
Abstract
A novel polymer–carbon (PTh–C) nanocomposites containing different percentages of polythiophene (10, 20 and 50% (w/w)) and carbon (Vulcan XC-72) was prepared by a facile solution dispersion method and used to support platinum nanoparticles. The effect of using different percentages of polythiophene in nanocomposites and subsequently prepared electrocatalysts was investigated. The resultant electrocatalysts were extensively characterized by physical (X-ray diffraction (XRD) and transmission electron microscopy (TEM)) and electrochemical (cyclic voltammetry (CV)) techniques. The TEM results showed that the fine Pt nanoparticles prepared by ethylene glycol (EG) method were distributed on the surface of the 50% PTh–C nanocomposites successfully. From the XRD patterns, the average size of dispersed Pt nanoparticles with the face-centered cubic (fcc) structure on 50% PTh–C, 20% PTh–C, 10% PTh–C and carbon were about 4.9, 5.2, 5.4 and 6.1 nm, respectively. The conductivity of PTh–C with different percentages of pure PTh was compared with the conductivity of the corresponding support of pure PTh. It is observed that the conductivity of 50% PTh–C nanocomposites is about 600 times higher than that of pure PTh. Finally, CV measurements of hydrogen and methanol oxidations indicated that Pt/50% PTh–C had a higher electrochemical surface area and higher catalytic activity for methanol oxidation reaction compared to other electrocatalysts. These measurements showed that the Pt/50% PTh–C electrocatalyst by the value of 3.85 had higher \(I_{\mathrm{f}}/I_{\mathrm{b}}\) ratio with respect to Pt/10% PTh–C and Pt/20% PTh–C by the values of 2.66 and 2.0, respectively.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Fuel cells have emerged as promising devices to address future energy needs for point-of-use applications including transport, stationary and portable applications due to their high power density and high energy conversion efficiencies with potentially less release of environmental pollutants, fuel options and fuel delivery options [1,2,3,4]. In particular, proton exchange membrane fuel cells (PEMFCs), including direct methanol fuel cells (DMFCs), are attractive candidates for commercial applications due to their relatively low-temperature operation (\(60{-}100{^{\circ }}\hbox {C}\)) and the quick start-up [5,6,7]. But they still face several key issues preventing the wide commercialization of PEM fuel cells such as their high cost and poor durability. Among the main components of a PEMFC, price of the electrocatalysts dominates the total fuel cell cost and play a critical role in durability of PEMFC materials [2, 8, 9]. Expensive Pt-based noble metal catalysts are the most popular catalysts for use in low-temperature fuel cells, particularly, PEMFC and DMFC, in many studies and commercial applications due to their low overpotential and high catalytic activity for fuel cell reactions, chemical stability in the harsh acidic environment, high exchange current density and superior work function [10, 11]. Electrocatalysis can be defined as the heterogeneous catalysis of electrochemical reactions by an electrode material. Catalysis is a surface phenomenon, therefore, catalysts are generally dispersed on the surface of somewhat larger particles that act as a supporter to maximize the exposed surface areas of the active components [12, 13]. To reduce the cost and improve the performance of a fuel cell, there are several strategies. One popular strategy is to develop suitable supports to achieve high catalyst dispersion, utilization, activity and stability [14,15,16]. A suitable catalyst support should have several important properties, including high electrical conductivity, large surface area, suitable porosity and high stability in acid media under fuel cell operational conditions. This way the platinum (Pt) metal catalysts are dispersed on high surface area supports, so that a very high portion of the surface area will be in contact with the reactant, therefore, the Pt loading amount can be reduced. Additionally, the support materials can also interplay with Pt nanoparticles and can affect the catalytic activity and durability [13, 17,18,19,20,21]. Among the different catalyst supports like carbons, oxides, alumina and silica, carbon materials, in particular, Vulcan XC-72 carbon blacks were the most widely used catalyst support due to their superior properties such as easy availability, low-cost, large surface area, high electrical conductivity, chemical inertness and pore structure. However, there are a few disadvantages regarding the use of carbon blacks as fuel cell catalyst support such as their low resistance to corrosion which causes performance losses in the fuel cell, low gas permeability and proton conductivity leading to low catalyst utilization and entity of many micropores results in a low accessible surface area for the deposition of metal particles [22,23,24,25]. On the other hand, conductive polymers (CPs) such as polyaniline (PANi), polypyrrole (PPy), polythiophene (PTh) and their derivatives such as carbon-substitute supports for fuel cell catalysts have received a considerable attention from researchers due to their suitable features like good electronic and proton conductivity, high surface area, suitable porosity, high stability, nontoxic effects and good resistance to corrosion in fuel cell operating conditions [26, 27]. Right around countless engaging conducting polymers, polythiophene has been quite compelling because of its wide range of electrochemical activity, higher electrical conductivity, improved mechanical properties, elevated charge transporter portability and different suitable headlines. Polythiophene can be viewed as a conjugated chain consisting of \(\hbox {sp}^{2}\hbox {p}_{x}\)-carbon atoms which have an analogous structure to cis-polyacetylenes and are stabilized by sulfur atoms. The relatively high polarizability of the sulfur atoms in the thiophene backbone will also influence the properties of a polythiophene. The lone pair of electrons on the polarized sulfur atom of thiophene allows them to readily bind to heavy metal ions. Thus, sulfur atoms would be the main sites for heavy metal ions [28,29,30,31]. However, on the basis of the histogram displaying the number of papers reporting PAni, PPy and PTh in ref. [27], few reports were studied on the usage of PTh as a catalyst support, whereas most of the reports belong to its derivatives such as poly(3-methylthiophene) (PMT) and poly(3,4-ethylenedioxythiophene) (PEDOT) [32,33,34]. In the vast field of materials science, recently the studies on nanocomposites have drawn great attention because the properties of composite materials can display synergistic improvements with respect to the individual components [17, 35, 36]. Although the presence of sulfur atoms in the polythiophene chains imparts high-affinity towards heavy metal ions, they may cause Pt particle aggregation [37]. So, in the present study, pure polythiophene has not been used as a Pt electrocatalyst support and the composition of nanocomposites varied 10–50% PTh by weight. A motivation behind this study was to synthesize PTh–C nanocomposites as electrocatalyst supports which have higher catalytic activity towards hydrogen and methanol oxidation. Hence, in this study, we prepared a series of polythiophene-carbon nanocomposites with different PTh–carbon weight ratio through a facile solution dispersion method. Furthermore, dispersed Pt nanoparticles on resulting supports were prepared by an ethylene glycol (EG) reduction method. The prepared supporting materials were characterized by Fourier transform infrared (FTIR), scanning electron microscopy (SEM), atomic force microscopy (AFM), X-ray diffraction (XRD) and conductivity measurement. Also, the resulting electrocatalysts were investigated by physical (XRD and transmission electron microscopy (TEM)) and electrochemical (cyclic voltammetry (CV)) measurements.
2 Experimental
2.1 Materials
Materials used in this work were thiophene, ammonium persulfate (APS), triethanolamine (TEA), sodium dodecyl sulfate (SDS), electrocatalyst precursor salts \(\hbox {H}_{2}\hbox {PtCl}_{6}\cdot 6\hbox {H}_{2}\hbox {O}\), methanol, ethanol, n-methyl-3-phenylpiperazine (NMP), dimethylformamide (DMF), acetone, tetrahydrofuran (THF), chloroform (\(\hbox {CHCl}_{3}\)), ethyl acetate, hexane and any other organic solvents from Merck company. All reagents were used without further purification and used as received. Vulcan XC-72 carbon powder from fuel cell earth was heat-treated at \(600{^{\circ }}\hbox {C}\) in air. Deionized (DI) water was employed throughout this work. Thiophene monomer was freshly purified by a simple distillation process for subsequent use.
2.2 Synthesis of PTh–C nanocomposites
The synthesis of PTh–C nanocomposites involves the following steps: At the first step, nanostructured PTh was prepared by chemical oxidative polymerization. 1.296 g SDS and 9.37 ml TEA were mixed with 30 ml of deionized water in a reaction vessel containing a magnetic mixer. These reactant mixtures were dispersed and sonicated using an ultrasonic processor for 1 h until the initial turbid solution became clear. Simultaneously, 8.26 g APS was dissolved in 20 ml deionized water and was added into the clear mixed solution after sonication (the molar ratio of APS to thiophene monomer was 1.14). The preliminary polymerization process was identified by the colour change of the reaction mixture. The polymerization was carried out for 24 h at \(70{^{\circ }}\hbox {C}\). Consequently, the resulting dark brown precipitate was collected by centrifugation and to separate the oligomers and impurities, product was washed several times with deionized water and methanol and dried by freeze dryer for 24 h. At the second step, three nanocomposites of polythiophene–carbon were prepared by a facile solution dispersion method containing 10, 20 and 50 wt% PTh. A calculated amount of freshly prepared PTh and carbon were added into 10 ml DMF. After stirring for 15 min, the resulting dark solution was refluxed at \(140^{\circ }\hbox {C}\) for 2 h. At the same time, a mixture of ice and deionized water was prepared. While the reflux process finished, the resulting solution was poured into the beaker containing ice water. Thereafter, the mixture was heated at \(100{^{\circ }}\hbox {C}\) until the complete evaporation of the solvent. The resulting dark powder was PTh–C nanocomposites.
2.3 Electrocatalyst preparation
The electrocatalyst was prepared by EG (ethylene glycol) method in which ethylene glycol was selected as both the solvent and reducing agent. For the catalyst preparation, 25 ml ethylene glycol containing a calculated amount of \(\hbox {H}_{2}\hbox {PtCl}_{6}\cdot \hbox {H}_{2}\hbox {O}\) (0.025 g) was stirred for 30 min. 0.1 M NaOH was added to adjust the pH of the solution to \(\sim \!\!\!13\), and then, the required amount of freshly prepared supports (0.04 g) was added to the solution under magnetic stirring for 1 h (followed by stirring for 1 h). Thereafter, the resulting suspension was refluxed at \(160{^{\circ }}\hbox {C}\) for 3 h to ensure that Pt was completely reduced from its metal salt. The solution was cooled down to room temperature and magnetically stirred for 12 h. Subsequently, the pH value of the solution was adjusted to 3 by 0.1 M \(\hbox {H}_{2}\hbox {SO}_{4}\), and stirring was continued for 24 h. Next, the resulting black solid phase was filtered and washed copiously with deionized water. Finally, the product was dried in an oven at \(70^{\circ }\hbox {C}\) and catalyst with 20% Pt loading was obtained.
2.4 Preparation of electrodes
First of all, the glassy carbon (GC) electrode (\(d = 2~\hbox {mm}\)) was polished with a \(1.0~\upmu \hbox {m}\), followed by a 0.3 and \(0.05~\upmu \hbox {m}\) alumina slurry and rinsed with deionized water to give it a mirrored finish. Catalyst inks containing 3.0 mg of prepared electrocatalyst, \(10~\upmu \hbox {l}\) of a Nafion solution (5 wt%, fuel cell earth), 1.5 ml isopropanol and deionized water were prepared using an ultrasonic bath for 1 h to obtain homogeneous dispersions. The electrode modified by the catalyst was fabricated by casting about \(5~\upmu \hbox {l}\) of this catalyst ink (loading: \(0.01~\hbox {mg Pt cm}^{-2}\)) onto the whole surface of the GC electrode and was dried at room temperature.
2.5 Physical characterization
Physical–chemical and electrochemical characterizations were carried out on the supporting materials and resultant electrocatalysts. The samples’ chemical structures were investigated by FTIR spectroscopy. Infrared spectra were recorded in the range of \(400{-}4000~\hbox {cm}^{-1}\) with the KBr pellet technique (Shimadzu 4100 spectrometer). The crystal structure of the samples was analysed using powder XRD (Bruker D8-advanced) with \(\hbox {CuK}\upalpha \) radiation (\(\lambda = 1.541874\) Å) operating at 40 kV and 30 mA. The \(2\theta \) angles were scanned from 5 to \(90^{\mathrm{o}}\) at \(5{^{\circ }}~\hbox {min}^{-1}\). The mean platinum particle size was estimated from Scherrer’s equation on the (1 1 1) peak. SEMs were carried out on a KYKY-EM 3200 microscope with an operating voltage of 26 kV. TEM was performed with a PHILIPS EM 208 microscope operating at 80 kV. To determine the conductivity of the samples, 4-point probe measuring system was performed.
2.6 Electrochemical measurements
To determine the activity and effectiveness of resultant electrocatalysts, electrochemical measurements are necessary. A conventional three-electrode electrochemical cell on a potentiostat/galvanostat (IVIUMSTAT.XR) set was employed to evaluate the resultant electrocatalysts by CV technique. The catalyst-modified electrode was used as working electrode. A Pt wire was served as the counter electrode and an Ag/AgCl (saturated in KCl) was used as the reference electrode. The electrochemical surface area (ESA) of the resultant electrocatalysts was determined by CV measurements carried out in \(0.5~\hbox {M H}_{2}\hbox {SO}_{4}\) aqueous solution as electrolyte. The scan range was from − 0.24 to 1.2 V in reference to Ag/AgCl and the scan rate was \(50~\hbox {mV s}^{-1}\). The ESA was determined by integrating the hydrogen adsorption/desorption peaks. CV measurements of methanol oxidation were performed in 0.5 M \(\hbox {H}_{2}\hbox {SO}_{4}\) and \(2~\hbox {M CH}_{3}\hbox {OH}\) solution with a scan rate of \(50~\hbox {mV s}^{-1}\) between − 0.1 and 1.0 V in reference to Ag/AgCl. High purity \(\hbox {N}_{2}\) was bubbled before and during the measurements to deaerate the electrolytes.
3 Results and discussion
3.1 Morphology and structure of obtained products
The chemical structures of carbon (C), pure polythiophene (PTh) and PTh–C nanocomposites were determined by FTIR spectroscopy (figure 1), which has furnished significant information about the formation of nanostructured PTh and nanocomposites. The FTIR transmission spectrum of the pure PTh exhibited characteristic vibration at \(2916.16~\hbox {cm}^{-1}\) for C–H stretching vibration band. The bands at 1674.09 and \(1404.08~\hbox {cm}^{-1}\) corresponded to C=C asymmetric and symmetric stretching vibrations of thiophene ring, respectively. The vibration bands observed at 1211.23 and \(1041.48~\hbox {cm}^{-1}\) are due to C–H bending and C–H in-plane deformation. Furthermore, the characteristic bands of PTh are seen at \(763~\hbox {cm}^{-1}\) (C–S bending) and \(694~\hbox {cm}^{-1}\) (C–S–C ring deformation stretching). The band at \(3402.20~\hbox {cm}^{-1}\) originated from O–H stretching of water in KBr. Results of FTIR studies clearly indicated polymerization of the monomer. The spectra of the nanocomposites exhibit most of the characteristic peaks of the pure PTh at the same position, without any new peaks which can be justified considering the weak interaction of PTh with carbon. As the amount of PTh in nanocomposites decreases, the intensity of characteristic peaks decreases, reaching its lowest value in 10% PTh–C nanocomposites. This result shows that polymer does not have a significant effect on the 10% PTh–C nanocomposites.

FTIR spectrum of carbon (C-Vulcan), pure polythiophene (PTh) and PTh–C nanocomposites.
SEM was performed to investigate the dimensions and the morphology of obtained products. The SEM micrographs of pure PTh and 50% PTh–C nanocomposites are shown in figure 2. Both micrographs exhibit a homogeneous colour. SEM micrograph of pure PTh shows an interesting fibre-like-structure with a diameter in the range of 85–100 nm, while micrographs of 50% PTh–C nanocomposites at magnifications of 20,000 and 30,000 clearly indicate that the fibre-like-structures are surrounded by agglomerated grain structures. The SEM analysis revealed the interaction of carbon particles with polymer and indicated carbon black (Vulcan XC-72) compound formation with PTh via solution dispersion method.

SEM micrographs of (a) nanostructured polythiophene, (b and c) 50% PTh–C nanocomposite at magnifications of 20,000 and 30,000.
3.2 Electrical conductivity of the pure PTh and 50% PTh–C nanocomposites supports
The electrical conductivity of the pure PTh and 50% PTh–C nanocomposites measured by the four-point probe technique showed that the conductivity of pure PTh is about \(3.39 \times 10^{-6}\hbox { S cm}^{-1}\), while the conductivity of 50% PTh–C nanocomposites is about \(19.79 \times 10^{-4}\hbox { S cm}^{-1}\). It is observed that the conductivity of PTh–C nanocomposites is about 600 times higher than that of pure PTh. This is expected since carbon is a common polymer additive used for enhancing physical properties, such as conductivity [38]. This result confirms that the carbon modified the electrical properties of PTh.
3.3 PTh dispersibility test
Conductive polymers are generally intractable and their solubility in common solvents is poor [39, 40]. For this purpose, known amounts of pure PTh (5 mg) were added into beakers and then, various organic solvents were introduced. Digital photos were taken after 24 h (see supplementary information). The dispersion state showed that the solubility of prepared PTh in the polar solvents is better than in the non-polar solvents. Among all the nine solvents tested, prepared PTh showed the highest dispersibility in NMP and DMF.
3.4 XRD analysis and TEM imaging
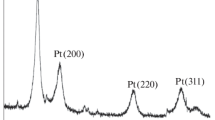
The XRD patterns of the resultant electrocatalysts are shown in figure 3. The diffraction peaks at about 39.7, 46.2, 67.8, 81.2 and \(85.7{^{\circ }}\) can be ascribed to the (111), (200), (220), (311) and (222) crystal planes of face-centered cubic (fcc) platinum. The average Pt particle sizes for the five electrocatalysts can be estimated from the Pt(111) peak using the Debye–Scherrer equation [41]:
In the equation, L is the average particle size, \(\lambda \) the wavelength of the incident X-ray (\(\hbox {CuK}\upalpha = 1.5406\) Å), \(\theta _{\mathrm{max}}\) the angle of the maximum peak and \(\beta _{2\theta }\) is the half width at peak maximum (FWHM). The average size of dispersed Pt nanoparticles on 50% PTh–C, 20% PTh–C, 10% PTh–C and carbon is calculated about 4.9, 5.2, 5.4 and 6.1 nm, respectively. Several studies have indicated that the Pt particles with the smaller size increase the activity of the catalyst. This result might be due to the higher specific surface area of the catalyst; however, the Pt particles with too small size can decrease the specific surface area of the catalyst, because they are unstable and easily oxidized by different oxidizing agents. These results confirm that there is an optimal size range for Pt particles with high catalytic activity.

XRD patterns of dispersed Pt nanoparticles on carbon (C), pure polythiophene (PTh) and PTh–C nanocomposite.
To confirm the particle size obtained by XRD, Pt/50% PTh–C were observed by TEM. TEM images and the corresponding histogram of Pt nanoparticle diameter of Pt/50% PTh–C are reported in figure 4. It was clearly observed that the well-dispersed platinum nanoparticles on the surface of the 50% PTh–C nanocomposites with size ranging from 4.7 to 6.3 nm were prepared by EG method successfully, which is in good accordance with XRD data.

(Top) TEM images of dispersed Pt nanoparticles on 50% PTh–C nanocomposite at different magnifications and (down) corresponding histogram of Pt nanoparticle diameter of Pt/ 50% PTh–C.
3.5 Electrochemical analysis
3.5.1 Electrochemical activity of electrocatalysts
Figure 5 shows the comparison of cyclic voltammograms of Pt/C, Pt/10% PTh–C, Pt/20% PTh–C and Pt/50% PTh–C electrocatalysts in nitrogen-saturated \(0.5~\hbox {M H}_{2}\hbox {SO}_{4}\) measured at a scan rate of \(50~\hbox {mV s}^{-1}\). Multiple hydrogen and oxygen adsorption/desorption peaks with different intensities and double-layer region can be clearly observed for all the electrocatalysts. As shown in figure 5, the Pt/50% PTh–C electrocatalyst had higher current as compared with that in other three electrocatalysts, due to the intriguing properties of 50% PTh–C as a support material such as larger surface area, higher dispersion of the platinum nanoparticles and higher electron conductivity.

Cyclic voltamograms of Pt/C, Pt/ PTh–C nanocomposite, and Pt/PTh in nitrogen saturated 0.5 M H\(_2\)SO\(_4\) measured at a scan rate of \(50~\hbox {mV}\,\hbox {s}^{-1}\).
The ESA of Pt catalyst on the modified electrode can be estimated from the area under the hydrogen adsorption peak during the electroadsorption and electrodesorption of hydrogen on Pt sites according to the following equation [42]:
where \(Q_{\mathrm{H}}\) is the amounts of charge exchanged during hydrogen adsorption and desorption (\(\hbox {mC cm}^{-2}\)), [Pt] the loading of Pt (\(\hbox {mg cm}^{-2}\)) on the studied electrode and C the required coulombic charge to oxidize a monolayer of hydrogen on Pt and its value is \(0.21~\hbox {mC cm}^{-2}\). The ESA results are summarized in table 1. Results show that ESA of Pt/50% PTh–C electrocatalyst (\(98~\hbox {m}^{2}\) \(\hbox {g}^{- 1}\hbox {Pt}\)) was larger than that of other three electrocatalysts. This result is believed to be due to the unique surface properties of the 50% PTh–C nanocomposites and higher dispersion and optimum size of Pt nanoparticles of the Pt/50% PTh–C.
3.5.2 Electrocatalytic performance of electrocatalysts for MOR
Figure 6 shows the electrocatalytic activity of Pt/10% PTh–C, Pt/20% PTh–C and Pt/50% PTh–C electrocatalysts towards methanol oxidation evaluated by CV in \(2~\hbox {M CH}_{3}\hbox {OH} + 0.5~\hbox {M H}_{2}\hbox {SO}_{4}\) measured at a scan rate of \(50~\hbox {mV s}^{-1}\) between − 0.1 and 1.0 V. As shown in the CV curves, there are two oxidation peaks belonging to methanol oxidation, one at about 0.65 V in the forward scan is attributed to the methanol electrooxidation on Pt and the other at about 0.45 V in the backward scan is ascribed to the additional oxidation of the adsorbed carbonaceous species to \(\hbox {CO}_2\). The ratio of forward oxidation peak current (\(I_{\mathrm{f}}\)) to the backward oxidation peak current (\(I_{\mathrm{b}}\)) is used to indicate the tolerance of Pt catalysts towards the poisonous species at the electrode surface. The values of \(I_{\mathrm{f}}\), \(I_{\mathrm{b}}\) and the current peak ratio of \(I_{\mathrm{f}}\) to \(I_{\mathrm{b}}\) (\(I_{\mathrm{f}}/I_{\mathrm{b}}\),) are listed in table 2. The results showed that the Pt/50% PTh–C electrocatalyst had higher \(I_{\mathrm{f}}/I_{\mathrm{b}}\) ratio than that of the Pt/10% PTh–C and Pt/20% PTh–C. Thus, Pt/50% PTh–C is presented as an ideal electrocatalyst support material with promising hydrogen and MOR catalytic activity.

Cyclic voltamograms of Pt/10% PTh–C, Pt/20% PTh–C and Pt/50% PTh–C in \(0.5~\hbox {M H}_2\hbox {SO}_4 + 1~\hbox {M CH}_3\hbox {OH}\) solution measured at a scan rate of \(50~\hbox {mV}~\hbox {s}^{-1}\).
4 Conclusions
In summary, we have demonstrated the successful synthesis of PTh–C nanocomposites with different weight percentages of PTh by a facile and efficient solution dispersion method. The results of FTIR spectroscopy, SEM, AFM, XRD and conductivity measurement clearly proved formation of PTh–C nanocomposite and indicated that the conductivity of PTh–C nanocomposites is about 600 times higher than that of pure PTh, which confirmed that the carbon successfully modified the electrical properties of PTh. Additionally, Pt/C, Pt/PTh–C nanocomposites and Pt/PTh electrocatalysts were prepared by EG reduction method. The resultant electrocatalysts were extensively characterized both by physical (XRD and TEM) and electrochemical (CV) techniques. XRD patterns indicated that the prepared electrocatalysts were present in the face-centered cubic (fcc) structure and the average size of dispersed Pt nanoparticles on 50% PTh–C, 20% PTh–C, 10% PTh–C and carbon is calculated about 4.9, 5.2, 5.4 and 6.1 nm, respectively. The TEM observations for Pt/50% PTh–C is consistent with the XRD results and indicated that the Pt nanoparticles were deposited on the supports successfully. The electrochemical characterization results revealed that Pt/50% PTh–C modified electrode displayed a higher ESA and higher catalytic activity for MOR when compared with the other electrocatalysts. It seems that these results are due to the intriguing characteristics of 50% PTh–C nanocomposites such as larger surface area, higher dispersion, the optimum size of the platinum nanoparticles and higher electron conductivity. According to these findings, Pt/50% PTh–C is presented as an ideal electrocatalyst support material with promising hydrogen and MOR catalytic activity.
References
Carrette L, Friedrich K A and Stimming U 2000 Chem. Phys. Chem. 1 162
Wang Y, Chen K S, Mishler J, Cho S C and Adroher X C 2011 Appl. Energy 88 981
Ellis M W, Von Spakovsky M R and Nelson D J 2001 Proc. IEEE 89 1808
Chen Z, Xu L, Li W, Waje M and Yan Y 2006 Nanotechnology 17 5254
Smitha B, Sridhar S and Khan A 2005 J. Membr. Sci. 259 10
Prater K B 1996 J. Power Sources 61 105
Wang S, Jiang S P, White T, Guo J and Wang X 2009 J. Phys. Chem. C 113 18935
Shao Y, Zhang S, Wang C, Nie Z, Liu J, Wang Y et al 2010 J. Power Sources 195 4600
Chung C G, Kim L, Sung Y W, Lee J and Chung J S 2009 Int. J. Hydrog. Energy 34 8974
Yaldagard M, Jahanshahi M and Seghatoleslami N 2013 World J. Nano Sci. Eng. 3 121
Sharma S and Pollet B G 2012 J. Power Sources 208 96
Beden B, Léger J-M and Lamy C 1992 in: J O M Bockris, B E Conway and R E White (eds) Modern aspects of electrochemistry (New York: Plenum Press) p 97
Wang Y-J, Wilkinson D P and Zhang J 2011 Chem. Rev. 111 7625
Sharma S and Pollet B G 2012 J. Power Sources 208 96
Wang J, Yin G-P, Zhang J, Wang Z and Gao Y 2007 Electrochim. Acta 52 7042
Adhikari A, Radhakrishnan S and Patil R 2009 Synth. Met. 159 1682
Antolini E 2010 Appl. Catal. B: Environ. 100 413
Unni S M, Dhavale V M, Pillai V K and Kurungot S 2010 J. Phys. Chem. C 114 14654
Vedrine J C, Dufaux M, Naccache C and Imelik B 1978 J. Chem. Soc. Faraday Trans. 1: Phys. Chem. Condens. Phases 74 440
Biloul A, Coowar F, Contamin O, Scarbeck G, Savy M, Van den Ham D et al 1990 J. Electroanal. Chem. Interfac. Electrochem. 289 189
Zhou J, Zhou X, Sun X, Li R, Murphy M, Ding Z et al 2007 Chem. Phys. Lett. 437 229
Dicks A L 2006 J. Power Sources 156 128
Antolini E 2009 Appl. Catal. B: Environ. 88 1
Maillard F, Simonov P A and Savinova E R 2009 in: P Serp and J Figueiredo (eds) Carbon materials for catalysis (New York: John Wiley & Sons, Inc.) chapter 12 p 429
Hezarjaribi M, Jahanshahi M, Rahimpour A and Yaldagard M 2014 Appl. Surf. Sci. 295 144
Wallace G G, Teasdale P R, Spinks G M and Kane-Maguire L A 2008 Conductive electroactive polymers: intelligent polymer systems, third edn (New York: CRC Press)
Antolini E and Gonzalez E 2009 Appl. Catal. A: General 365 1
Roncali J 1992 Chem. Rev. 92 711
Kattimani J, Sankarappa T, Praveenkumar K, Ashwajeet J and Ramanna R 2014 Int. J. Adv. Res. Phys. Sci. 1 17
Lee J M, Lee S J, Jung Y J and Kim J H 2008 Curr. Appl. Phys. 8 659
Sulub R, Martinez-Millan W and Smit M A 2009 Int. J. Electrochem. Sci. 4 1015
Schopf G and Koßmehl G 1997 Adv. Polym. Sci. 129 3
Cao Y, Wang P and Qian R 1985 Die Makromolekulare Chemie 186 1093
Tsakova V 2008 J. Solid State Electrochem. 12 1421
Gomez-Romero P 2001 Adv. Mat. 13 163
Fischer H 2003 Mat. Sci. Eng: C 23 763
Roy S, Christensen P, Hamnett A, Thomas K and Trapp V 1996 J. Electrochem. Soc. 143 3073
Takada T, Nakahara M, Kumagai H and Sanada Y 1996 Carbon 34 1087
Kumar D and Sharma R 1998 Eur. Polym. J. 34 1053
Gangopadhyay R and De A 2000 Chem. Mat. 12 608
Lin R, Cao C, Zhang H, Huang H and Ma J 2012 Int. J. Hydrog. Energy 37 4648
Koponen U, Kumpulainen H, Bergelin M, Keskinen J, Peltonen T, Valkiainen M et al 2003 J. Power Sources 118 325
Acknowledgements
We wish to express our sincere gratitude to the Nanotechnology Research Institute of Babol University for its scientific and financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nasrollahzadeh, M., Jahanshahi, M., Yaldagard, M. et al. Synthesis, characterization and comparison of polythiophene–carbon nanocomposite materials as Pt electrocatalyst supports for fuel cell applications. Bull Mater Sci 41, 85 (2018). https://doi.org/10.1007/s12034-018-1599-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12034-018-1599-x