
Abstract
Colorectal cancer (CRC) is a highly prevalent cancer worldwide, but treatment outcomes can vary significantly among patients with similar clinical or historical stages. This study aimed to investigate the differences in immune cell abundance associated with malignant progression in CRC patients. We utilized data from patients with CRC obtained from The Cancer Genome Atlas as our training set. To assess immune cell infiltration levels, an immune cell risk score (ICRS) was calculated. Furthermore, we performed network analysis to identify effective T cell-related genes (ETRGs) and subsequently constructed an effective T cell prognostic index (ETPI). The performance of the ETPI was evaluated through external validation using four Gene Expression Omnibus datasets. Additionally, a nomogram analysis and drug sensitivity analysis were conducted to explore the clinical utility of the ETRGs. We also examined the expression of ETRGs in clinical samples. Based on the ICRS, we identified activated CD4+ and CD8+ T cells as protective factors in terms of prognosis. Six ETRGs were identified to develop the ETPI, which exhibited remarkable prognostic performance. In the external validation of immunotherapy, the low ETPI group demonstrated a significantly lower recurrence rate. To optimize therapeutic strategies, we developed a nomogram. Notably, patients with different ETPI values exhibited varying responses to tumor pathway inhibitors. Finally, we observed higher protein expression of certain ETRGs in normal tissues compared to tumors. Our findings suggest that the ETPI may contribute to the precise selection of patients based on tumor microenvironment and key genomic landscape interactions, thereby optimizing drug benefits and informing clinical strategies in future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is the third most common type of malignant tumor in both men and women, with an estimated 1.9 million new cases expected in 2023 [1]. CRC is the second leading cause of cancer-related deaths worldwide [2]. As a heterogeneous disease, CRC is marked by numerous molecular alterations that disrupt various signaling pathways, contributing to its malignancy, invasiveness, and progression [3]. Clinically, tumor staging as defined by the American Joint Committee on Cancer (AJCC) remains the standard guideline for risk stratification in individual patients. However, it is worth noting that many patients with similar clinical and historical stages may exhibit significantly different prognoses. This discrepancy may be attributed to the fact that the current guideline does not take into account the molecular biologic characteristics of the tumors [4,5,6,7].
The tumor microenvironment (TME) is a complex network of cells and biomolecules that play a crucial role in the initiation, progression, and metastasis of tumors. In addition to tumor cells, the TME includes a variety of infiltrating cell types, such as fibroblasts, immune cells, inflammatory cells, interstitial cells, microvessels, and biomolecules. Among these cell types, immune cells are particularly important as they participate in the tumor immune response and regulate tumor growth and metastasis by modulating the TME at the cellular level. CD4+ and CD8+ T cells are two common types of immune cells that infiltrate tumors and are considered important anti-tumor cells with different densities observed in solid tumors.
Many cancer types, including both primary and metastatic forms, exhibit short disease-free survival (DFS) and overall survival associated with low infiltration of CD8+ T cells, CD3+ T cells, and CD45RO+ memory T cells. This pattern is observed in CRC, squamous cell carcinomas (SCCs), and other adenocarcinomas [8, 9]. However, it is important to consider that tumor progression in these cancers may be influenced by a higher density of CD8+ T cells, which could contribute to advanced malignancy [10]. CD4+ T cells, encompassing a broad range of functions and categories, such as T helper 1 (Th1) cells, T helper 2 (Th2) cells, T helper 17 (Th17) cells, regulatory T (Treg) cells, and T follicular helper (TFH) cells, have variable prognostic values. These cells interact with secreted cytokines, proinflammatory cytokines, and TME. The increased presence of tumor-infiltrating lymphocytes has been associated with favorable treatment outcomes, regardless of other notable clinicopathological factors, such as stretches of DNA microsatellite instability (MSI) [11]. MSI, one of the three distinct pathways in genomic instability in CRC, is a result of a defect in the DNA mismatch repair (MMR) system [12]. Additionally, deficient DNA mismatch repair (dMMR) activates an anti-tumor immune response in TME.
Therefore, the abundance of CD4+ T and CD8+ T cells indicates patients’ prognosis throughout CRC development. Anti-cancer immune factors cannot effectively treat the so-called “cold” tumors that lack infiltration of CD4+ and CD8+ T cells, leading to resistance to immune checkpoint blockade (ICB) monotherapy, neoadjuvant chemotherapy, and worse clinical outcomes [13]. Although there are many studies related to CD4+ and CD8+ T cells in other categories of cancers, from the point of bioinformatics, few studies on the clinical application and cellular gene interactions in the field of colorectal neoplasms have been conducted. Hence, transitioning from the conventional approach to diagnosing and treating CRC, there is an urgent need to leverage genomics and cell-level analyses to uncover key genes involved in the interaction between CD4+ and CD8+ T cells. Developing a prediction model that integrates the molecular characteristics of these lymphocytes aligns with the current trajectory of clinical advancement.
In this study, our objective was to utilize multiple gene expression datasets to analyze variations in immune cell abundance associated with the malignant progression in patients with CRC. Additionally, we developed a personalized risk stratification index and investigated its clinical value in prognostic estimation, with a specific focus on differences in infiltrated lymphocyte populations and key genes. Furthermore, a comparative study involving immunohistochemistry (IHC) was conducted to detect these key genes. This study has the potential to aid in the identification of specific CRC patients, optimizing treatment plans, and improving outcomes.
Materials and Methods
Publicly Available Gene Expression and Clinical Data Collection and Processing
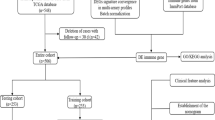
In total, bulk-RNA sequencing data in count and fragments per kilobase million (FPKM) forms of 554 patients with CRC were accessed from The cancer genome atlas (TCGA) (https://portal.gdc.cancer.gov/) for the construction of immune cell risk score (ICRS) and selection of effective T cell-related genes (ETRGs), which were used to generate effective T cell prognostic index (ETPI), as a training set. Clinical data encompassing completely cured survival information of 728 patients from the TCGA Colon and Rectal Cancer cohort (COADREAD) were downloaded from the UCSC Xena database (https://xenabrowser.net/datapages/). The FPKM value was transformed into transcripts per million (TPM) for subsequent analysis. Progression-free interval event (PFI) and PFI time were used to assess patients with cancer progression or death in TCGA.
A total of 414 patients with CRC, who had progression-free interval (PFI) times exceeding 30 days, were included in the screening process for pre-processing. For validation of the individualized index of risk stratification (ETPI), we selected GSE14333 and GSE38832 datasets from the gene expression omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/), which provided disease-free survival (DFS) information for 226 and 92 patients with CRC, respectively. Furthermore, we utilized GSE39582 dataset, which included recurrence-free survival (RFS) information for 519 patients, to evaluate the performance of the signature in predicting recurrence. GSE78220 was further collected for the evaluation of immunotherapy benefits and included 10 partial responders (PR), 4 complete responders (CR) to pembrolizumab, and 13 patients with progressive melanoma.
Construction and Validation of the ICRS
Single-sample gene set enrichment analysis (ssGSEA) for infiltration assessment of 28 types of tumor-infiltrating immune cells was performed, and the cells for PFI prediction were screened via ssGSEA followed by least absolute shrinkage and selection operator (LASSO) analysis. LASSO is a linear regression method based on L1 regularization. L1 regularization significantly increases the stability of modeling and achieves the purpose of selecting the most characteristic cell types. To remove the multicollinearity effect of immune cells that contribute little to the prognosis, it is required to establish ICRS to evaluate the comprehensive immune cell status in patients with CRC. The ICRS was further developed based on 10 types of immune cells, and the most accurate prognostic value after processing the gene set variation analysis (GSVA) algorithm was obtained via the expression profile of specific immunological marker genes [14]. The corresponding coefficients were identified by multivariate Cox analysis using the following formula: ICRS = \(\sum Coef\times {x}_{i}\), where Coef is the mean of the risk coefficient and \({x}_{i}\) is the richness of immune cells indicating prognosis in CRC.
We established divided groupings based on high and low scores using the optimal cut-off value of ICRS. This cut-off value was determined using the receiver operating characteristic curve (ROC) method in the ggrisk package from R software (version 4.1.1). We selected the cut-off point that showed a significant difference between the two groups. Subsequently, K–M analysis revealed survival curves across the CRC cohort. A composite heatmap was generated by integrating the single-sample gene set enrichment analysis (ssGSEA) scores of 10 types of cells and clinical characteristic parameters of individual subgroups based on ICRS.
After classification using the overall immune status of individuals based on ICRS, the abundance of 28 types of immune cells in the high- and low-score groups was calculated using the ssGSEA algorithm. To compare the differences in the immune atlas landscape between the two groups, an unpaired t test was conducted to compare the proportions. We analyzed the infiltration of 28 types of immune cells between the high-score and low-score groups and found that only activated CD4+ and CD8+ T cells were more abundant in the low-score group. This suggests that these two cell types may act as protective factors in the prognosis of the disease. Further explorations of the infiltration differences between groups with these two immune cells were performed.
Identification of Hub Genes Related to the Abundance of Activated CD4+ and CD8+ T Cells
Patients from the CRC cohort were collected based on their ssGSEA score and divided into two groups. One group exhibited high infiltration of both activated CD4+ and CD8+ T cells, while the other group showed a low abundance of both types of lymphocytes.
The DESeq R package in R software (4.1.1) was used to compare the genomic expression profiles and detect differentially expressed genes (DEGs) between the two groups. Then, to minimize the potential biologic function insight, the DEGs between groups were separated by ssGSEA analysis. Gene ontology (GO) enrichment and Kyoto encyclopedia of genes and genomes (KEGG) analyses were used to calculate the top enrichment categories in biologic behaviors depending on the P value. The DEGs that met the screening parameters, including a log2 fold change (|logFC|) greater than 0.585 and a false discovery rate (FDR) below 0.05, were subjected to the weighted gene co-expression network analysis (WGCNA) procedure. The soft threshold β was set to 3, which provided a power value for the development of the co-expression network. Next, the gene expression data were transformed into a topological overlap matrix (TOM). A dynamic shearing module was used to identify the results of gene clustering. Subsequently, a hierarchical clustering dendrogram was generated based on module eigengene (ME) calculation and correlations between MEs and ssGSEA scores of the two cell types. An integrated heat map was built to identify the most favorable MEs.
Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, https://string-db.org/) was used to establish a protein–protein interaction (PPI) network to determine the relationships among genes from MEs. The hub genes and key modules were depicted in Cytoscape (version 3.5.1; http://www.cytoscape.org). Subsequently, a degree cut-off (top 10% ranked) was applied, and Cytohubba, a plugin of Cytoscape software, was utilized to identify hub genes associated with the abundance of activated CD4+ and CD8 + T cells. The genes identified in the PPI network underwent further analysis using ClueGO, a plugin within Cytoscape, with kappa statistics applied to GO terms, KEGG, and REACTOME categories.
Consensus Development and Evaluation of ETPI
The glmnet R package was utilized to conduct LASSO regression analysis on hub genes, aiming to identify the ETRGs for evaluating the infiltration of activated CD4+ and CD8+ T cells in CRC. Subsequently, ETPI was defined using the following formula: ETPI = \(\sum Gene expression \times Coef\) where gene expression: expression value of the ETRGs and Coef: risk coefficients of abundance of activated CD4+ and CD8+ T cells in CRC.
The patients were classified into high- and low-score groups after processing the optimal cut-off value based on the maximum Youden index in survminer R package when conducting the K–M and ROC curve analyses to evaluate the validity of ETPI. A heatmap depicting the differential expression among ETRGs was integrated with the clinical parameters in the CRC cohort.
To assess the independent prognostic significance of ETPI, univariate and multivariate Cox regression analyses were conducted. Stromal and immune scores in the CRC cohort were normalized and calculated using the ESTIMATE algorithm. Subsequently, a boxplot was utilized to illustrate the distribution trend of stromal and immune scores in the high and low ETPI groups. The maftools R package was employed to analyze differences between the high and low ETPI groups using waterfall charts to depict somatic mutation distribution. Somatic mutation analysis was conducted to calculate the tumor mutational burden (TMB) for each patient. The relationship between the index and TMB was assessed through Pearson correlation analysis.
External Validation of ETPI
To verify the rationality and stability of ETPI, three GEO datasets (GSE14333, GSE38832, and GSE39582) were selected as external verification cohorts, including the gene expression matrices of 226 patients with CRC with DFS and gene expression levels of 92 patients with CRC and 519 patients with CRC with RFS, respectively. The same formula was applied to generate the ETPI for validation. The K–M analysis was employed to demonstrate the correlation between ETPI and both DFS and RFS statuses of patients in the GEO external validation cohorts.
Subsequently, we investigated the potential importance of ETPI in immunotherapy. We chose GSE7822027 to examine the objective response rate (ORR) of patients with advanced melanoma who underwent pembrolizumab treatment. The dataset included 10 patients who exhibited a partial response (PR) and four patients who achieved a complete response (CR). To analyze this data, we employed Fisher’s precision probability test.
Construction and Validation of a Nomogram for Patients with CRC
To fully leverage the interaction between our signature and clinical indicators, we assessed MSI status through univariate and multivariate Cox regression analyses, alongside clinical parameters and ETPI, as independent prognostic factors. Following the establishment of the nomogram, accuracy was evaluated using calibration curve plots. Subsequently, decision curve analysis (DCA) was conducted to ascertain the clinical benefit in medical practice.
Immune Checkpoint and Drug Sensitivity Analysis
Immune checkpoint analysis was performed by comparing the high and low ETPI groups from the TCGA CRC cohort. The drug analysis datasets were accessed from the genomics of drug sensitivity in cancer (GDSC) (https://www.cancerrxgene.org/) and CellMiner (http://discover.nci.nih.gov/cellminer/) databases to explore the drug sensitivity of ETRGs in patients with CRC via Oncopredict R package. Subsequently, the potential relationship between the expression profile of ETRGs and drug sensitivity was determined using the Pearson correlation test.
Validation of the Protein Expression Levels of the ETRGs
The protein expression levels of the ETRGs in both CRC and normal tissues were verified, subsequently. The data of immunohistochemistry (IHC) analysis were obtained from the Human Protein Atlas database (HPA database, https://www.proteinatlas.com). The HPA database provides IHC results and contains data on proteomics-based analysis of tumor and normal tissues.
Statistical Analysis
In total, data analyses and plotting were performed using R software (4.1.1). Cox regression and K–M analyses were conducted using the survival R package. Pearson’s correlation coefficient was used to assess the correlation between the two types of continuous variables. Continuous variables were compared using the Wilcoxon rank-sum test or t test. The optimal cut-off value was formulated using the survminer package. The time-dependent area under the ROC curve (AUC) for survival variables was calculated using the timeROC package. All statistical tests were two-sided. P < 0.05 indicates a statistically significant difference.
Results
Construction and Validation of the ICRS
Based on the ssGSEA and LASSO regression analysis (Fig. 1A, B) of the abundance of 28 types of infiltrating immune cells, ICRS was constructed according to 10 cell types with the following formula: infiltration status of activated CD4+ T cell × (− 1.13039891) + infiltration status of activated CD8+ T cell × (− 0.76299930) + infiltration status of activated dendritic cells × (− 0.15277958) + infiltration status of central memory CD8+ T cell × 2.46604479 + infiltration status of eosinophil × 0.08038523 + infiltration status of gamma delta T cell × (− 3.03445654) + infiltration status of immature dendritic cells × 2.66387634 + infiltration status of mast cells × 0.68973920 + infiltration status of plasmacytoid dendritic cells × 0.90481339 + infiltration status of type1 T helper cells × 2.17922932. The clinical information, PFI, and the abundance of 16 types of immune cells in each patient from the high- and low-score groups, determined by an optimal cut-off value, are illustrated in Supplementary Fig. 2.

Construction and validation of ICRS. A LASSO coefficient profiles of immune cells. B Cross-validation of selection of tuning parameters in LASSO regression analysis. C K–M curves of PFI in divisions of patients with CRC based on ICRS. D ROC curves of ICRS for 1, 3, and 5 years in TCGA training cohort. E, F Forest plots of univariate and multivariate Cox regression analyses of ICRS and clinical parameters in TCGA training cohort. G Integrated heatmap of ICRS, age, sex, clinicopathological parameters, AJCC stages, and ssGSEA scores of 10 types of immune cells. ICRS the immune cell risk score, LASSO least absolute shrinkage and selection operator, K–M Kaplan–Meier, PFI progression-free interval event, CRC colorectal cancer, TCGA the cancer genome atlas, ROC receiver operating characteristic, ssGSEA the single-sample gene set enrichment analysis
In addition, K–M analysis revealed an unfavorable PFI in the high-score group (Fig. 1C). The results illustrated the dismal infiltration of activated CD4+ and CD8+ T cells in the high-score group compared with the low-score group in the training set. ROC analysis was performed to assess the efficiency of ICRS with the area under receiver operating characteristic curves (AUCs) for 1-, 3-, and 5 year PFI time prediction, which were 0.62, 0.69, and 0.73, respectively (Fig. 1D). Based on the training cohort, univariate and multivariate Cox regression analyses were performed to identify whether ICRS was an independent prognostic factor for CRC (P < 0.05) (Fig. 1E, F). The forest plot derived from univariate and multivariate Cox regression analyses demonstrated that the hazard ratio (HR) of the ICRS was greater than 1, with a P value less than 0.05. This finding indicates that ICRS is an independent factor with negative impacts on the survival and prognosis of patients with CRC.
The heatmap displaying clinical parameters and infiltration abundance of selected immune cells revealed that only activated CD4+ and CD8+ T cells had lower infiltration levels in the high-score group, which experienced a shorter PFI time. The abundance of other immune cells increased in correlation with poor prognosis and advanced cancer stages (Fig. 1G). Furthermore, to depict the tumor immune microenvironment between high- and low-score groups, a boxplot was used to display the difference in the abundance of 28 types of immune cells, which showed that the density of 18 immune cells was significantly different in the distribution of immunological proportion (Fig. 2A). In addition, among the 18 types of immune cells mentioned above, only the activated CD4+ and CD8+ T cells presented more infiltrating conditions in the low-score group. These results imply that activated CD4+ and CD8+ T cells might play important roles in the prediction of disappointing tumor progression in CRC.

Immune infiltration and survival analysis of ICRS. A Comparison of 28 types of tumor-infiltrating immune cells between the high- and low-score groups. B K–M curves of PFI in patients with CRC with both high and low infiltration of activated CD4 + and CD8 + T cells. ICRS the immune cell risk score, LASSO least absolute shrinkage and selection operator, K–M Kaplan–Meier, PFI progression-free interval event, CRC colorectal cancer, TCGA the cancer genome atlas, ROC receiver operating characteristic, ssGSEA the single-sample gene set enrichment analysis
Combined Analysis of Activated CD4 + and CD8 + T Cell Infiltration in Prognostic Assessment
Patients from the training set were screened using the ssGSEA scores related to the abundance of activated CD4+ and CD8+ T cells. To further explore the significance of these two types of T cells in immune infiltration, we used the optimal cut-off value to classify patients into high- and low-infiltrating divisions based on the ssGSEA score of activated CD4+ and CD8+ T cells, respectively. K–M analyses showed that samples with low tumor-infiltrating activated CD4+ T cells and those with low tumor-infiltrating activated CD8+ T cells had poor prognoses (see Supplementary Fig. 1A–D). Thus, we performed a combined survival analysis by integrating these subgroups (both high-infiltrating activated CD4+ and CD8+ T cells and both low-infiltrating activated CD4+ and CD8+ T cells), revealing that the differences between the integrated groups appeared to be more remarkable (Fig. 2B).
Identification of Hub Genes Related to the Abundance of Activated CD4+ and CD8+ T Cells
To further investigate the two groups with the most significant survival differences identified in the combined prognostic analysis, we identified 2107 DEGs by comparing the gene expression profiles (Supplementary Fig. 3A). We then conducted WGCNA to identify gene modules most relevant to the ssGSEA score of activated CD4+ and CD8+ T cells. The genes were clustered into 10 modules labeled with different colors, as depicted in Supplementary Fig. 3B. The yellow module exhibited the highest correlation between MEs and the corresponding modules, indicating a strong potential for developing candidate hub genes associated with the ssGSEA score of activated CD4+ and CD8+ T cells (Supplementary Fig. 3B).
To explore the core functions of candidate hub genes, a degree algorithm was used to screen hub genes. A total of 553 genes within the yellow module were utilized to create a PPI network. This network, composed of 405 nodes and 2738 edges, was constructed using the STRING database and subsequently visualized using Cytoscape software. The CytoHubba plugin in Cytoscape was used to sort the previous MEs in descending order according to the degree algorithm score, screened by the top 10% to identify the hub genes. In total, 41 hub genes were selected based on the PPI network analysis, showing interactions with each other in the network (Fig. 3A).

Identification of hub genes and biologic function enrichment analyses. A The PPI network of 41 hub genes related to infiltration of activated CD4 + and CD8 + T cells; B–D GO, KEGG, and REACTOME enrichment analyses of hub genes, according to different gene enrichment regions, shown as a pie chart in the form of percentages. B In GO analysis, regulation of T cell activation (55.36%) is dominant. C In KEGG analysis, hub genes were enriched in Th17 cell differentiation (42.9%). D In REACTOME enrichment analysis, costimulation by the CD28 + family is the top enrichment pathway (57.9%). PPI protein–protein interaction, GO gene ontology, KEGG Kyoto encyclopedia of genes and genomes
Biological Functional Enrichment Analyses
The count data of DEGs between the group with a high abundance of activated CD4+ and CD8+ T cells and the group with low infiltration of these cell types was analyzed using GO enrichment and KEGG pathway analyses to elucidate their potential functions and biologic characteristics (Supplementary Fig. 3C–F).
KEGG pathway analysis identified significantly abundant pathways, including cytokine-cytokine receptor interaction, Th17 cell differentiation, and Th1 and Th2 cell differentiation (Supplementary Fig. 3C, D). The GO enrichment analysis revealed elevated expression in biological process (BP) terms related to the response of IFN-γ, cytokine-mediated signaling pathway, and regulation of T cell activation. In terms of cellular component (CC), enrichments were observed in terminologies associated with the plasma membrane and MHC class II protein complexes. Molecular function (MF) analysis highlighted enriched activities related to immune receptors, receptor ligands, and signaling receptor activation (Supplementary Fig. 3E, F). The differences in the biologic functions mentioned above have the potential to inform the development of intervention therapies targeting low abundance of activated CD4+ and CD8+ T cells, potentially reducing PFI time.
In addition, employing the STRING database platform, we assessed the enrichment of 405 nodes from PPI analysis. The results indicated that the selected genes were primarily enriched in functions associated with immune receptor activity, MHC protein binding, cytokine receptor activity, and other fundamental biologic functions relevant to the behaviors of tumor-infiltrating T cells. To functionally decipher the top 10% of genes, ClueGO was used for functional enrichment analysis. The analysis indicated that “regulation of T cell activation” (55.36%) was the most dominant, with significantly enriched GO terms, followed by “leukocyte mediated cytotoxicity” (14.3%) (Fig. 3B). The predominant KEGG categories were “Th17 cell differentiation” (42.9%) and “Graft-versus-host disease” (39.3%). In the REACTOME pathway terms, “Costimulation by CD28 family” and “Phosphorylation of SLP-76 by p-SYK” were the top enriched pathways, with 57.9% and 19.7%, respectively (Fig. 3C, D).
Development and Evaluation of ETPI
The 41 hub genes associated with the immune abundance of activated CD4+ and CD8+ T cells in CRC underwent LASSO regression analysis to construct the ETPI (Fig. 4A, B). Finally, six effective ETRGs were identified, including FOXP3, IL15, PDCD1, MX1, CASP1, and IDO1. The formula for the prognostic risk assessment of ETPI was defined as follows: ETPI = (− 0.10605548) × Exp (FOXP3) + (− 0.11924243) × Exp (IL15) + (0.061298204) × Exp (PDCD1) + (0.00430608) × Exp (MX1) + (− 0.01306998) × Exp (CASP1) + 0.001462837 × Exp (IDO1), where “Exp” represents the expression profile of the individual of ETRGs. Patients in the low ETPI group had a more favorable PFI than those in the high ETPI group, as demonstrated by the K–M analysis plot (Fig. 4C). ROC analysis evaluated the prognostic discrimination of the ETPI, revealing 1-, 3-, and 5 year AUCs of 0.67, 0.69, and 0.74, respectively (Fig. 4D). The univariate and multivariate Cox regression analyses were performed to show that ETPI was an independent risk factor for CRC (P < 0.05) (Fig. 4E, F). Furthermore, patients in the combined analysis of the TCGA training cohort were assigned into two groups with 181 and 179 patients in high- and low-score groups, respectively. A heatmap integrating the expression of the six ETRGs and clinical characteristics was generated to explore the differences between the two groups (Fig. 4G). A poor prognosis was observed in the high ETPI group, which exhibited higher expression levels of ETRGs and a higher TNM stage, both with statistical significance.

Development and validation of ETPI. A LASSO coefficient profiles of 41 hub genes. B Cross-validation for selection of tuning parameters in LASSO regression. C K–M curves of PFI in patients with CRC in ETPI score-based TCGA cohort. D ROC curves of prognostic signatures for 1, 3, and 5 years in TCGA cohort. E, F Forest plots of univariate and multivariate Cox regression analyses of ETPI and clinical features in TCGA cohort. G Differences in clinicopathologic characteristics and expression level of six ETRGs between the high- and low ETPI groups. ICRS the immune cell risk score, LASSO least absolute shrinkage and selection operator, K–M Kaplan–Meier, PFI progression-free interval event, CRC colorectal cancer, TCGA the Cancer Genome Atlas, ROC receiver operating characteristic, ETPI the effective T cell prognostic index, ETGR effective T cell-related genes
Furthermore, our analysis of the box plots revealed significant statistical differences between the high and low ETPI groups concerning the Immune and ESTIMATE items. However, no such differences were observed in terms of stromal score (Fig. 5A). The correlation curve demonstrated that the immune score and ETPI were negatively correlated (Fig. 5B). Patients in the low ETPI group tended to have a higher distribution of MSI-H status (Fig. 5C). Moreover, patients in the high ETPI group exhibited a higher TMB, with a mutation rate of 97.22% (altered in 175 of 180 samples), compared to 94.64% (altered in 159 of 168 patients) in the low ETPI group (Fig. 5D, E).

Immune status and MSI assessment based on ETPI. A The boxplots related to individual ESTIMATE scores based on the ETPI division. B The correlation curve suggesting a negative correlation between ETPI and immune scores. C The boxplots showing the relationships between ETPI scores and MSI status, with a significant difference in comparison of MSI-H and MSS/MSI-L. D, E The waterfall plots of somatic mutation features were established and analyzed based on the ETPI score, showing more mutation in the high ETPI group (97.22% in the high ETPI group vs. 94.64% in the low ETPI group). Each column represents an individual patient; the upper bar plot on the waterfall plots shows TMB; the percentage numbers on the right side indicate the mutation frequency in each gene: the right bar plot shows the proportion of each variant type. ETPI the effective T cell prognostic index, MSI microsatellite instability, TMB tumor mutation burden. *P < 0.05; **P < 0.01; ***P < 0.001; ns no significance
External Validation of ETPI
To validate the efficacy of ETPI, we utilized 837 patients from CRC cohorts (GSE14333, GSE38832, and GSE39582) obtained from the GEO database as external validation cohorts for further analysis. The same formula was applied to calculate the ETPI of the validation cohorts using the expression values of the six ETRGs. In the GSE14333 dataset, patients were divided into two groups, with 92 patients exhibiting high ETPI relative to 134 with low ETPI, demonstrating significantly more favorable PFI time in the latter (Supplementary Fig. 4A). In the GSE38832 dataset, 51 patients with high ETPI experienced shorter DFS times, while 41 patients with low ETPI tended to have better survival outcomes (Supplementary Fig. 4B). Additionally, in the GSE39582 cohort, 154 patients with high ETPI showed poor DFS time compared to 365 patients with favorable DFS time in the low ETPI group (Supplementary Fig. 4C).
For the evaluation of ORR in immunotherapy, CR, PR, and patients resistant to pembrolizumab application were compared using Fisher’s precision probability test and illustrated via boxplots (P < 0.05) after regrouping based on ETPI. The results revealed that patients achieving a complete response to pembrolizumab were more likely to obtain low ETPI scores, whereas those resistant to pembrolizumab, indicative of malignant progression in melanoma, tended to be classified into the high ETPI group (Supplementary Fig. 4D, E).
Construction and Validation of a Nomogram
Subsequently, univariate and multivariate Cox regression analyses were employed to identify risk factors meeting the threshold (P < 0.05) in the training dataset. Seven candidates emerged as significant predictors of PFI time, leading to the generation of a nomogram. This nomogram incorporated microsatellite status, ETPI, and significant clinical parameters including TNM stage, age, and sex. Notably, patients with CRC exhibited a higher likelihood of shorter PFI time with an increase in the total score according to the nomogram’s prediction of disease progression (Fig. 6A). The results showed that the nomogram had better evaluation and prediction of survival outcomes in CRC, with the 1-, 3-, and 5 year calibration curves revealing high fitness between prediction and actuality (3- year calibration curve in Fig. 6B, 1- and 5-calibration curves in Supplementary Fig. 5B, C). DCA curves showed that the nomogram possessed a more specific preponderance than other clinical parameters (Fig. 6C).

Construction of a nomogram based on ETPI. A A nomogram combining ETPI with clinicopathological features (age, sex, TNM stages, and MSI). The total score was generated by integrating the scores of each parameter, and the possibility that the patient’s PFI is less than 5 years, 3 years, and 1 year was evaluated by the ruler at the bottom of the diagram. B Calibration plot showing that nomogram-predicted survival probabilities correspond closely to the actually observed proportions for 3 year prediction. C DCA of the nomogram and other clinicopathological features. ETPI the effective T cell prognostic Index, MSI microsatellite instability, DCA decision curve analysis
Immune Checkpoint and Drug Sensitivity Analysis
The expression levels of the majority of 38 types of genes related to immune checkpoints in the low ETPI group were significantly higher than those in the high ETPI group (see Supplementary Fig. 5A), suggesting that patients with low ETPI might benefit from immune checkpoint inhibitor (ICI) therapy.
We analyzed the IC50 (50% inhibition concentration) of 198 drugs from GDSC for high- and low ETPI CRC samples. After cleaning up the abnormal values, the four commonly used chemotherapy medications, including 5-fluorouracil, irinotecan, oxaliplatin, and paclitaxel, in the low ETPI group had significantly lower IC50 values than those in the high ETPI group (Fig. 7A). This suggests that patients with low ETPI may derive greater benefit from chemotherapy. Inhibitors of traditional tumor activation pathways and immune checkpoint blockades were also assayed to explore the differences between the two groups. We tend to screen for target drugs that were clinically suggested to be resistant or highly resistant, such as JAK-8517, and sensitive targeted drugs, such as AZD8186, SB505124, and BI-2536. In boxplots, patients were shown to possess a significantly lower ETPI with a decline in the IC50 values in JAK-8517 (JAK1/2 inhibitor) and AZD8186 (PI3Kalpha, PI3Kbeta inhibitor), compared to the decline in IC50 along with an increase in ETPI in SB505124 (TGFBR1/ACVR1B/ACVR1C inhibitor) and BI-2536 (PLK1/2/3 inhibitor) (Fig. 7A).

Drug sensitivity analysis. A Boxplots for comparing traditional chemotherapy agents and tumor pathway inhibitors between the two subtypes based on ETPI. Drugs including 5-fluorouracil, irinotecan, oxaliplatin, and paclitaxel exhibited significantly lower IC50 in the low ETPI group than in the high ETPI group. Tumor pathway inhibitors, including JAK-8517, AZD8186, SB505124, and BI-2536, are illustrated. B Correlation curves between five selected immunotherapy agents and three ETRGs according to the top six drug correlation coefficients. C Boxplots showing the difference in z-score between divisions of patients with CRC according to the expression levels of CASP1, PDCD1, and IL15. ETPI the effective T cell prognostic index; ETGR effective T cell-related genes; *P < 0.05; **P < 0.01; ***P < 0.001; ns no significance
Moreover, drugs approved by the Food and Drug Administration (FDA) in the Cellminer database were analyzed for correlation with the six ETRGs. Based on the order of drug correlation coefficients, we identified the top six candidates and plotted correlation curves between five drugs and three genes (CASP1, PDCD1, and IL15) from ETRGs. Significant positive correlations were found between CASP1 expression and the z-scores of vemurafenib (a selective inhibitor of BRAF) and encorafenib (a BRAF inhibitor). Similarly, a positive relationship was observed between PDCD1 expression and the z-scores of irinotecan (a topoisomerase I inhibitor) and imatinib (a selective inhibitor of BCR/ABL, v-Abl, PDGFR, and c-kit). Additionally, the z-scores of vemurafenib and afatinib decreased with increased expression of IL15 and CASP1 (Fig. 7B).
Using box plots, we stratified the CRC cohort into high and low expression groups based on the expression levels of the three selected ETRGs and evaluated the difference in z-scores between the two groups. For CASP1, the high expression group exhibited significantly higher z-scores of vemurafenib and encorafenib (P < 0.05), while no significant differences were observed in the other medications (Fig. 7C).
Validation of Protein Expression Profiles of ETRGs in Clinical Samples
The protein expression levels of the ETRGs were determined using IHC methods from the HPA database (Supplementary Figs. 6, 7). Staining for protein expression of FOXP3 and PDCD1 were not detected in both normal rectum and rectal cancer tissues. However, CASP1 was expressed at a high level in glandular cells in rectal normal tissues, whereas the malignant tissues showed low CASP1 staining in IHC results. In normal glandular cells of colon, MX1 also possessed a high protein expression profile but the results showed a medium MX1 level in rectal tumor. Moreover, IL15 was expressed at a medium level in glandular cells in normal tissues with an adverse situation in colon cancer. Comparing the expression profiles of ETRGs between normal and tumor tissues suggests that the genes identified in our study may act as protective factors in prognosis.
Discussion
Although there has been a persistent decrease in overall mortality and incidence of CRC among the elderly, recent years have seen a rise in incidence among young and middle-aged adults [1]. This trend presents challenges due to vague conventional approaches, which may result in inappropriate prescription-making and surveillance [15]. Consequently, there is an urgent need for diverse treatment directions related to immunology, as personalized assessment approaches have demonstrated superior outcomes.
Over the past two decades, immunotherapy has garnered significant attention for its ability to enhance responses in refractory solid tumors like lung cancer and melanoma [16]. Notably, immunotherapy shares a common trait with conventional treatments such as radiotherapy and chemotherapy—they all exert some influence on the immune system. Research has delved into the densities of specific lymphocytes, like CD8+ T cells, within the core or periphery of CRC tumors, resulting in the development of a digital score with prognostic value throughout tumor progression [17]. At a more granular level, understanding the genomic landscape of CRC has been a focal point, particularly in identifying druggable targets, especially in metastatic stages [18]. Alterations in RAS and BRAF genes, along with MSS, have emerged as gold standards in molecular features guiding CRC therapy development [19]. Molecular classification in CRC, based on genetic mutations and transcriptomics, including key gene expressions and the TME, has led to more effective treatment strategies such as consensus molecular subtypes [20]. Hence, the abundance and interaction of tumor-infiltrating lymphocytes and other immune cells in CRC patients play a crucial role in making precise decisions regarding modern complex treatments. To bridge the gap between immune cells and the genomic landscape, we developed a related prognostic index, ETPI, linked to activated CD4+ and CD8+ T cells and validated its clinical value for prognosis, prediction, and drug benefit in CRC.
In this study, we collected data on key genes related to immune cells that varied across 28 categories [21]. We then calculated the infiltration status of the 28 types of immune cells in the CRC cohort. Each cell-owned individual score in every patient reflects its density. To eliminate multicollinearity, we further developed ICRS based on the screened 10 immune cell types using LASSO regression analysis, which performed a more suitable assessment of the immune status of patients with CRC. The training cohort was classified into high- and low-score groups, with significant differences in the PFI time. The immune cell landscape was reperformed in a new division based on ICRS.
Ten types of immunocytes in ICRS have been widely observed. For example, the CD8+ central memory cell is considered the prime predictive factor in melanoma therapy using anti-CTLA4 [14], but is currently not reported in CRC. The high immature proportion of tumor-infiltrating dendritic cells indicates a shorter overall survival in CRC [22], while the plasmacytoid dendritic cell seems to be a negative prognostic indicator in primary breast cancer [23]. As for the type1 T helper cell (Th1 helper cell, also belonging to the CD4+ T cell), it was shown that a favorable survival outcome is strongly associated with infiltration of the Th1 helper cell in many cancer types. However, it has been reported that in non-small cell lung cancer (NSCLC), the high frequency of Th1 helper cells might indicate the opposite survival value [24]. Similar to NSCLC, we found that patients with higher ICRS obtained more intensive Th1 helper cells and presented unfavorable PFI times in CRC. Th1 helper cells seem to act as a risk factor for tumor progression. Of particular interest, our investigation revealed that individuals with high ICRS scores displayed reduced levels of intensely activated CD4+ and CD8+ T cells, correlating with poorer PFI outcomes. Conversely, this cohort exhibited an opposing trend in the abundance of other immune cell types. This observation suggests that colorectal cancer patients presenting solely with heightened tumor infiltration of activated CD4+ and CD8+ cells may experience a more moderate malignant progression or even a more favorable survival prognosis as assessed by ICRS.
To bridge the relationship between cellular and genomic levels upon differences in the two types of T cells, survival analysis revealed great differences between patients with both high abundance of activated CD4+ and CD8+ T cells, compared to those with low densities of activated CD4+ and CD8+ T cells. Therefore, we selected these two groups of patients for further analysis. Regarding the biologic function enrichment of DEGs, the CC terminologies were mapped to the MHC class II protein complex. Previous studies have shown that the loss of MHC class expression leads to functional hindrance of the MHC-T cell receptor complex and affects T cell control of cancer [25]. CD4+ T cells also restrict tumor progression without the assistance of MHC class II, T cell receptors, and cytokines [26]. Activated CD8+ T cells may have a bystander-killing activity [27]. Furthermore, the top 10% of the 41 screened genes were predominantly implicated in the regulation and differentiation of T cells in various biologic functions. Noteworthy enrichment was observed in molecular functions, such as immune receptor, receptor ligand, and signaling receptor activator activities. These findings underscore the intricate molecular dynamics governing immune cell interactions, which in turn play a pivotal role in shaping survival outcomes, alongside variations in the abundance of activated CD4+ and CD8+ T cells in CRC.
Following the previous analysis, we established ETPI (constructed using the ETRGs, including FOXP3, IL15, PDCD1, MX1, CASP1, and IDO1) as a signature to evaluate the prognosis of patients with CRC and subsequently performed drug sensitivity analysis according to the expression profiles of ETRGs. Our index worked independently of age, sex, T, N, M, and AJCC staging systems and had relative advantages in the performance of prognostic prediction. FOXP3, IL15, CASP1, and IDO1 were significantly correlated with survival outcomes and disease deterioration according to univariate Cox regression analysis.
FOXP3 is a critical gene that regulates CD4+ CD25+ regulatory T cells. In our study, higher expression of FOXP3 in individuals with CRC showed an unfavorable survival time. Both activated CD4+ T cells and CD8+ T cells mainly exist in the peripheral blood, together with high FOXP3 expression in regulatory T cells, exerting anti-tumor effects [28]. These results are in line with those of previous studies [29].
Among the whole cytokine–cytokine receptor family, the decreasing profile of IL15 in CRC is probably one of the key mechanisms of the low density of tumor-infiltrating T cells, indicating a higher risk of relapse and poor survival outcomes [30]. Our findings align with previous studies indicating that patients within the high ETPI group exhibit decreased expression levels of IL15, suggesting a less favorable prognosis.
It has been widely observed that caspase 1 is one of the key components of canonical and noncanonical inflammasomes in inflammatory bowel diseases and some types of tumors [31]. The T cell-intrinsic caspase 1 accelerates colitis-associated tumorigenesis and promotes CD4+ T cell (Th17) differentiation, independent of inflammasome activation [32]. However, the role that caspase 1 plays in CRC remains to be elucidated. As one of the independent prognostic factors in ETRGs, CASP1 was expressed at low levels in the high ETPI group, with an unoptimistic survival time. The lack of CASP1 might lead to unsatisfactory anti-tumor activities or contribute to malignant progression since it possibly facilitates the proliferation and infiltration of T helper cells in CRC.
As a risk factor for CRC, IDO1 induces the proliferation of Treg cells and arrests the development of G1-phase T cells in tumors through kynurenine catalysis [33]. However, in the present study, there was no significant difference in the genomic level of IDO1 between the classifications divided by ETPI. However, it also has the potential to be applied in immunotherapy.
IHC analysis of protein expression of the ETRGs showed results consistent with those of previous genomic studies of patient samples based on the HPA database. The results showed strong staining of ETRGs (CASP1, MX1, IDO1, and IL15) in glandular and epithelial cells in normal tissues, implying that these genes had potential protective roles in CRC. However, in the IHC analysis, FOXP3 and PDCD1 showed rather low levels in normal tissues, which might be caused by the low sample qualities and quantities.
In a previous study, certain patients exhibiting MSI-H or dMMR status showed promising responses to PD1 monoclonal antibody therapy. However, fewer than 5% of cancer patients with MSI-H or dMMR status experience a superior prognosis and exhibit increased infiltration of anti-malignancy immune cells within solid tumors [16]. Interestingly, our study found that patients with CRC possessing lower ETPI levels displayed a relatively higher prevalence of MSI-H status, suggesting the potential utility of ETPI as a tool for identifying microsatellite status.
The drug sensitivity analysis identified four common medications—5-fluorouracil, irinotecan, oxaliplatin, and paclitaxel—due to ETPI’s efficacy in categorizing CRC patients for potential responsiveness to current chemotherapies. ETPI also demonstrated robust performance in discerning patients sensitive to both traditional and novel tumor pathway inhibitors. Moreover, an evaluation using an external cohort of advanced melanoma patients highlighted the signature’s predictive value in immunotherapy response. ETPI holds promise as a surrogate for prognostic assessment and drug benefit selection, offering valuable insights for clinical treatment strategies, especially in guiding decisions regarding immunotherapy, based on medication sensitivity analysis. At the cellular level, CD8+ T cell density and PDL1 expression serve as predictors for response to PDL1 or PD1-targeted immunotherapies across various cancers [13]. Additionally, depletion of peripheral CD4+ T cells suggests ineffectiveness of combined therapy with anti-CTLA4 and anti-PD1 [34]. Furthermore, ETPI, constructed from relatively stable genes in the human genome, may enhance the feasibility of anti-tumor therapies based on genomic and tumor-infiltrating immune cells.
Despite demonstrating promising survival outcomes and clinical significance in CRC, this study still has several limitations that warrant consideration. Firstly, the datasets utilized were sourced from retrospective public databases, highlighting the need for prospective multicenter validations and in vivo/in vitro experiments to further validate the findings. Secondly, the focus on key genomic phenotypes in CRC cohorts via PPI analysis may have inadvertently overlooked obscure genes with potential functional roles in tumor-infiltrating immune cells. Future studies should explore downstream pathway analyses at cellular and subcellular levels. Lastly, while the research focused on constructing the TME, further investigation into malignant intrinsic features based on tumor ecosystem dynamics is essential for a comprehensive understanding.
Conclusion
In conclusion, we identified key characteristics of activated CD4+ and CD8+ T cells by generating enrichment scores for their abundance in CRC. By analyzing transcriptome differences between groups with varying infiltration levels of these T cells, we developed a promising prognostic index, ETPI. This index, based on ETRGs associated with the infiltration of CD4+ and CD8+ T cells in CRC, demonstrated strong predictive capabilities for tumor progression, recurrence, and survival outcomes. By utilizing this index, we classified CRC patients into subtypes with high and low ETPI, each characterized by distinct TME and MSI statuses. Drug sensitivity analysis based on the constituent genes of ETPI indicated that our index could effectively identify patients likely to benefit from chemotherapy and immunotherapy. Furthermore, a nomogram was developed, demonstrating greater specificity than other clinical parameters. Hence, this study provides a novel perspective on immune infiltration in CRC and predicted potential biomarkers and molecular interaction relevant to immunotherapy and prognosis. ETPI could assist in the precise selection of CRC patients for specific therapies based on key genomic interactions, potentially optimizing treatment strategies in future clinical practice.
Data Availability
Derived data supporting this research are available from the corresponding author upon reasonable request.
References
Siegel, R. L., Wagle, N. S., Cercek, A., Smith, R. A., & Jemal, A. (2023). Colorectal cancer statistics, 2023. CA: A Cancer Journal for Clinicians, 73(3), 233–254.
Miller, K. D., Nogueira, L., Devasia, T., Mariotto, A. B., Yabroff, K. R., Jemal, A., Kramer, J., & Siegel, R. L. (2022). Cancer treatment and survivorship statistics, 2022. CA: A Cancer Journal for Clinicians, 72(5), 409–436.
Vogelstein, B., Fearon, E. R., Hamilton, S. R., Kern, S. E., Preisinger, A. C., Leppert, M., Nakamura, Y., White, R., Smits, A. M., & Bos, J. L. (1988). Genetic alterations during colorectal-tumor development. The New England Journal of Medicine, 319(9), 525–532.
Cadiz, F., Gormaz, J. G., & Burotto, M. (2018). Breast cancer staging: Is TNM ready to evolve? JCO Global Oncology, 4, 1–3.
Hattori, A., Takamochi, K., Oh, S., & Suzuki, K. (2019). New revisions and current issues in the eighth edition of the TNM classification for non-small cell lung cancer. Japanese Journal of Clinical Oncology, 49(1), 3–11.
Loibl, S., & Gianni, L. (2017). HER2-positive breast cancer. Lancet, 389(10087), 2415–2429.
Nagtegaal, I. D., Quirke, P., & Schmoll, H. J. (2011). Has the new TNM classification for colorectal cancer improved care? Nature Reviews. Clinical Oncology, 9(2), 119–123.
Galon, J., Costes, A., Sanchez-Cabo, F., Kirilovsky, A., Mlecnik, B., Lagorce-Pagès, C., Tosolini, M., Camus, M., Berger, A., Wind, P., & Zinzindohoué, F. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science, 313(5795), 1960–1964.
Fridman, W. H., Pagès, F., Sautès-Fridman, C., & Galon, J. (2012). The immune contexture in human tumours: Impact on clinical outcome. Nature Reviews Cancer, 12(4), 298–306.
Giraldo, N. A., Becht, E., Pages, F., Skliris, G., Verkarre, V., Vano, Y., Mejean, A., Saint-Aubert, N., Lacroix, L., Natario, I., & Lupo, A. (2015). Orchestration and prognostic significance of immune checkpoints in the microenvironment of primary and metastatic renal cell cancer. Clinical Cancer Research, 21(13), 3031–3040.
Ogino, S., Nosho, K., Irahara, N., Meyerhardt, J. A., Baba, Y., Shima, K., Glickman, J. N., Ferrone, C. R., Mino-Kenudson, M., Tanaka, N., & Dranoff, G. (2009). Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clinical Cancer Research, 15(20), 6412–6420.
Pino, M. S., & Chung, D. C. (2010). The chromosomal instability pathway in colon cancer. Gastroenterology, 138(6), 2059–2072.
Tumeh, P. C., Harview, C. L., Yearley, J. H., Shintaku, I. P., Taylor, E. J., Robert, L., Chmielowski, B., Spasic, M., Henry, G., Ciobanu, V., & West, A. N. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 515(7528), 568–571.
Tietze, J. K., Angelova, D., Heppt, M. V., Reinholz, M., Murphy, W. J., Spannagl, M., Ruzicka, T., & Berking, C. (2017). The proportion of circulating CD45RO+ CD8+ memory T cells is correlated with clinical response in melanoma patients treated with ipilimumab. European Journal of Cancer, 75, 268–279.
Salazar, R., & Tabernero, J. (2014). New approaches but the same flaws in the search for prognostic signatures. Clinical Cancer Research, 20(8), 2019–2022.
Ganesh, K., Stadler, Z. K., Cercek, A., Mendelsohn, R. B., Shia, J., Segal, N. H., & Diaz, L. A., Jr. (2019). Immunotherapy in colorectal cancer: rationale, challenges and potential. Nature Reviews Gastroenterology & Hepatology, 16(6), 361–375.
Pagès, F., Mlecnik, B., Marliot, F., Bindea, G., Ou, F. S., Bifulco, C., Lugli, A., Zlobec, I., Rau, T. T., Berger, M. D., & Nagtegaal, I. D. (2018). International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. The Lancet, 391(10135), 2128–2139.
Yaeger, R., Chatila, W. K., Lipsyc, M. D., Hechtman, J. F., Cercek, A., Sanchez-Vega, F., Jayakumaran, G., Middha, S., Zehir, A., Donoghue, M. T., & You, D. (2018). Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell, 33(1), 125–136.
Van Cutsem, E., Cervantes, A., Adam, R., Sobrero, A., Van Krieken, J. H., Aderka, D., Aguilar, E. A., Bardelli, A., Benson, A., Bodoky, G., & Ciardiello, F. (2016). ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Annals of Oncology, 27(8), 1386–1422.
Ciardiello, F., Ciardiello, D., Martini, G., Napolitano, S., Tabernero, J., & Cervantes, A. (2022). Clinical management of metastatic colorectal cancer in the era of precision medicine. CA A Cancer Journal for Clinicians, 72(4), 372–401.
Charoentong, P., Finotello, F., Angelova, M., Mayer, C., Efremova, M., Rieder, D., Hackl, H., & Trajanoski, Z. (2017). Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Reports, 18(1), 248–262.
Sandel, M. H., Dadabayev, A. R., Menon, A. G., Morreau, H., Melief, C. J., Offringa, R., van der Burg, S. H., Janssen-van Rhijn, C. M., Ensink, N. G., Tollenaar, R. A., & van de Velde, C. J. (2005). Prognostic value of tumor-infiltrating dendritic cells in colorectal cancer: Role of maturation status and intratumoral localization. Clinical Cancer Research, 11(7), 2576–2582.
Sisirak, V., Faget, J., Gobert, M., Goutagny, N., Vey, N., Treilleux, I., Renaudineau, S., Poyet, G., Labidi-Galy, S. I., Goddard-Leon, S., & Durand, I. (2012). Impaired IFN-α production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Research, 72(20), 5188–5197.
Jian, H., Fangrong, S., Haitao, H., Chunhua, L., & Guangbo, Z. (2017). Th1high in tumor microenvironment is an indicator of poor prognosis for patients with NSCLC. Oncotarget, 8(8), 13116–13125.
Gettinger, S., Choi, J., Hastings, K., Truini, A., Datar, I., Sowell, R., Wurtz, A., Dong, W., Cai, G., Melnick, M. A., & Du, V. Y. (2017). Impaired HLA Class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discovery, 7(12), 1420–1435.
von Geldern, M., Simm, B., Braun, M., Weiß, E. H., Schendel, D. J., & Falk, C. S. (2006). TCR-independent cytokine stimulation induces non-MHC-restricted T cell activity and is negatively regulated by HLA class I. European Journal of Immunology, 36(9), 2347–2358.
Schietinger, A., Philip, M., Liu, R. B., Schreiber, K., & Schreiber, H. (2010). Bystander killing of cancer requires the cooperation of CD4+ and CD8+ T cells during the effector phase. Journal of Experimental Medicine, 207(11), 2469–2477.
Zhang, L., Yu, X., Zheng, L., Zhang, Y., Li, Y., Fang, Q., Gao, R., Kang, B., Zhang, Q., Huang, J. Y., & Konno, H. (2018). Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature, 564(7735), 268–272.
Salama, P., Phillips, M., Grieu, F., Morris, M., Zeps, N., Joseph, D., Platell, C., & Iacopetta, B. (2009). Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. Journal of Clinical Oncology, 27(2), 186–192.
Jakobisiak, M., Golab, J., & Lasek, W. (2011). Interleukin 15 as a promising candidate for tumor immunotherapy. Cytokine & Growth Factor Reviews, 22(2), 99–108.
Saleh, M., & Trinchieri, G. (2011). Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nature Reviews Immunology, 11(1), 9–20.
Gao, Y., Deason, K., Jain, A., Irizarry-Caro, R. A., Dozmorov, I., Coughlin, L. A., Rauch, I., Evers, B. M., Koh, A. Y., Wakeland, E. K., & Pasare, C. (2020). Transcriptional profiling identifies caspase-1 as a T cell–intrinsic regulator of Th17 differentiation. Journal of Experimental Medicine, 217(4), e20190476.
Mellor, A. L., & Munn, D. H. (2008). Creating immune privilege: Active local suppression that benefits friends, but protects foes. Nature Reviews Immunology, 8(1), 74–80.
Germano, G., Lu, S., Rospo, G., Lamba, S., Rousseau, B., Fanelli, S., Stenech, D., Le, D. T., Hays, J., Totaro, M. G., & Amodio, V. (2021). CD4 T cell–dependent rejection of beta-2 microglobulin null mismatch repair–deficient tumors. Cancer Discovery, 11(7), 1844–1859.
Acknowledgements
The authors gratefully acknowledge the data shared in TCGA, GEO, and UCSC Xena databases.
Funding
This study was supported by no grants.
Author information
Authors and Affiliations
Contributions
Chengru Chen and Peng Zou contributed to the design of the study, data collection, and analysis. Chengru Chen drafted and revised the manuscript. Peng Zou was responsible for data cleaning. Xiaobin Wu provided guidance for research and reviewed the article. All authors contributed to the article and have approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors have declared that there were no commercial relationships and conflicts of interest in the research.
Ethical Approval
This study did not require ethics approval since the transcriptome and protein expression data were obtained from publicly available databases.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
12033_2024_1237_MOESM1_ESM.tif
Supplementary file1 (TIF 12454 kb)—Supplementary Figure 1 Divisions of patients with CRC related to the abundance of activated CD4+ and CD8+ T cells. (A, B) Patients with high infiltration of activated CD4+ T cells had better survival outcomes. (C, D) Patients with high infiltration of activated CD8+ T cells had better survival outcomes. (E) Percentage composition of selected patients based on the infiltration of activated CD4+ and CD8+ T cells. (F) K–M curves of survival analysis pertaining to PFI in patients based on the different abundance of activated CD4+ and CD8+ T cells. CRC, colorectal cancer; K–M, Kaplan–Meier; PFI, progression-free interval
12033_2024_1237_MOESM2_ESM.tif
Supplementary file2 (TIF 13924 kb)—Supplementary Figure 2 Survival analyses based on high and low infiltration of 16 immune cell types in patients with CRC. CRC, colorectal cancer
12033_2024_1237_MOESM3_ESM.tif
Supplementary file3 (TIF 16085 kb)—Supplementary Figure 3 Key gene modules screening and biologic function enrichment analysis. (A) Volcano plot on the infiltration of activated CD4+ and CD8+ T cell-related DEGs between groups with both high and low infiltration of activated CD4+ and CD8+ T cells in TCGA cohort. The red dots on the left side of the 0 axis represent downregulated genes, and the right-side red dots represent upregulated genes. (B) Correlation analysis between MEs and ssGSEA score of activated CD4+ and CD8+ T cells. The yellow module has the most significant correlation. (C, D) KEGG enrichment analyses. Using q < 0.05, the top ten enrichment items are displayed as a bar chart and bubble chart according to q value and count value. (E, F) GO analysis. The top 5 gene enrichment items (BP, CC, and MF) in the three catalogs were identified according to the q value, which is shown as a bar chart and bubble chart. DEGs, differentially expressed genes. KEGG, Kyoto encyclopedia of genes and genomes, GO, gene ontology, MEs, module eigengenes, ssGSEA, the single-sample gene set enrichment analysis
12033_2024_1237_MOESM4_ESM.tif
Supplementary file4 (TIF 7471 kb)—Supplementary Figure 4 External validation of ETPI. (A, B) K–M curves of DFS time showed that the low ETPI groups had more favorable survival outcomes in GSE14333 and GSE38832. (C) K–M curves of RFS time showed that the low ETPI groups had better survival outcomes in GSE39582. (D) A bar plot for promotions of progression disease (PD), partial remission (PR), and complete remission (CR) upon pembrolizumab treatment for melanoma, indicating that the low ETPI group showed high ORR to immunotherapy in GSE78220. (E) The boxplots indicated that patients with low ETPI tended to exhibit high ORR to pembrolizumab treatment based on Fisher’s precision probability test. ETPI, the effective T cell prognostic index, K–M, Kaplan–Meier, DFS, disease-free survival, RFS, recurrence-free survival, ORR, objective response rate
12033_2024_1237_MOESM5_ESM.tif
Supplementary file5 (TIF 11865 kb)—Supplementary Figure 5 Immune checkpoint inhibitor analysis and calibration curves of the nomogram. (A) The expression level of 38 genes related to immune checkpoint inhibitors in high and low ETPI groups. (B, C) 1- and 5-year calibration curves of the nomogram. ETPI, the effective T cell prognostic index, *P < 0.05; **P < 0.01;***P < 0.001; ns, no significance
12033_2024_1237_MOESM6_ESM.tif
Supplementary file6 (TIF 35866 kb)—Supplementary Figure 6 The protein expression of ETRGs. The protein expression levels of CASP1, FOXP3, IDO1, and IL15. (A, B) CASP1 is expressed at a high level in normal tissues and showed low staining in tumor tissues. (C, D) Non-detection of FOXP3 expression in both normal and tumor tissues. (E, F) IDO1 expression is higher in normal tissues than in tumor tissues. (G, H) IL15 expression is higher in normal tissues than in tumor tissues. T, tumor tissues, N, normal issues, ETGR, effective T cell-related genes
12033_2024_1237_MOESM7_ESM.tif
Supplementary file7 (TIF 20549 kb)—Supplementary Figure 7 The protein expression of ETRGs. The protein expression levels of MX1 and PDCD1. (A, B) MX1 is expressed at a high level in normal tissues but with low staining in tumor tissues. (C, D) Non-detection of PDCD1 expression in both normal and tumor tissues. ETGR, effective T cell-related genes
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, C., Zou, P. & Wu, X. Development and Validation of an Immune Prognostic Index Related to Infiltration of CD4+ and CD8+ T Cells in Colorectal Cancer. Mol Biotechnol (2024). https://doi.org/10.1007/s12033-024-01237-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12033-024-01237-z