_{x}{\text{Sb}}\) alloys. Thus, a comprehensive study for a better understanding of the structural, electronic and optical properties of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys for different x values within an accurate method is required. Hence, in the present work, we study the structural, electronic and optical properties of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) (x = 0, 0.25, 0.50, 0.75 and 1) alloys within density functional theory (DFT) [10,11] by utilizing the all electron full potential linearized augmented plane wave (FP-LAPW) technique [12,13]. These calculations can be viewed as a prediction, which can be tested against experiment in the future works.
This paper is organized as follows: in section 2, we give a brief description of computational method. The results of the structural, electronic and optical properties are discussed in section 3. The results are also compared with other existing experimental data and theoretical results. Finally, the summary of main results and conclusion are given in section 4.
2 Computational details
Present theoretical simulations are carried out based on density functional theory DFT [10,11] by utilizing the FP-LAPW [12,13] technique as implemented in WIEN2k package [14,15]. The structural properties were performed using Wu and Cohen (GGA-WC) potential [16]. To overcome the shortcoming of underestimating the energy band gap with GGA approach, we have computed the electronic and optical characteristics of the present materials by applying modified Becke and Janson functional with local density approximation (mBJ–LDA) [17]. The basis function was expanded to \({R}_{{\text{mt}}}\times {K}_{{\text{max}}}=8\), where \({R}_{{\text{mt}}}\) represents the smallest muffin-tin radius of all atomic spheres in the unit cell and \({K}_{{\text{max}}}\) indicates the plane wave cutoff. The Fourier expansion of the charge density was set with a maximum of \({G}_{{\text{max}}}=12\), and the wave functions in the muffin-tin spheres were expanded to \({l}_{{\text{max}}}=10\). The muffin-tin radius was selected in such a way that there was no charge leakage from the core. Besides, 3000 k-points were used to calculate the structural and electronic properties in the irreducible Brillouin zone. Additionally, 10,000 k-points were used for the optical properties because the calculation of optical properties requires a dense mesh of eigenvalues and their corresponding eigenvectors [17].
3 Results and discussion
3.1 Structural properties
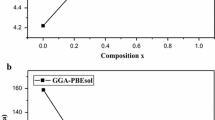
Both GaSb and InSb are semiconductors exhibiting zinc blende structure [18,19]. In this study, \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys (x = 0, 0.25, 0.50, 0.75 and 100) were modelled with ordered structures described in terms of periodically repeated \(1\times 1\times 1\) supercells with eight atoms and then, replacing one by one Ga atom with In atom to get the alloys with 25, 50, 75 and 100% concentrations. Structural optimizations were performed by minimizing total energy with respect to the unit cell volume and the results of total energy vs. unit-cell volume were fitted with Murnaghan’s equation of states [20]. The optimized lattice constants are given in table 1 with corresponding theoretical and experimental data available in the literatures. For the parent compounds, GaSb and InSb, the calculated lattice parameters are in good agreement with experimental data and previous theoretical works. A similar level of accuracy is expected for \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys (x = 0.25, 0.50 and 0.75), where experimental and theoretical data for the specific concentration considered in this study are missing. The calculated lattice constants are plotted as a function of x in figure 1. The lattice constants of these alloys follow closely the Vegard's linear law. As expected, due to larger atomic radius of In compared to Ga, the lattice parameter of these alloys increases with the increase in x. Equation (1) shows the calculated lattice parameters of these alloys that are fitted by the first order polynomials:

Calculated lattice constant vs. composition (x) for GaSb, InSb and \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) (x = 0.25, 0.50 and 0.75) alloys. The solid curve is linear fit to the data points.
To study the stability of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys, the formation energies of these alloys are calculated. The formation energy per formula unit of these alloys as a function of x is defined as:
where \(E\left({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\right)\), \(E\left({\text{GaSb}}\right)\) and \(E\left({\text{InSb}}\right)\) are total energy per formula unit of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys, \({\text{GaSb}}\) and \({\text{InSb}},\) respectively. Using the above definition, the formation energy of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys is calculated. The calculated formation energy of these alloys is given in table 2. The results of these calculations show that the formation energy of these alloys for all values of x is negative. Negative formation energy of these alloys means that \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys are thermodynamically stable.
3.2 Electronic structure
The electronic properties of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys (x = 0, 0.25, 0.50, 0.75 and 1) were investigated by calculating the energy band structure. The electronic band structures of the considered alloys were calculated by following the high symmetry k-points path using mBJ–LDA potential. The results of these calculations are shown in figure 2. The calculated electronic band structures indicate that \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys (x = 0, 0.25, 0.50, 0.75 and 1) demonstrate direct band gap semiconductor nature as the valence band maximum and the conduction band minimum both are located at gamma point, which can be seen in figure 2.

Calculated band structure of (a) GaSb, (b) InSb, (c) \({{\text{Ga}}}_{0.75}{{\text{In}}}_{0.25}{\text{Sb}}\), (d) \({{\text{Ga}}}_{0.50}{{\text{In}}}_{0.50}{\text{Sb}}\), (f) \({{\text{Ga}}}_{0.25}{{\text{In}}}_{0.75}{\text{Sb}}\) alloys using mBJ-LDA functional.
Table 3 presents the calculated band gap (\({E}_{{\text{g}}})\) along with previously reported theoretical and experimental results. For the parent compounds, GaSb and InSb, the calculated energy band gaps are in good agreement with experimental data and previous theoretical works. A similar level of accuracy is expected for \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys (x = 0.25, 0.50 and 0.75), where experimental and theoretical data for the specific concentration considered in this study are missing. Figure 3 shows the variation in band gap with respect to the concentration x. As shown in figure 3, In incorporated into GaSb leads to a dramatic band gap reduction and the band gap value varies in non-linearity with increasing In composition. Equation (2) shows dependence of the obtained band gap on the concentration, x. The results are fitted to a quadratic equation:

Calculated band gap vs. composition (x) for GaSb, InSb and \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) (x = 0.25, 0.50 and 0.75) alloys. The solid curve is quadratic fit to the data points.
3.3 Optical properties
The study of the optical properties of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys is very important because of their potential use in the optoelectronic devices. In this study, the optical properties of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys were calculated using mBJ–LDA potential. The linear response of a material to an external electromagnetic field could be described using the complex dielectric function \(\varepsilon \left(\omega \right)\) [23,24]. The dielectric function \(\varepsilon \left(\omega \right)\) depends on the electronic band structure of a crystal and its investigation via optical spectroscopy is an effective way for the determination of the overall band structure behaviour of a solid [23,24]. It means that the electronic band structure calculations of a solid can be utilized for acquiring the dielectric function and examination of the optical function aids to provide a better elucidation for the electronic band structure. As stated earlier, in this study, the calculations have been done using FP-LAPW method. The procedure for the determination of the linear dielectric function \(\varepsilon \left(\omega \right)\) using the FP-LAPW method is described in detail by Abt et al [25] and Ambrosch-Draxl and Sofo [26]. Complex dielectric function \(\varepsilon \left(\omega \right)\) can be divided into two parts: real part \({\varepsilon }_{1}\left(\omega \right)\) and imaginary part \({\varepsilon }_{2}\left(\omega \right)\) as given in equation (3):
The imaginary part of the complex dielectric function can be calculated using equation (4):
where \(\hslash \omega\) is the energy of incident photon, \(|{k}_{n}>\) are the eigenfunctions with eigenvalues \({E}_{{k}_{n}}\), p is the momentum operator and \(f\left({k}_{n}\right)\) is the Fermi distribution function and the integral is over the first Brillouin zone [26]. The real part of the complex dielectric function can be obtained using the imaginary part as given in equation (5):
where p is the principals of integral. Other optical properties can be calculated by the knowledge of the complex dielectric function [27].The calculated real part, \({\varepsilon }_{1}\left(\omega \right)\) and imaginary part, \({\varepsilon }_{2}\left(\omega \right)\) of complex dielectric function are shown in figures 4 and 5. The \({\varepsilon }_{2}\left(\omega \right)\) values show the interband transition for the semiconductor materials and describe the complete response of materials to disturbances caused by electromagnetic radiation. Furthermore, \({\varepsilon }_{2}\left(\omega \right)\) is directly related to the electronic band structure of materials and describes their absorptive behaviour. The onset point in the \({\varepsilon }_{2}\left(\omega \right)\) spectra, as shown in figure 4, is observed at a point for the \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys, which are related to the corresponding band gap values as given in table 2. The major peaks (b and c) of \({\varepsilon }_{2}\left(\omega \right)\) are also shown in figure 4. The energy values of these peaks are closely related to the corresponding band gap energy values in the calculated electronic band structure.

Calculated imaginary dielectric function, \({\varepsilon }_{2}\left(\omega \right)\) of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys with respect to In content (x) using mBJ–LDA potential.

Calculated real dielectric function,\({\varepsilon }_{1}\left(\omega \right)\) of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys with respect to In content (x) using mBJ–LDA potential.
The most important quantity in the real part of dielectric function is the static dielectric function that is zero frequency limit, \({\varepsilon }_{1}\left(0\right)\). The calculated static dielectric function of GaSb, InSb and \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) (x = 0.25, 0.50 and 0.75) are given in table 4. The values of \({\varepsilon }_{1}\left(0\right)\) for the GaSb and InSb agree well with the result obtained in the previous studies. The comparison of our calculated static dielectric constant of GaSb (\({\varepsilon }_{1}\left(0\right)=12.461\)) and InSb (\({\varepsilon }_{1}\left(0\right)=13.693\)) with room temperature measurements, \({\varepsilon }_{1}\left(0\right)=15.70\) for GaSb [29] and \({\varepsilon }_{1}\left(0\right)=16.8\) for InSb [29] shows that we get similar increase when going from GaSb to InSb, but the values are underestimated. The increase of static dielectric constant from GaSb to InSb is related to the reduction in the band gap and the deviation to the experimental values may be related to the neglect of temperature effects in our theoretical results. For \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) (x = 0.25, 0.50 and 0.75) alloys, experimental results for the static dielectric constants to compare with, are still missing. The calculated static dielectric constants are plotted as a function of x in figure 6. The results of static dielectric constant vs. x composition are fitted to the following equation:

Calculated static dielectric constant vs. composition (x) for GaSb, InSb and \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) (x = 0.25, 0.50 and 0.75) alloys. The solid curve is cubic fit to the data points.
As can be seen from figure 5, for all x values, the \({\varepsilon }_{1}\left(\omega \right)\) decreases in energy \(\cong 4\) eV and becomes negative in the special energy range. Hence, the incident photon is attenuated and the electromagnetic wave transition through \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys is nearly zero in this energy range.
4 Conclusions
In this research, we have performed ab initio DFT calculations using FP-LAPW method to investigate the structural, electronic and optical properties of \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys. We have calculated the concentration dependence of the lattice parameter of considered alloys. A linearity following Vegard’s law with lattice parameter has been shown. In addition, we have computed electronic band structure of considered alloys. The results show that In incorporation into GaSb reduces the band gap dramatically. The results also show that there is a non-linear dependence of the band gap on composition x in \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys. Regarding optical properties, the real and imaginary parts of the dielectric function have been calculated. \({\varepsilon }_{1}\left(0\right)\) increases with an increase in concentration of In, consistent with reduction in energy band gap. The onset point and major peaks in the \({\varepsilon }_{2}\left(\omega \right)\) spectra were investigated for the \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys and shown that they are related to the corresponding band gap values. For all values of x, \({\varepsilon }_{1}\left(\omega \right)\) decreases in energy \(\cong 4\) eV and become negative for higher energy values. Hence, the incident photon is attenuated and the electromagnetic wave transition through \({{\text{Ga}}}_{1-x}{{\text{In}}}_{x}{\text{Sb}}\) alloys is nearly zero in this energy range.
References
Streicher M, Corregidor V, Catarino N, Alves L C, Franco N, Fonseca M et al 2016 Nucl. Instrum. Methods Phys. Res. B 371 278
Sun J, Han M, Gu Y, Yang Z X and Zeng H 2018 Adv. Opt. Mater. 6 1800256
Kimura D, Balasekaran S, Inada H, Iguchi Y and Kimata M 2018 SEI Tech. Rev. 86 41
Paajaste J, Suomalainen S, Koskinen R, Härkönen A, Guina M and Pessa M 2009 J. Cryst. Growth 311 1917
Yang B, Tao G and Li X 2019 United States Patent Application US 16 234
Liu Q, Wang J, He G, Yang D, Zhang W and Liu J 2020 Vacuum 174 109177
Inatomi Y, Sakata K, Arivanandhan M, Rajesh G, Nirmal Kumar V, Koyama T et al 2015 npj Microgravity 1 1
Li D, Lan C, Manikandan A, Yip S, Zhou Z, Liang X et al 2019 Nat. Commun. 10 1664
Gadkari D B 2020 Int. J. Eng. Res. Appl. 10 7
Hohenberg P and Kohn W J 1964 Phys. Rev. 136 B864
Kohn W and Sham L J 1965 Phys. Rev. 140 A1133
Singh D and Nordstorm L 1994 ebook of planwaves, pseudopotential, and the LAPW method (Berlin: Springer)
Mun Wong K, Alay-e-Abbas S M, Fang Y, Shaukat A and Lei Y 2013 J. Appl. Phys. 114 034901
Blaha P, Schwarz K, Madsen G K, Kvasnicka D and Luitz J 2001 Wien2k 60 1
Schwarz K and Blaha P 2003 Solid state calculations using Wien2k Comput. Mater. Sci. 28 259
Wu Z and Cohen R E 2006 Phys. Rev. B 73 235116
Tran F and Blaha P 2009 Phys. Rev. Lett. 102 226401
Adachi S 1985 J. Appl. Phys. 58 R1
Madelung O 2004 Semiconductors: data handbook (Springer)
Murnaghan F D 1944 Proc. Natl. Acad. Sci. 30 244
Gagui S, Ghemid S, Meradji H, Zaidi B, Amimour B, Tahir S A et al 2020 Optik 219 165253
Namjoo S, Rozatian A S, Jabbari I and Puschnig P 2015 Phys. Rev. B 91 205205
Smith D Y, Shiles E, Inokuti M and Palik E 1985 Handbook of optical constants of solids 1 369
Smith N V 1971 Phys. Rev. B 3 1862
Abt R, Ambrosch-Draxl C and Knoll P 1994 Phys. B: Condens. Matter 194 1451
Ambrosch-Draxl C and Sofo J O 2006 Comput. Phys. Commun. 175 1
Wooten F 1973 Am. J. Phys. 41 939
Wooten Frederick 1972 Optical properties of solids (Academic Press)
Dixon J R Jr and Furdyna J K 1980 Solid State Commun. 35 195
Acknowledgement
The authors acknowledge access to HPC resources at University of Hormozgan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Noorafshan, M., Heydari, S. Study of ab initio calculations of structural, electronic and optical properties of ternary semiconductor \({\mathbf{G}\mathbf{a}}_{1-\mathbf{x}}{\mathbf{I}\mathbf{n}}_{\mathbf{x}}\mathbf{S}\mathbf{b}\) alloys. Bull Mater Sci 47, 88 (2024). https://doi.org/10.1007/s12034-024-03177-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12034-024-03177-5