
Abstract
The P3HT grafted on CNTs to form the P3HT-g-CNTs nanocomposites was synthesized and their morphologies, structure have been characterized via the sedimentation test, scanning electron microscopy (SEM), X-ray diffraction (XRD) and transmission electron microscopy (TEM). The results showed that the P3HT- g-CNTs has a better thermal stability than that of the P3HT/CNTs blend. The nanocomposite based on P3HT- g-CNTs and doped spinel LiNi0.5Mn1.5O4 (LNMO) have been fabricated via mixing process. The structure and morphologies of LNMO/P3HT- g-CNTs nanocomposites have also been performed by SEM, XRD and TEM. The electrochemical performance of LNMO/P3HT- g-CNTs nanocomposites as cathode materials of lithium-ion batteries were investigated by cyclic voltammetry and electrochemical impedance spectroscopy and exhibited the high diffusion of lithium ions in the charge–discharge process.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Electronic devices based on conjugated polymers and carbon nanotubes (CNTs) are being developed at a fast pace due to their unique electrical and optical properties. Such materials exhibited their low cost and processing advantages because organic electronic devices are based on thin films, flexibility, light weight, semitransparency as well as large-area application [1–4]. On the other hand, since discovery in 1991 [5], CNTs have been extensively studied for their unique properties such as 1D tubular structure, high electrical and thermal conductivities, mechanical, optical properties and extremely large surface area.
Among various energy and power technologies, rechargeable Li-ion batteries (LIBs) are one of the most efficient energy storage devices based on electrochemical energy storage and conversion [6–11]. Compared to traditional rechargeable batteries such as lead–acid and Ni–Cd batteries, rechargeable LIBs provide many advantages including high voltage, high energy-to-weight ratio, i.e., high energy density, long cyclic life, no memory effect and slow loss of charge [12,13]. Owing to these advantages, LIBs are currently the most popular battery type of powering portable electronic devices. Although, LIBs have shown remarkably commercial success, the electrodes and their component materials are still the target of interesting research for enhancement of electrochemical performance of battery devices. The main disadvantages of LIBs are low power density as well as low ion diffusion and charge transfer during the process of charge–discharge at high rates as a consequence of high polarization [14–16]. Therefore, the development of new electrodes having a large surface area, a short diffusion and high electric/thermal conductions is necessary to overcome the disadvantages of conventional materials. Recently, there have been many reports presenting the structures combining CNTs and nanoparticles such as V2O5 and SnO2 via a hydrothermal process [17]. Such obtained V2O5–SnO2/CNTs composites display fast Li+ transformation and high electronic conductivity. Recently, Shao et al[18] has reported the synthesis of SnO2-based composite coaxial nanocables with multi-walled CNTs and polypyrrole (PPy), SnO2@CNTs@SnO2@PPy, which exhibit a reversible capacity as high as 600 mAh g−1 and a coulombic efficiency close to 100%, which is potential for practical application in lithium-ion batteries. More recently, Zhao et al[19] also reported the synthesis of a novel CNTs@SnO2@PPy coaxial nanocable as superior anode material with SnO2 particle size of 2–3 nm, moreover this composite exhibited a high capacity of 823 mAh g−1 after 100 cycles. Even at a high rate of 3000 mA g−1, a reversible capacity of 480 mAh g−1 still remained.
In terms of preparation of hybrid materials based on conjugated polymers with CNTs, the main challenge for incorporation of CNTs with conjugated polymers is the dispersion and stabilization of CNTs in polymer matrices, which implies to the CNTs aggregation because of strong Van der Waals interactions between the CNTs sidewalls. For this target, the conjugated molecules have been modified to get functional group which can be grafted onto CNTs leading to a good dispersion of CNTs in solution and in bulk [20–26]. Among those conjugated materials, regioregular poly(3-hexylthiophene) (P3HT) has attracted significant interest due to its outstanding electronic and optical properties, moreover its good solubility in common organic solvents and has a self-assemble by π– π interactions to form crystalline nanowires, or fibrils, which favour charge carrier transport [27–29]. Therefore, the combination of CNTs and P3HT would be interest materials for potential cathode materials in LIBs.
Here, we presented the strategy to increase the dispersion of P3HT-g-CNTs in solution to form the nannocomposite, which further used to combine with doped spinel LiNi0.5Mn1.5O4 (LNMO) for fabrication of cathode materials as LNMO/P3HT-g-CNTs nanocomposites in LIBs. We used a regioregular P3HT prepared by Grignard metathesis (GRIM) with well-known end-groups. The controlled synthesis of regioregular P3HT is mostly carried out by the GRIM method, that allows for the synthesis of P3HT with well-controlled regioregularity (>99%) and average molecular weight (M n), narrow polydispersity index (D) (1.1–1.3), and defined end-groups (H/Br). The structure and morphology of the electrode were characterized using XRD, SEM and TEM. Finally, the efficiency of the electrode materials using LNMO/P3HT-g-CNTs nanocomposites was evaluated by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS).
2 Experimental
2.1 Synthesis of P3HT with H/Br end group
A dried 500 ml three-neck flask was flushed with nitrogen and was charged with 2-bromo-3-hexyl-5-iodothiophene (15 g, 40 mmol). After three azeotropic distillations by toluene, anhydrous tetrahydrofuran (THF) (220 ml) was added under nitrogen atmosphere, the mixture was stirred at 0°C for 1 h. i-PrMgCl (2 M solution in THF, 19.14 ml, 38.28 mmol) was added via a syringe and the mixture was continuously stirred at 0°C for 1 h. The reaction mixture was allowed to cool down to 0°C. The mixture was transferred to a flask containing a suspension of Ni(dppp)Cl2 (760 mg, 1.4 mmol) in THF (25 ml). The polymerization was carried out for 24 h at 0°C followed by fast addition of 5 M HCl solution. The reaction was stirred for 15 min and extracted with chloroform. The polymer was precipitated from cold methanol and washed several times with hexane. The polymer was characterized by1H NMR and GPC [28] (yield: 70%).
FTIR (cm−1): 721, 819, 1376, 1454, 1510, 2853, 2922, 2953, 3056.1H NMR (300 MHz, CDCl3), d (ppm): 6.96 (s, 1H), 2.90 (t, 2H), 1.79 (q, 2H), 1.52 (q, 6H), 0.94 (t, 3H). GPC, M n, 6000 g mol−1. D = 1.18. Maldi-ToF (m/z): 1410, 1576, 1742, 1908, 2074, 2240, 2406, 2572, 2738, 2904, 3070, 3236. UV–Vis (CHCl3): \(\lambda _{{\max }} =\) 450 nm.
2.2 CNTs purification
150 mg of CNTs were immersed in 100 ml of the mixture of H2 SO 4 (95%) and HNO3 (65%). The reaction was performed in sonication bath for 4 h. The CNTs were washed with deionized (DI) water several times, filtered and then dried to constant weight before characterization.
2.3 Synthesis of P3HT-graft-CNTs (P3HT-g-CNTs)
Br-P3HT-H (M n = 6000, D = 1.18) (100 mg, 0.01 mmol) was dissolved in anhydrous toluene (30 ml) under N2 in the presence of CNTs–COOH (100 mg). The mixture was sonicated for 15 min before adding 1 mg of tetrabutylammonium hydrosulfate (TBAHSO4) and 18 mg of potassium fluoride (KF). The reaction was carried out at reflux for 72 h and the product was recovered by precipitation in cold methanol. Then the obtained material was dried under vacuum at 60°C for 24 h before characterization.
2.4 Synthesis of spinel LNMO materials
Li2 CO 3 (2.5 g, 67.6 mmol), MnCO3 (23.32 g, 0.202 mol) and Ni(NO3)2⋅6H2O (19.65 g, 67.6 mmol) were prepared by mixing and decomposed at 600°C for 24 h. Then, the mixture was grounded and made into pellets, which were heated at 900°C for 24 h to obtain LNMO.
2.5 Synthesis of LNMO/P3HT-g-CNTs nanocomposites
Spinel of LNMO (9.8 g) materials and purified P3HT-g-CNTs (0.2 g) were mixed together. Then, the binder of 60% PTFE dispersed in water (1 g) and 15 ml of absolute ethanol were added to the mixture. The mixture was stirred for 15 min, then the mixture was sonicated for 30 min, and the solvent was evaporated at 80°C for 24 h. Then, the mixture was coated on aluminum substrates of about 0.1 mm in thickness as cathode materials, which were dried at 130°C under vacuum for 24 h.
2.6 Structure and morphology analysis
1H NMR spectra were recorded in deuterated chloroform (CDCl3) with tetramethylsilane (TMS) as an internal reference on a Bruker Avance 300 MHz. FTIR (thermo fisher scientific-model Nicolet 6700, frequency range of 4000–500 cm−1). Spectra were recorded using BIO-RAD Excalibur spectrometer equipped with an ATR Harrick Split PeaTM. Size exclusion chromatography (SEC) measurements were performed on a polymer PL-GPC 50 gel permeation chromatograph system equipped with an RI detector, with THF as the eluent at a flow rate of 1.0 ml min−1. Molecular weight and molecular weight distribution were calculated with reference to polystyrene standards. UV–Vis absorption spectra of polymers in solution and polymer thin films were recorded on a Shimadzu UV-2450 spectrometer over a wavelength range of 300–700 nm. Wide angle powder X-ray diffraction (XRD) patterns were recorded at room temperature on a Bruker AXS D8 Advance diffractometer using CuK α radiation (k = 0.15406 nm), at a scanning rate of 0.05° s−1. SEM images were acquired using a field-emission scanning electron microscope (FESEM) (NOVA NANOSEM450, FEI) equipped with an Oxford energy dispersive X-ray (EDX) analysis system. TEM images were performed on a FEI Tecnai F20 microscope equipped with an Oxford EDX analysis system and with an acceleration voltage of 200 kV.
2.7 Electrochemical characterization
CV was performed on MGP2 (Biologic, France) using ECLab software. The electrochemical cell including three electrode cells: a platinum electrode covered with a composite material as a working electrode (WE), an Ag/0.1 mol l−1, AgNO3/0.01 mol l−1, tetrabutylammonium perchlorate (TBAP) in acetonitrile as reference electrode (RE), and a Pt wire as counter electrode (CE). The electrolyte solution consists of 0.75 mol l−1 lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) dissolved in 1:1 mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) [22–24]. The standard voltage of the reference electrode is 0.542 V vs. standard hydrogen electrod (SHE) at 25°C.
The impedance measurement (EIS) was performed in Swagelok-type batteries at room temperature. The cell consisting of two electrode films as positive and negative electrodes was assembled. The electrolyte was a 1 mol l−1 solution of LiClO4 in EC-DMC (1:1). The EIS was carried out using an Autolab 302 N (MetroOhm).
3 Results and discussion
Purified CNTs were first treated with a mixture of H2 SO 4 (95%)/HNO3 (65%) to clean the surface. Moreover, this treatment process also has functionalized reacting groups such as hydroxyl, carboxyl and carbonyl groups on the surface of CNTs. These functional groups not only facilitated the dispersion of the CNTs in water but also acted as centres for incorporation of metal-oxide particles. The structures and morphologies of CNTs were characterized by SEM. Figure 1 shows the TEM images of purified CNTs (figure 1b), which are much clearer compared with the as-prepared one (figure 1a). These results suggest that the CNTs were successfully purified using the acid treatment process. It is also observed that the acid treatment process causes the peeling of graphite layers on surface of untreated CNTs, and therefore, the diameter of purified CNTs is quite smaller than the diameter of untreated CNTs. The functional groups such as hydroxyl and carboxyl groups of CNTs were characterized by FTIR. The presence of hydroxyl groups and carbonyl groups corresponds to the appearance of broad peaks at 3425 and 1736 cm−1, respectively. It should be noted that the presence of hydroxyl and carbonyl groups can enhance the dispersion of CNTs as well as reactivity with other functional groups of polymers [30–33].

TEM images of CNTs (a) before and (b) after acid treatment.
The synthesis of P3HT-g-CNTs hybrid materials is described in scheme 1. Regioregular poly(3 hexylthiophene) (rr-P3HT) was synthesized using the GRIM polymerization method obtaining an α-bromo-P3HT with average molecular weight of 6000 g mol−1 and dispersity index of 1.18. Then P3HT was reacted with carboxyl groups of CNTs in the presence of TBAHSO4/KF catalytic system for 24 h under reflux condition. The obtained product have been precipitated in cold methanol then dried under vacuum at 60°C for 24 h.

Synthesis of P3HT-g-CNTs hybrid materials.
The success of the reaction has been attested by the appearance of a vibrational band at 1730 cm−1 corresponding to ester linkage between P3HT and CNTs. To prove that the grafting effectively occurred and that the obtained nanocomposites are stable in solution, the sedimentation tests have been performed. We prepared three samples in THF including: a mixture of P3HT and CNTs (P3HT/CNTs), a simple CNTs and a the covalently grafted system (P3HT-g-CNTs). In the case of P3HT/CNTs, 20 mg of mixture P3HT and CNTs (weight ratio 1:1) was added into 10 ml of THF, then the solution was sonicated for 1 h. It is also similar to the case of a simple CNTs (20 mg) and P3HT-g-CNTs (20 mg). The comparative sedimentation evolutions of the P3HT/CNTs, a simple CNTs and P3HT-g-CNTs have been characterized via recording the H d/H t ratio vs. time, where H d is the height of the sedimentation front and H t is the height of total solution. After sonication of these solutions, these samples have been kept stationary for estimation of sedimentation. Within 1 h, the CNTs aggregate and leave a clear solution (figure 2), the physical blend of P3HT/CNTs also aggregated after 2 h. On the other hand, P3HT-g-CNTs solution exhibited extreme efficiency in increasing the CNTs dispersion in solution, the supernatant remaining black after 24 h (figure 2).

Results of the sedimentation test for the three materials: CNTs, P3HT/ CNTs and P3HT-g-CNTs.

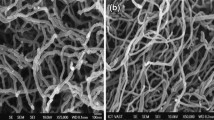
SEM images of (a) CNTs and (b) P3HT-g-CNTs nanocomposites.
To determine the structure of P3HT-g-CNTs nanocomposites, morphological studies were performed by SEM and TEM on the obtained materials. The samples were prepared by drop casting a 0.25 mg ml−1 in chloroform solution. SEM results show a 1D tubular structure for both the CNTs, and P3HT-g-CNTs nanocomposites (figure 3).
To reach more insights into the morphologies of P3HT-g-CNTs nanocomposites, the TEM results exhibited clearly the 1D tubular structure of the P3HT-g-CNTs nanocomposites as seen in figure 4. Moreover, the thickness of P3HT-g-CNTs is larger than the bare CNTs one. This phenomenon can be explained that the CNTs have been covered by P3HT layer to form nanocomposites structure.

TEM images of (a) bare CNTs and (b) P3HT-g-CNTs nanocomposites.
Moreover, to determine the content of CNTs in the P3HT-g-CNTs nanocomposites and to recognize any change in the degradation of the P3HT-g-CNTs, thermogravimetric analyses (TGA) were performed on blend of P3HT/CNTs and P3HT-g-CNTs materials from room temperature to 900°C at a 20°C min−1 heating rate, including the blend P3HT/CNTs and P3HT-g-CNTs nanocomposites. As seen in figure 5, in the case of the blend P3HT/CNTs, the mass lost is about 10% at 300°C, while the P3HT-g-CNTs shows the mass loss of 2%. This result suggests that the P3HT-g-CNTs has a better thermal stability than that of the P3HT/CNTs due to their nanocomposites structure. Moreover, both of the blend P3HT/CNTs and P3HT-g-CNTs exhibited complete degradation at 900°C.

TGA curves of the blend P3HT/CNTs and P3HT-g-CNTs.
Spinels of LiNi0.5Mn1.5O4 (LNMO) was synthesized by solid-state reaction. The morphology of the LNMO particles exhibits irregular cubes and exhibit grain sizes from 2 to 20 μm, which were analysed by SEM in figure 6a. Furthermore, the LNMO structures were characterized by XRD analysis as shown in figure 6b. The diffraction peaks at 2𝜃 = 18, 36.5, 38, 44, 48, 58 and 64° relate to the doped spinel structure of LNMO materials [34,35].

SEM image of (a) LNMO particles and (b) XRD patterns of the doped spinel structure LNMO materials.
The schematic illustration of the formation of the LNMO/ P3HT-g-CNTs nanocomposites via the mixing process is shown in figure 7. The P3HT-g-CNTs, PTFE dispersed in deionized water and LNMO materials were stirred in ethanol for 15 min, then the mixture was sonicated for 30 min and the solvent was evaporated under vacuum. Finally, the obtained mixture was coated on aluminum substrates of about 0.1 mm in thickness as cathode materials, which were then dried at 130°C under vacuum for 24 h to form LNMO/ P3HT-g-CNTs nanocomposites. The morphologies of LNMO/ P3HT-g-CNTs (2 wt%) nanocomposites are characterized by SEM in figure 8a. The SEM results show a 1D tubular structure of CNTS in the LNMO/P3HT-g-CNTs nanocomposites structure after mixing process. A typical high-magnification transmission electron microscopy (TEM) image (figure 8b) further shows that LNMO distributed randomly on the CNTs surface. It should be noted that the particle size distribution of LNMO is much smaller than the original particle size of LNMO. This phenomenon can be explained that the preparation of LNMO/P3HT-g-CNTs nanocomposites via mixing process eliminated the particles aggregation. Therefore, the obtained particle size distribution of LMNO in its nanocomposites is around 50 nm. These results confirm that LNMO particles were deposited on the surface of the LNMO/P3HT-g-CNTs nanocomposites. More interestingly, the LNMO/P3HT-g-CNTs nanocomposites exhibit an interpenetrating network structure that offers a benefit of charge transfer in cathode materials.

Schematic illustration of the synthesis of LNMO/P3HT-g-CNTs nanocomposites.

(a) SEM images of LNMO/P3HT-g-CNTs and (b) TEM image of LNMO/ P3HT-g-CNTs nanocomposites.
The structure of LNMO/P3HT-g-CNTs (2 wt%) nanocomposites was also determined by XRD. The XRD diagram in figure 9 shows the most important peaks related to CNTs at 25° and all characteristic peaks corresponding LNMO at 18, 36.5, 38, 44, 48, 58 and 64°, respectively.

XRD pattern of the LNMO/P3HT-g-CNTs nanocomposites.
Li+ intercalation/de-intercalation LNMO/P3HT-g-CNTs nanocomposites as cathode electrode materials was characterized by CV. Figure 10 shows the CV curves of LNMO/ P3HT-g-CNTs (2 wt%) compared to LNMO/VC (vulcan carbon ≈ 2 wt%) as a standard material for comparison. The CV curves were carried out with a potential region of 0.1–1.3 V vs. Ag/AgNO3 and a scanning rate of 0.2 mV s−1 in an 0.75 mol l−1 LiTFSI/EC:DMC (1:1 by volume) electrolyte solution. As seen in figure 10, the oxidation and reduction peaks in those curves relate to the Ni3+/Ni4+ redox couple, indicating the lithium intercalation/de-intercalation via the oxidation–reduction process. The intensity of Ni3+/Ni4+ oxidation and reduction peaks of the LNMO/P3HT-g-CNTs nanocomposites is higher than that of the LNMO/VC materials. This result proved that the higher efficiency of LNMO/P3HT-g-CNTs nanocomposites is better than LMNO/ VC materials.

CV curves of LNMO/P3HT-g-CNTs nanocomposites with 2 wt% CNTs and LNMO/VC with 2 wt% vulcan carbon.
The electronic conductivity of LNMO/P3HT-g-CNTs as cathode nanocomposites compared to LNMO/VC was characterized by EIS. As seen in the Nyquist plot spectra, at a high content of vulcan carbon, the charge transfer resistance (R ct) of LNMO/VS (2 wt%) decreases significantly and the diffusion resistance corresponding Warburg (W) at low frequencies seems to dominate (figure 11). On the other hand, the Warburg value of LNMO/P3HT-g-CNTs (2 wt%) is much lower than that of LNMO/VC (2 wt%) (figure 11). These results indicate that the electrochemical reaction of LNMO/P3HT-g-CNTs nanocomposites is better than that of LNMO/VC materials as a result of high diffusion of lithium ions in the charge–discharge process.

Nyquist plot of LNMO/P3HT-g-CNTs (2 wt%) and LNMO/VC (2 wt%) nanocomposite electrode at room temperature.
Charge–discharge profile of the LNMO/P3HT-g-CNTs was characterized at the current density C/10 using Swagelok-type cell. The charge–discharge capacity of LNMO/ P3HT-g-CNTs (10 wt%) exhibited the value of 145 Ah kg−1 that much higher than LNMO/VC (10 wt%) (70 Ah kg−1) and LNMO/10 wt% CNTs (90 Ah kg−1) (figure 12). These results suggested that the replacement of vulcan carbon by CNTs gave the efficiency of charge–discharge capacity due to high diffusion of lithium ions.

1st charge–discharge cycle at C/10 rate of nanocomposite electrodes: LNMO/10 wt% VC, LNMO/10 wt% CNTs and LNMO/10 wt% P3HT-g-CNTs.
The cycling performance of LNMO/P3HT- g -CNTs (10wt%) nanocomposites and LNMO/CNTs (10 wt%) and LMNO/ vulcan carbon (10 wt%) were shown in figure 13. After 40 cycles, the capacity retentions were 80% for LNMO/P3HT-g-CNTs (10 wt%), 78% for LNMO/CNTs (10 wt%) and 75% for LMNO/vulcan carbon (10 wt%), respectively. These results proved that P3HT-g-CNTs agent mixed in the composite electrode reduce slightly the cycle efficiency due to enhancement of Li+ transport rate and electron into the spinel structure. It is well-known that LMO bulk material exhibited the poor cycle ability at the 4 voltage plateau because of Jahn–Teller distortion of the (MnO6) octahedron caused by the Mn3+ cation at the 16d site. The P3HT-g-CNTs addition forming the conducting network enhanced the electrical connections among LiNMO particles therefore Li+ intercalation/de-intercalation rate into/out of the spinel structure enhanced.

Cycling performance of nanocomposites electrodes at C/10 rate: LNMO/10 wt% VC, LNMO/10 wt% CNTs and LNMO/ 10 wt% P3HT-g-CNTs.
4 Conclusions
We have successfully synthesized the hybrid materials of P3HT-g-CNTs via direct coupling reaction of P3HT and CNTs in the presence of TBAHSO4 and KF as catalytic system. The P3HT-g-CNTs has been characterized via the sedimentation test, SEM, TEM and TGA analyses. We also have successfully prepared the cathode materials based on LNMO/P3HT-g-CNTs via mixing process. The LNMO/ P3HT-g-CNTs nanocomposites has been characterized and proved a remarkably higher efficiency of and Ni2+/Ni4+ redox couples in comparison to traditional cathodes based on LNMO/vulcan carbon. With this proof of concept, LNMO/ P3HT-g-CNTs nanocomposites could act as promising cathode materials for the development of potential power lithium rechargeable batteries.
References
Tasis D, Tagmatarchis N, Bianco A and Prato M 2006 Chem. Rev. 106 1105
Andrews R, Jacques D, Qian D and Rantell T 2002 Acc. Chem. Res. 35 1008
Li G, Shrotriya V, Huang J, Yao Y, Moriarity T, Emery K and Yang Y 2005 Nat. Mater. 4 864
McCullough R D 1998 Adv. Mater. 10 93
Iijima S 1991 Nature 354 56
Arico A S, Bruce P, Scrosati B, Tarascon J M, Van W and Van Schalkwijk W 2005 Nat. Mater. 4 366
Jiang C, Hosono E and Zhou H 2006 Nano Today 1 28
Wallace G G, Chen J, Mozer A J, Forsyth M, MacFarlane D R and Wang C Y 2009 Mater. Today 12 20
Li H, Wang Z, Chen L and Huang X 2009 Adv. Mater. 21 4593
Chen J, Minett A I, Liu Y, Lynam C, Sherrell P, Wang C and Wallace G G 2008 Adv. Mater. 20 566
Myung S T, Komaba S, Hirosaki I N, Yashiro H and Kumagai N 2004 Electrochim. Acta 49 4213
Taberna P L, Mitra S, Poizot P, Simon P and Tarascon J M 2006 Nat. Mater. 5 567
Lu L, Wang J Z, Zhu X B, Gao X W and Liu H K 2011 , J. Power Sources 196 7025
Etacheri V, Marom R, Elazari R, Salitraa G and Aurbach D 2011 Energy Environ. Sci. 4 3243
Santhanagopalan S, Guo Q, Ramadass P and White R E 2006 J. Power Sources 156 620
Doerffel D and Abu Sharkh S 2006 J. Power Sources 155 395
Guo Q, Sun Z, Gao M, Tan Z, Zhang B and Su D S 2013, J. Energy Chem. 22 347
Shao Q G, Chen W M, Wang Z H, Qie L, Yuan L X, Zhang W X et al 2011, Electrochem. Commun. 13 1431
Zhao Y, Li J, Wang N, Wu C, Dong G and Guan L 2012 , J. Phys. Chem. C 116 18612
Zhao Y L and Stoddart J F 2009 Acc. Chem. Res. 42 1161
Ehli C, Rahman G M A, Jux N, Balbinot D, Guldi D M, Paolucci F et al 2006 J. Am. Chem. Soc. 128 11222
Guldi D M, Rahman G M A, Zerbetto F and Prato M 2005 Acc. Chem. Res. 38 871
Lou X, Daussin R, Cuenot S, Duwez A S, Pagnoulle C, Detrembleur C et al 2004, Chem. Mater. 16 4005
Zou J, Liu L, Chen H, Khondaker S I, McCullough R D, Huo Q and Zhai L 2008 Adv. Mater. 20 2055
Kang Y K, Lee O S, Deria P, Kim S H, Park T H, Bonnell D A et al 2009 Nano Lett. 9 1414
Liu J, Zou J and Zhai L 2009 Macromol. Rapid Commun. 30 1387
Yokoyama A, Miyakoshi R and Yokozawa T 2004 Macromolecules 37 1169
Yokozawa T and Yokoyama A 2009 Chem. Rev. 109 5595
Sheina E E, Liu J, Iovu M C, Laird D W and McCullough R D 2004 Macromolecules 37 3526
Park S K, Mahmood Q and Park H S 2013 Nanoscale 5 12304
Wu W, Jiang W, Zhang W, Lin D and Yang K 2013 Environ. Sci. Technol. 47 8373
Li X, Lan L, Ai M, Guo Y, Cai Q and Yang X 2014 Colloids Surf. B 123 753
Gao C, He H, Zhou L, Zheng X and Zhang Y 2009 Chem. Mater. 21 360
Arrebola J C, Caballero A, Hernan L and Morales J 2008, J. Power Sources 183 310
Arrebola J C, Caballero A, Camer J L G, Hernan L, Morales J and Sanchez L 2009 Electrochem. Commun. 11 1061
Acknowledgements
This research was supported by the Department of Science and Technology (DOST), Ho Chi Minh City, Vietnam.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
VAN LE, T., NGUYEN, T.A., THI NGUYEN, N.M. et al. Synthesis and characterization of nanocomposites based on poly(3-hexylthiophene)-graft-carbon nanotubes with LiNi0.5Mn1.5O4 and its application as potential cathode materials for lithium-ion batteries. Bull Mater Sci 39, 1177–1184 (2016). https://doi.org/10.1007/s12034-016-1264-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12034-016-1264-1