
Abstract
The effect of isochronal annealing on the phase transformation in iron oxide nanoparticles is reported in this work. Iron oxide nanoparticles were successfully synthesized using an ash supported technique followed by annealing for 2 h at various temperatures between 300 and 700°C. It was observed using X-ray diffraction (XRD) and transmission electron microscopy (TEM) that as-grown samples have mixed phases of crystalline haematite (α-Fe2O3) and a minor phase of either maghemite (γ-Fe2O3) or magnetite (Fe3O4). On annealing, the minor phase transforms gradually to haematite. The phase transformation is complete at annealing temperature of 442°C as confirmed by differential scanning calorimetric (DSC) analysis. The unresolved phases in XRD were further analysed and confirmed to be maghemite from the X-ray absorption near edge structure (XANES) studies. The magnetic measurements showed that at room temperature nano- α-Fe2O3 is weak ferromagnetic, and its magnetization is larger than the bulk value. The mixed phase sample shows higher value of magnetization because of the presence of ferromagnetic γ-Fe2O3 phase.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The study of particle size, phase transformation and microstrain of nanomaterials is of crucial importance both from fundamental and application points of view. In particular, transition metal oxide nanoparticles (NPs) have attracted considerable interest as they exhibit properties which differ widely from the bulk phases.[1] Iron NPs are important owing to their significant applications in the magnetic memories, ferro-fluids, precursor for catalyst, bio-medical applications and in the synthesis of carbon nanotube, etc.[2–7]
It has been observed that structural properties of nanomaterials depend on their fabrication techniques. The structural parameters that can be controlled by modifying fabrication conditions include size, shape, phase composition, microstrain, etc. A large variation in size and shape of NPs has been reported in earlier studies by the optimization of fabrication techniques.[8,9]The change in particle size enables tailoring of the materials to make it suitable for a wide range of applications including magnetic ferrofluids and catalysts.[10] Out of various suitable methods for synthesis of NPs reported in the literature,[11,12]ash supported method[11] is found to be particularly interesting, as it is simple, low cost and environmental friendly. The technique can also be upgraded for industrial production of NPs. The critical aspects, however, are the control over particle size, size distribution, crystalline quality and change in chemistry by post-growth processes. Annealing is an attractive post-growth processing option in these cases. A relevant study performed by Vales et al[12] showed that in the case of iron oxide (Fe2O3) NPs prepared by the sol–gel method, the particle size increases with an increase in the annealing temperature. It is also reported that the phase composition of Fe2O3 NPs changes with change in annealing temperature. In this respect, several stable phases of iron oxide like α-Fe2O3, γ-Fe2O3 and ε-Fe2O3 are also reported by Vales et al.[12]
Phase transition and magnetic properties of iron oxide NPs have been studied by Guo et al.[13] They reported that iron oxide sample undergo transition from ferromagnetic to ferrimagnetic behaviour when NaOH concentrations are gradually increased. They found that iron oxide samples at different NaOH concentrations show obviously morphologies-dependent feature.
It is very well known that the iron oxide NPs show different magnetic properties for various phases. Jeong et al[14] have reported that Fe2O3 NPs exhibit super-paramagnetism with temperatures of 119.5 and 94.3 K. In yet another study by Kim et al,[15] Fe3O4 NPs prepared by the co-precipitation method, exhibited an average size of 15 nm. These particles were found to be in super-paramagnetic phase. Similarly, in case of Fe2O3 thin films, magnetite to haematite phase transformation was observed with the increase in the deposition temperature from 500 to 700°C and it was observed that Fe3O4 is ferromagnetic by Xu et al.[16] Hence the authors concluded that the magnetic properties are very much dependent on the phase of the particle as well as on the shape and size. Xu et al[17] reported magnetic size effects and phase transformations in their studies. They observed that nanophase of γ-Fe2O3, with a range of particle sizes and these are obtained by the controlled annealing of sonochemically synthesized Fe2O3 NPs. Annealing of γ-Fe2O3 in the air resulted in an indirect transformation in α-Fe2O3 via intermediate Fe3O4 phase and the magnetic properties were found to be strongly different from those of the bulk γ-Fe2O3 phase.
Looking into these changes, identification of proper phase, therefore, seems to be very important. Thus, in the present paper, we have undertaken phase identification studies of iron oxide NPs synthesized by a method known as the ash supported method. In this method we get NPs in the form of residue and the cost involved in the preparation is much less as compared to other NPs synthesis methods. The analysis of the iron oxide NPs has been done using synchrotron X-ray diffraction (SXRD), X-ray absorption near edge structure (XANES), high-resolution transmission electron microscopy (HRTEM), vibration sample magnetometer (VSM) and differential scanning calorimetry (DSC).
2 Experimental
To prepare iron oxide NPs, the initial reactants were taken to be iron nitrate Fe (NO3)3⋅9H2O as a precursor and methanol as a solvent. The chemicals used are analytical grade, procured from MERCK. Iron nitrate (10.1 g) was dissolved in 100 ml methanol and a Wattman filter paper was soaked in this 0.25 M methanolic solution of iron nitrate. This soaked filter paper was burnt in the air and iron oxide NPs were obtained in the form of residue. These iron oxide NPs are designated as, as-prepared sample (sample #1). This sample was divided into five parts designated as sample #2 to sample #6. Samples #2, #3, #4, #5, #6 were annealed in air for 2 h at temperatures 300, 400, 500, 600 and 700°C, respectively. The samples were annealed at various temperatures in order to study the phase transformation of iron oxide NPs and the effect of annealing on size and strain of NPs. A sample is also formed using Wattman filter papers soaked only in methanol solvent. Again, these filter papers were burnt in the air and the residue was obtained designated as ash particles. This was carried out to see that there is no artefact coming from filter paper or methanol. The NPs, thus synthesized, were analysed by X-ray diffraction (XRD) using angle-dispersive X-ray diffraction (ADXRD) beamline (BL-12) on Indus-2 synchrotron radiation source, India.[18] The beamline is based on a Si (111)-based double-crystal monochromator. The photon beam (0.3 mm × 0.3 mm) of energy ∼11 keV was used. An image plate area detector (Mar 345dTb) was used for recording the XRD pattern. The data were reduced to standard I-2 using fit 2D software. The wavelength and the sample to detector distance were accurately calibrated using XRD pattern of LaB6 NIST standard obtained on the same set-up. The LaB6 data were also used for the estimation of instrumental broadening for Williamson–Hall plot. For further phase confirmation and estimation of average sizes of the NPs, transmission electron microscopy (TEM) was performed using Phillips, CM200 instrument equipped with W-filament operated at 200 kV.
Iron K-edge XANES spectra were recorded on the same (ADXRD) beamline in transmission mode, using a pair of ionization chambers. The measured energy resolution (E/ ΔE) of the beamline at ∼8 keV (Cu-K edge) is ∼7000 and the energy repeatability is ∼100 meV. The normalization of the XANES spectra was done for comparison among samples annealed at different temperatures. The differential scanning calorimetric (DSC) measurements of ash particles and as-grown sample were carried out using TA Instruments MDSC model 2910 at a scan rate of 10°C min −1. The magnetic properties were analysed using vibration sample magnetometer (VSM, PPMS from Quantum design, 14 T VSM).
3 Results
3.1 Structural characterization
3.1.1 XRD analysis
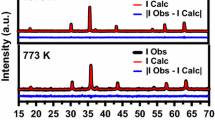
Figure 1 shows SXRD pattern of samples #1–#6 performed on ADXRD beamline on Indus-2 synchrotron source at Indore, India, using photon energy of 11 keV. An analysis of XRD patterns shows that as-prepared NPs (sample #1) exhibit polycrystalline nature and contain at least two crystalline phases. The peaks marked as A in figure 1 are either due to γ-Fe2O3 (JCPDS 15-0615) phase or magnetite (Fe3O4) phase (JCPDS #11-0614; Fd3m). It is difficult from the diffraction peaks of these samples, to differentiate between two phase’s γ-Fe2O3 and Fe3O4 as the d-spacing of the two phases are nearly equal and NPs show broad XRD peaks. Intensities of these peaks (marked A in figure 1) are much smaller than the major peaks and therefore, these peaks are from a minor phase. The peaks marked as B are assigned to α-Fe2O3 phase (JCPDS # 24-0072) having rhombohedral symmetry (space group: R-\(\overline 3\) [148]). These peaks are from the major phase. On isochronal annealing of as-prepared samples, the intensities of the peaks marked A, corresponding to γ-Fe2O3 or magnetite (Fe3O4) tend to decrease with the increase in annealing temperature, while intensities of the peaks marked B corresponding to α-Fe2O3 become stronger. This is in agreement with the reported annealing studies of Bora et al,[19] on iron oxide NPs synthesized by wet chemical route. However, in the present paper the minor phase has been attributed to γ-Fe2O3. It is assumed that it is not possible to assign these peaks uniquely because the d-values are too close and the peaks are broad (particles being nano). Beyond annealing temperature of 400°C, peaks A completely disappear and only peaks corresponding to phase B remain. To determine precisely the temperature at which minor phase disappears, we performed DSC measurements on as-prepared sample and on ash particles. The first derivative of the signal (shown in figure 2) clearly shows that minor phase transforms into major phase at 442°C. DSC scan of ash particles (figure 3) does not show any signal.

X-ray diffraction patterns for samples #1–#6 of iron oxide annealed in air at various temperatures. Samples #1–#3 show mixed phase, whereas #4–#6 show single phase. γ-Fe2O3 or Fe3O4 and α-Fe2O3 phases are represented by symbols A and B, respectively.

Differential scanning calorimetry plot of as-prepared sample (derivative) as a function of increasing temperature.

Differential scanning calorimetry plot of ash particles and as-prepared sample as a function of increasing temperature.
Size, shape and microstrain play important role in determining the phase transformation and magnetic properties of iron oxide NPs.[19–21]We have undertaken a diffraction peak analysis to estimate average particle size and microstrain. We will comment on the shape of the synthesized NPs, while discussing TEM results (section 3.1.2). We have employed Williamson–Hall (W–H) analysis to get average particle size and strain in the samples. According to the W–H method the individual contribution to the broadening of diffraction peaks can be expressed by the following equation:[22]
In the above equation β measured is the experimentally measured FWHM of the broad peak, β instrumental is the instrumental broadening calculated from the diffraction broadening of the NIST LaB6 standard sample
where β is the FWHM of the Bragg peaks (in radians), 2𝜃 the Bragg angle and λ the wavelength of the X-ray (=0.11211 nm), L the particle size in nm and ε the microstrain. The obtained data have been used to construct a linear plot of β 2 cos 2 𝜃 as a function 4 sin 2 𝜃 ((see equation (4)), as shown in figure 4. The intercept on y-axis and the slope of the best fit straight line through data points give (0.9 / L)2 and 4 ε 2, respectively. This gives the particle size and the microstrain. In figure 5, we have plotted particle size and microstrain as a function of annealing temperature. It is seen that the particle size decreases first from 74 ± 4 nm (for sample #1) to 40 ± 4 nm for sample #3 and then increases monotonously up to 168 ± 4 nm for sample #6. The obtained particle sizes have been further confirmed by TEM (discussed in section 3.1.2). We find that the microstrain is about 5 times larger in mixed phase compared to those in single phase. After phase transformation, the strain in single phase is independent of annealing temperature.

Williamson–Hall Plot for as-prepared (#1) and annealed samples (#2–#6). The intercept on the y-axis gives the particle size and the slope of the best fit straight line gives the microstrain for all samples.

Particle size and strain variations with varying annealing temperatures.
It is important to note that the NPs transform from a mixed phase (mixture of α-Fe2O3 and γ-Fe2O3 or Fe3O4) to a single phase (α-Fe2O3) at annealing temperatures of 440°C. Also, the sizes of the particles first decreases up to sample #3 (annealed at 400°C) then it increases due to transformation of mixed phase (major + minor) in to single phase (major phase). As shown in figure 5, we see an inflection point around the size of about 40 nm. In the literature,[23] it has been reported that the critical size for size induced α- to γ-Fe2O3 phase transformation is 40 nm. The analysis suggests that the phase transformation may be particle size induced. Also a large drop in microstrain is observed, for samples annealed at more than 400°C. For precise identification of the minor phase (γ-Fe2O3 or Fe3O4), we have employed XANES measurements on the samples (to be discussed in section 3.1.3).
3.1.2 Transmission electron microscopy (TEM)
Figure 6a shows typical TEM characterization of ash particles. Electron diffraction pattern shows the presence of pure amorphous phase. The corresponding images do not give any signature showing the presence of NPs. Figure 6b–d shows micrographs, high-resolution images and electron diffraction patterns of sample #1 (as prepared), samples #3 and #6 annealed at 400 and 700°C, respectively. Average particle size obtained for samples #1, #3 and #6 are 25 ± 10, 17.6 ± 10 and 72 ± 10 nm, respectively. Particle size was measured using image J software. More than fifty particles were included in each case. The results are in qualitative agreement with those obtained using XRD analysis. Particle sizes were also measured taking dark field images and the results were in reasonably good agreement with the particle sizes obtained using W–H plot from XRD peak widths. Corresponding selected area electron diffraction patterns for all these samples are shown in figures 6b–d. Diffraction pattern analysis shows that samples #1 and #3 (as-prepared and annealed at 400°C) have a mixed phase of α-Fe2O3 and γ-Fe2O3/Fe3O4. However, analysis of electron diffraction patterns for sample annealed at 700°C show only α-Fe2O3. The d-values obtained from HR images are 3.69 Å for (as-prepared) sample #1, 2.17 and 3.7 Å for samples #3 and #6, respectively.

TEM micrograph, high-resolution (HR) image and electron diffraction of samples (a) no diffraction rings for ash particles as expected, (b) #1—as grown, (c) #3—400°C and (d) #6—700°C.
In XRD result, we observed that the minor phase (γ-Fe2O3 or Fe3O4) disappears after 400°C. It is also confirmed from DSC results that phase transformation temperature is 442°C. The electron diffraction pattern for all samples again confirms that minor phase is γ-Fe2O3 or Fe3O4 converted to α-Fe2O3 phase.
3.1.3 Spectroscopic characterization (XANES)
As discussed in section 3.1, XRD of NPs cannot distinguish between γ-Fe2O3 and Fe3O4 phases because of close d-values of the two phases and large FWHM. However, Fe is in 3 + and 8/3 + oxidation states in the two phases, respectively. The presence of Fe3O4 in the mixed phase is likely to increase average oxidation state of Fe in the mixture. XANES is a proven technique to estimate average oxidation state of transition metal, in a compound. We have, therefore, undertaken spectroscopic characterization (using XANES) of the samples to resolve the issue. Figure 7 shows Fe-K edge XANES spectra of samples #1–#6 along with that of a standard Fe (NO2)3 (with known oxidation state of Fe). All the curves have been normalized and vertically shifted for clarity. The XANES spectra of transition metal oxide samples have four features marked ‘P’, ‘A’, ‘W’ and ‘B’ as shown in the figure. The pre-edge (marked ‘P’) is located at about 10 eV below the main absorption edge (marked ‘A’) and is attributed to dipole forbidden transition 1s →3d.[24]This feeble quadrupole transition is activated by the mixing of 3d–4p orbitals of the metal and also by the mixing of 3d–2p orbitals of the metal and the legand. This transition (1s →3d) is more intense when Fe ions are in tetrahedral coordination compared to those with octahedral coordination.[24] A part of Fe in Fe3O4 (having bivalency of 2 and 3) is coordinated and hence gives more intense pre-edge (1s →3d) transition than that in pure Fe2O3 phase, which is purely octahedrally coordinated. For our samples, the intensity of the pre-edge is very feeble and the same for all the samples, including the samples which are in single α-Fe2O3 phase. This indicates that the minor phase is γ-Fe2O3 and not Fe3O4. However, the concentration of Fe3O4 phase is small as shown from the XRD results,we will further confirm this from the analysis of the energy position of the main absorption edge. This discussion indicates that the mixed phase samples #1–#3 have γ-Fe2O3 as minor phase.

Fe-K edge XANES spectra of samples #1–#6 and that of the standard Fe(NO2)3 sample.
The main absorption shoulder (marked ‘A’ in the figure) is assigned to the dipole forbidden transition 1s →4s.[24]Finally, the feature marked ‘W’ is called the white line and is assigned to 1s →4p dipole transition. The curve beyond ‘B’ is EXAFS region and represents nearer neighbour chemistry of the Fe atom. The energy position of the absorption edge (marked ‘A’ in figure 7) indicates the average oxidation state of Fe in the sample.[25]The edge gets blue shifted with increase in the oxidation state of absorbing atom. To exactly find the energy position of the absorption shoulder, the XANES spectra were differentiated and the maxima of the peaks were taken as the energy value. For our samples, we find that the edge energies thus obtained are tabulated in table 1. From the table, we find that edge energies of all the samples are found to be in the range 7127.5 ± 0.5 eV. So the edge energies of all the samples are within the error bars of the measurements. Hence, the average oxidation state of all the samples as + 2. These corroborate the fact that minor phase in samples # 1–# 3 is γ-Fe2O3.
3.1.4 Magnetic measurements
To study the magnetic properties, M–H measurements for samples #1 and #5 were carried out by applying the magnetic field of 2.5 T at room temperature (figure 8). These samples were chosen because these samples have approximately the same size but different phases and therefore, the change in magnetic properties can be attributed to the change in phase alone. It may be mentioned here that the magnetic properties of transition metal oxide NPs depend upon size in addition to other factors.[26] From figure 8, the magnetization of as-prepared iron oxide (#1) was found to be ∼5.67 emu g −1 and for sample annealed at 600°C (#5) was ∼3.7 emu g −1. It may be mentioned that sample #1 is in mixed phase of α-Fe2O3 and γ-Fe2O3, whereas, sample #5 is in single-phase α-Fe2O3. Bulk α-Fe2O3 is canted antiferromagnetic (AF) at lower temperature and shows weak ferromagnetic behaviour above 260 K (Morin temperature). The weak ferromagnetism in this case is because of canting of AF aligned spins. In the literature, M s value for bulk α-Fe2O3 is 2.1 emu g −1.[27,28]Bulk γ-Fe2O3 is ferromagnetic with M s = 76 emu g −1. It is interesting to note that the M s for γ-Fe2O3 NPs have been reported to be both lower 13.35 emu g −1[20] and higher 106 emu g −1[21]than the bulk value, depending upon the shape and size of the particles. For our samples, the magnetization is not saturated at higher magnetic field. Therefore, the obtained values are not saturation magnetization (M s) but these values are maximum magnetization (\(M_{\max }\)). The sample #5 (single-phase α-Fe2O3) particles show maximum magnetization value of 3.7 emu g −1, which is higher than the reported bulk value. This may be because of higher canting angle for NPs than reported 5° canting angle for bulk α-Fe2O3. We found that even higher applied magnetic field the magnetization has not saturated specially for the mixed phase (as-prepared) sample. This clearly shows that the maximum magnetization of mixed phase (α-Fe2O3 and γ-Fe2O3) is much higher than the single phase (α-Fe2O3). Hence we conclude that the minor phase in our sample is γ-Fe2O3, which is ferromagnetic.

M–H curve of sample #1 (as-prepared) and sample #5 (annealed at 600°C).
4 Discussion
Phase transformation studies in iron oxide NPs have been reported earlier as a function of annealing temperature[17] have investigated sonochemically prepared Fe2O3 NPs. They find that annealing in vacuum at temperatures between 240 and 450°C produces γ-Fe2O3, with average particle size between 4 and 14 mm. On further annealing of γ-Fe2O3 in air gives α-Fe2O3 phase. Chernyshova et al[23] studied size-dependent structural transformations using FTIR, Raman spectroscopy, XRD and TEM. They have found that the phase transformation α-Fe2O3 to γ-Fe2O3 depends upon size. They have discussed their results by considering a general model taking into account spinel defect and absorbed species (hydroxyles in their case). The critical size of NPs at which phase transformation takes place depends upon the environment (dry or hydroxyle). Bora et al[19] have also studied Fe2O3 samples synthesized by the wet chemical method. Using XRD, XANES and TEM techniques on the samples annealed up to 900°C in the air, they showed that at lower annealing temperatures the sample comprises of maghemite (γ-Fe2O3) and haematite (α-Fe2O3) phases and with increasing annealing temperature, γ-Fe2O3 transforms into α-Fe2O3 (above 350°C). TEM investigation showed prismatic morphology of the particles having stacking faults.
We observed that, as-prepared sample contains mixed phases of α-Fe2O3 (as a major phase) and γ-Fe2O3 or Fe3O4 (as a minor phase). On annealing in air the minor phase transforms to a major phase for annealing beyond 440°C. Table 2 summarizes phase transformation in iron oxide NPs studied by Chernyshova et al,[23] Xu et al,[17] Bora et al[19] and the present work. From these data one can say that although in most of the cases final phase at temperatures >450°C is α-Fe2O3, the sequence of phase transformation is different. In all the cases the method of preparation is also different. XANES measurements have been used to confirm that the minor phase is γ-Fe2O3. These results are in agreement with Bora et al[19] and Xu et al[17]. VSM studies show that maximum magnetization (\(M_{\max }\)) for α-Fe2O3 NPs is higher than its bulk value reported in the literature.[27,28]This may be because of the higher canting angle for NPs.
5 Conclusions
Iron oxide NPs have been synthesized by the simple, environmental friendly and cost effective ash-supported technique. Structural analysis using SXRD and electron diffraction techniques show that as-prepared sample consists of mixed phase of α-Fe2O3 (major phase) and a minor phase consisting of either γ-Fe2O3 or Fe3O4 phase. On annealing, the minor phase gradually transforms to the major phase. The phase transformation is nearly complete for annealing at ∼440°C. The result was corroborated by DSC scan and the exact phase transformation was found to be 442°C; in agreement with the XRD results.
From spectroscopic analysis using XANES, we confirm that the minor phase is γ-Fe2O3, for the first time. Williamson–Hall analysis of XRD data shows the particle size first decreases and then increases with the increase in annealing temperature. The minimum size being ∼40 nm, which is also the critical particle size for size-induced phase transformation. The sizes of nanoparticles were also confirmed by the TEM images and found to be in reasonably good agreement, with XRD results. Microstrain obtained by W–H analysis shows that the strain is about five times larger in mixed phase than in single-phase NPs. Magnetic measurements revealed that the maximum magnetization for nanophase α-Fe2O3 is higher than that reported for bulk. Also the presence of ferromagnetic γ-Fe2O3 phase in the mixed phase sample increases the magnetization value, as expected. Since the phase transformation temperature is ∼440°C, the magnetic phase is clearly γ-Fe2O3 and not Fe3O4.
References
Coey J M D 1971 Phys. Rev. Lett. 27 1140
Speliotis J E 1999 J. Magn. Magn. Mater. 193 29
Perez J M 2007 Nat. Nanotechnol. 2 535
Kim D K, Zhang Y, Voit W, Rao K V, Kehr J, Bjelke B and Muhammed M 2001 Scr. Mater. 44 1713
Han S, Yu T, Park J, Koo B, Joo J, Hyeon T, Hong S and Im J 2004 J. Phys. Chem. B 108 8091
Butter K, Philipse A P and Vroege J G 2002 J. Magn. Magn. Mater. 252 1
Ago H, Nakamura K, Imamura S and Tsuji M 2004 Chem. Phys. Lett. 391 308
Daou T J, Greneche J-M, Lee S-J, Lee S, Lefevre C, Sylvie B-C and Pourroy G 2010 J. Phys. Chem. C 114 8794
Sahoo S K, Agarwal K, Singh A K, Polke B G and Raha K C 2010 J. Eng. Sci. Technol. 2 118
Leslie-Pelecky D L and Rieke R D 1996 Chem. Mater. 8 1770
Chen P, Zhang H B, Lin G D, Hong Q and Tsai K R 1997 Carbon 35 1495
Vales V, Poltierova-Vejpravova J, Mantlikova A, Holy V, Trypekt V, Brazda P and Dolye S 2010 Phys. Status Solidi (c) 7 1399
Guo X, Zhong S, Zhang J, Wang W, Mao J J and Xie G 2010 J. Mater. Sci. 45 6467
Jeong J-R, Lee S-J, Kim J-D and Shin S-C 2004 Phys. Status Solidi (b) 241 1593
Kim D K, Kim S S, Choa Y-H and Kim H T 2007 Ind. Eng. Chem. 13 1137
Xu L, Zhang W, Ding Y, Peng Y, Zhang S, Yu W and Qian Y 2004 J. Phys. Chem. B 108 10859
Xu X N, Wolfus Y, Shaulov A and Yeshurun Y 2002 J. Appl. Phys. 91 4611
Sinha A K, Sagdeo A, Gupta P, Kumar Ashok, Singh M N, Gupta R K, Kane S R and Deb S K 2011 AIP Conf. Proc. 1349 503
Bora Dabjeet K, Artur B, Selma E, Olga S, Thomas G and Costable Edwin C 2012 Curr. Appl. Phys. 1213 817
Layek S, Pandey A, Pandey A and Verma H C 2010 Int. J. Eng. Sci. Technol. 2 33
Zhou S-M, Zhang X-T, Gong H-C, Zhang B, Wu Z-S, Du Z-L and Wu S-X 2008 J. Phys.: Condens. Matter 20 075217
Williamson G K and Hall W H 1951 Acta Metall. 1 22
Chernyshova I V, Hochella M F and Madden A S 2007 Phys. Chem. Chem. Phys. 9 1736
Sankar D, Sarode P R and Rao C N R 1983 Chem. Phys. 76 435
Wilke M, Farges F, Petit P-E, Brown G E and Martin F 2001 Am. Mineral. 86 714
Shakeel A, Hasanain S K, Nasia A and Nadeem M 2008 J. Appl. Sci. Res. 3 417
Dhiman P, Kumar A and Singh M 2012 Adv. Mater. Lett. 3 330
Shan G, Liu H, Xing J, Zhang G, Ma Z and Liu J 2002 EU-forum of nanotechnology, Beijing, China, December, p 68
Acknowledgements
We acknowledge the help provided by Mr MN Singh, RRCAT, Indore in XRD and XANES measurements, Dr. Awasthi and Mr Bhardwaj, UGC, DAE-CSR, Indore, for DSC measurement and Dr Premlata Gupta, for providing the annealing furnace. Thanks are also due to Dr ML Sharma, Director, Institute of Science and Laboratory Education, IPS Academy, for his valuable support and extending the permission to perform this work. I am also thankful to Dr Usha Singh for her valuable support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
DEOTALE, A.J., NANDEDKAR, R.V., SINHA, A.K. et al. Effect of isochronal annealing on phase transformation studies of iron oxide nanoparticles. Bull Mater Sci 38, 599–606 (2015). https://doi.org/10.1007/s12034-015-0914-z
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12034-015-0914-z