
Abstract
p53 is a tumor suppressor gene activated in response to cellular stressors that inhibits cell cycle progression and induces pro-apoptotic signaling. The protein level of p53 is well balanced by the action of several E3 ligases and deubiquitinating enzymes (DUBs). Several DUBs have been reported to negatively regulate and promote p53 degradation in tumors. In this study, we identified USP19 as a negative regulator of p53 protein level. We demonstrate a direct interaction between USP19 and p53 by pull down assay. The overexpression of USP19 promoted ubiquitination of p53 and reduced its protein half-life. We also demonstrate that CRISPR/Cas9-mediated knockout of USP19 in cervical cancer cells elevates p53 protein levels, resulting in reduced colony formation, cell migration, and cell invasion. Overall, our results indicate that USP19 negatively regulates p53 protein levels in cervical cancer progression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
p53 is a tumor suppressor gene that plays a crucial role in response to cellular stress. It has been found to regulate an array of genes that participate in multiple biological functions such as DNA damage repair, transient cell cycle arrest, cellular senescence, and apoptosis [1,2,3]. Inactivation or deficiency of p53 is associated with cancer progression, and tumors lacking p53 show more malignant characteristics such as genetic instability, increased invasiveness, and metastatic potential [4,5,6,7,8,9]. Activation/inactivation of p53 in response to cellular stress largely occurs through ubiquitination [10]. E3 ligases such as Mdm2 and TRIM65 promote the ubiquitination and proteasomal degradation of p53 [11,12,13]. Mdm2-mediates p53 ubiquitination and induces exporting p53 to the nucleus [14, 15]. There are several other E3 ligases such as COP1, ARF-BP1, Pirh2, MSL2, and Parc that regulate p53 stability and its localization [16].
Ubiquitin moieties covalently attached to the target protein are removed by deubiquitinating enzymes (DUBs) through a process known as deubiquitination. Ubiquitin-specific proteases (USPs) are the largest subfamily of DUBs that directly or indirectly regulate the stability and activation of p53 [10, 17,18,19,20,21]. The function of DUBs might be as important as E3 ligases in regulating protein turnover. However, the mechanisms regulating p53 deubiquitination and their implications in cancer remain enigmatic. DUBs such as USP7, USP10, USP11, USP24, USP29, USP42, and USP9X were reported to regulate p53 ubiquitination leading to its protein stabilization [22]. However, some DUBs such as USP2, USP4, USP5, USP15, and USP26 were reported to show opposite negative regulation of ubiquitinated p53 [23,24,25,26,27].
USP19 is known to play a critical role in regulating tumorigenesis and cancer dissemination [28]. There are reports indicating that USP19 negatively regulates cell proliferation and migration in renal cells and ovarian carcinomas [29, 30]. USP19 showed a positive regulation on ubiquitinated cellular inhibitors of apoptosis 1 (cIAP1) and cIAP2 leading to protein stabilization. Depletion of USP19 reduced cIAP1 and cIAP2 protein levels leading to caspase-activation and apoptosis [31]. Recently, a regulatory link was discovered between wild-type p53 and members of the IAP family [32, 33]. Attenuation of p53 and retinoblastoma protein was found to transactivate cIAP1 and cIAP2 genes and promote mammary carcinoma [34]. This dependence of IAP family members on p53 and USP19 prompted us to investigate if there is a regulatory link between USP19 and p53.
In the present study, we demonstrate that USP19 directly interacts with p53 in HeLa cells. Overexpression of USP19 repressed p53 whereas CRISPR/Cas9-mediated knockout of USP19 enhanced p53 levels. Furthermore, we found that USP19 reduces p53 protein turnover by promoting its degradation through the ubiquitin-proteasomal pathway. Loss of USP19 in HeLa cells reduced the proliferation and metastatic capacity of these cells. Overall, we conclude that depletion of USP19 promotes p53 protein levels and suppresses cervical cancer progression.
Materials and Methods
Cell Culture and Transfection
Human embryonic kidney cells (HEK293) and HeLa cells were purchased from the Korean Cell Line Bank (Seoul, South Korea) and maintained in DMEM (GIBCO BRL, Rockville, MD, USA) supplemented with 10% fetal bovine serum (GIBCO) and 1% penicillin and streptomycin (GIBCO) at 37 °C in a humidified atmosphere with 5% CO2. The cells were passaged regularly according to cell confluence.
Transfection
HEK293 cells were transfected using polyethyleneimine (Polysciences, Warrington, PA, USA) following the manufacturer’s recommendations. HeLa cells were transfected using Lipofectamine 3000 (Cat no. L3000001, Thermo Fisher Scientific).
Plasmids and sgRNAs
Flag-tagged USP19 (Addgene #78,597), Flag-tagged USP19CA (Addgene #36,307), HA-tagged ubiquitin (Addgene #18,712), and His-p53 (1-393) (Addgene #24,859) were purchased from Addgene (MA, USA). p53 was subcloned into a mammalian expression vector pcDNA-6X-Myc vector. To screen the DUB candidates, plasmid vectors encoding Cas9-2a-mRFP-2a-PAC (puromycin N-acetyl-transferase puromycin resistance gene) and single guide RNA (sgRNA) were purchased from Toolgen (Seoul, South Korea). The sgRNA target sequences were designed using a publicly available bioinformatics tool (www.broadinstitute.org), and the desired oligonucleotide sequences were cloned into vectors as previously described [35]. Briefly, oligonucleotides containing each target sequence were synthesized (Bioneer, Seoul, South Korea), and then T4 polynucleotide kinase was used to add terminal phosphates to the annealed oligonucleotides (Bio-Rad, CA, USA). The vectors were ligated with the annealed oligonucleotides after digestion with BsaI restriction enzyme.
Antibodies and Reagents
Mouse monoclonal antibodies against p53 (SC-126, 1:1000), Flag (Anti-DDDDK-tag, M185-3 L, 1:1,000) (MBL Life Science), ubiquitin (SC-8017, 1:1,000), HA (SC-7392, 1:1000), GAPDH (SC-32,233, 1:1,000), histidine (SC-8036) and normal mouse IgG (SC-2025, 1:1,000) were purchased from Santa Cruz Biotechnology. Rabbit polyclonal antibodies against USP19 (Proteintech, 25768-1-AP) were purchased from Proteintech. Protein A/G Plus Agarose beads (SC-2003, Santa Cruz Biotech), the proteasomal inhibitor MG132 (S2619, Selleckchem), the protein translation inhibitor cycloheximide (239,765, Merck), puromycin (12,122,530, Invitrogen), and DAPI (H-1200, Vector Laboratories) were purchased from the indicated manufacturers.
T7E1 Assay
The isolation of genomic DNA was performed using DNeasy Blood and Tissue Kits (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The desired regions of DNA consisting of the nuclease target site were first PCR-amplified using hemi-nested or nested primers, and PCR amplicons were then denatured by heating and annealed to generate heteroduplex DNA. Heteroduplex DNA was then treated with 5 units of T7 endonuclease 1 enzyme (New England Biolabs, MA, USA) for 20 min at 37 °C, and cleavage was assessed using 2% agarose gel electrophoresis. The mutation frequencies were assessed on the basis of band intensity, which was quantified using ImageJ software. The results were then presented as indel percentages using the following equation: mutation frequency (%) = 100 × (1 − [1 − fraction cleaved] 1/2) where the fraction cleaved is the total relative density of the cleavage bands divided by the sum of the relative density of the cleaved and uncut bands. Oligonucleotide sequences used for sgRNA plasmid construction are listed in Supplementary Table S1. The oligonucleotide sequences used to get T7E1 PCR amplicon of USP19 are presented in Supplementary Table S2. The expected cleavage sizes after the T7E1 assay are summarized in Supplementary Table S3.
Immunoprecipitation Assays
At 48 h post-transfection, cells were harvested and lysed in IP lysis buffer (25 mM Tris-HCl (pH 7.4), 150 mM sodium chloride, 1 mM EDTA, 1% NP-40, 5% glycerol, and 1 mM PMSF and protease inhibitor cocktail) before estimating the protein concentration using the Bradford assay (Thermo Scientific). Around 2–3 mg of lysate was immunoprecipitated with antibodies overnight and then incubated with 25 µL of protein A/G Sepharose beads at 4 °C for 3 h. Before SDS-PAGE, the beads were washed with lysis buffer, eluted, and boiled in a 2×SDS sample buffer (5X SDS sample loading buffer containing 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromophenol blue, and 0.125 M Tris-HCl [pH 6.8]). Immunoprecipitated proteins were detected by Western blot analysis with the indicated antibodies. Mouse IgG (CST- 58,802 S, 1: 5,000; Cell Signaling Technology) light chain-specific secondary antibody was used to prevent interference from heavy and light immunoglobulin chains in the binding assay.
Pull Down Assay
A pull down assay was performed as described previously [36, 37]. Briefly E. coli (BL21) was transformed with His-p53 plasmid. A single colony was inoculated in LB media supplemented with ampicillin (50 ug/mL) overnight. The next day, 0.1 O.D culture was inoculated and incubated at 37 oC. Once the culture reached 0.6 O.D, the cells were induced with isopropyl-β-D-thiogalactopyranoside (IPTG; Promega) (0.1 mM) at 37 oC for 4 h on the shaker incubator. The cell growth was arrested, and the cell culture was pelleted down. The pellet was resuspended in a cell lysis buffer (25 mM Tris-HCl (pH 7.4), 150 mM sodium chloride, 1 mM EDTA, 1% NP-40, 5% glycerol, 1 mM PMSF, and protease inhibitor cocktail) and then sonicated (40% duty, 10-s pulse, 30-s rest for 10 min on ice). The bacterial cell lysates expressing His-tagged p53 were purified using Nickel-NTA agarose beads (Cat. No 30,210, Qiagen). The purified His-tagged p53 protein immobilized on Nickel-NTA beads was then incubated with HEK293 cell lysate expressing Flag-USP19. The beads were washed with washing buffer (20 mM Tris–Cl, pH 8.0, 300 mM NaCl, 20 mM imidazole, and 1X protease inhibitor cocktail). The bound proteins were eluted using elution buffer (20 mM Tris–Cl, pH 8.0, 300 mM NaCl, 250 mM imidazole, and 1X protease inhibitor cocktail). The samples were then boiled using 5X SDS loading buffer for 5 min and immunoblotted with anti-Flag or anti-His antibodies.
Deubiquitination Assay
The effect of USP19 on p53 protein stability was determined by performing deubiquitination assays. Cells were treated 48 h post-transfection with the proteasome inhibitor MG132 for 6 h and then harvested. Cells were then lysed for 20 min in denaturing lysis buffer containing 150 mM sodium chloride, 1% Triton X-100, 1% sodium deoxycholate, 1% SDS, 50 mM Tris-HCl (pH 7.4), 2 mM EDTA, 1 mM PMSF, and protease inhibitor cocktail. Then, 3 mg of cell lysate was immunoprecipitated with the respective antibodies at 4 °C overnight and incubated with 25 µL of protein agarose beads for 3 h at 4 °C. The beads were washed with lysis buffer and eluted in 2X SDS sample loading buffer (5X SDS sample loading buffer containing 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromophenol blue, and 0.125 M Tris-HCl (pH 6.8) before being boiled at 95 to 100 °C for 5 min. The samples were then loaded onto SDS-PAGE gels and analyzed using Western blotting with the indicated antibodies.
Generation of USP19-Knockout HeLa Cells Using CRISPR-Cas9
HeLa cells were transfected with sgRNA1 and sgRNA2 targeting USP19 along with a Cas9 construct. Transfected cells were selected using puromycin selection (2 µg/mL) for 48 h after transfection. The following day, the cells were seeded into 96-well plates at a density of 0.25 cells per well. After 15 days, round single-cell colonies were selected, trypsinized, and re-plated into 24-well cell culture plates. Genomic DNA was isolated from single-cell clones and used to check the cleavage efficiency using the T7E1 assay. T7E1-positive clones were further cultured and stored in a liquid nitrogen tank for later use.
Duolink Assay
The potential physical interaction between USP19 and p53 was assessed using a Duolink in situ proximity ligation assay (PLA) kit (Cat. no. DUO92101, Sigma Aldrich). HeLa cells were fixed in 4% PFA for 10 min at room temperature and then blocked with blocking solution. Cells were treated with antibodies targeting USP19 and p53 for 1 h at 37 °C and then incubated with PLA probes for 1 h at 37 °C in a humidified chamber. Cells were then washed with Buffer A and ligation-ligase solution was added. Later, slides were incubated for 100 min in an amplified polymerase solution at 37 °C in the dark. Finally, cells were stained with mounting medium containing DAPI. A Leica fluorescence microscope was used to capture the fluorescence images (Leica, DM 5000B; Leica CTR 5000; Wetzlar, Germany).
Soft Agar Assay
Mock, USP19-KO, and USP19KO reconstituted with USP19 or USP19CS HeLa cells were subjected to an anchorage-independent colony formation assay. First, 1% agarose gel and 1X complete DMEM were mixed at a 1:1 ratio and plated onto 35 mm culture dishes, and the plates were incubated for 30 min at room temperature in a laminar hood. Cells were resuspended in 0.70% agarose with DMEM (1:1 ratio) and seeded at a density of 1 × 104 cells per well as a second layer. Cells were cultured for 14 days at 37 °C in a humidified atmosphere containing 5% CO2. Anchorage-independent colonies were stained using crystal violet dye (0.01% diluted in 20% methanol) and counted under a light microscope (IX71, Olympus, Tokyo, Japan).
Wound Healing Assay
Migration activity of mock, USP19-KO, and USP19KO reconstituted with USP19 or USP19CS HeLa cells was analyzed by wound healing assays. Cells were cultured to near 90% confluence, and scratches were made in the monolayers with a sterile 1 mL pipette tip in a definite array. The wounded cell layer was washed with PBS and incubated in complete medium. Wound closure was compared at specific time intervals by viewing plates under a light microscope. ImageJ software was used for quantification.
Transwell Cell Invasion Assay
The invasion ability of mock, USP19-KO, and USP19KO reconstituted with USP19 or USP19CS HeLa cells was analyzed using 0.8 μm Transwell chambers coated with Matrigel for 1 h at 37 °C (Corning, NY, USA) according to the manufacturer’s instructions. Briefly, 3 × 104 cells were suspended in 200 µL of serum-free DMEM medium and placed in 24-well chambers. Next, 750 µL of complete medium was added to each well of the 24-well plate, and the cells were incubated at 37 °C with 5% CO2 for 24 h. Cells from the upper surface of the chamber were scraped off, and cells on the lower surface were fixed using ice-cold methanol. Cells were then stained with crystal violet (0.05% in 20% methanol), and the average number of cells was counted under a light microscope.
Gene Expression Profiling Interactive Analysis
The DepMap portal containing RNA expression data was used to analyze the expression correlation between USP19 and p53 in this study.
Statistical Analysis
The results were documented as the means and standard deviations from three independent experiments. Experiments involving three groups were examined by one-way analysis of variance followed by Dunnett’s post-hoc test. All statistical analyses were performed using GraphPad Prism 9 software (CA, USA).
Results
USP19 Negatively Regulates p53 Protein Levels
To determine the effect of USP19 on p53 protein levels, we transfected increasing concentrations of Flag-USP19 along with a constant amount of Myc-p53 in HEK293 cells. We found that a dose-dependent increase of USP19 resulted in a gradual decrease in p53 protein level (Fig. 1A). A catalytic mutant USP19, which lacks deubiquitinating activity, in which the conserved catalytic cysteine residue is replaced with alanine at position 506 (USP19CA) was transfected in increasing concentrations along with a constant amount of Myc-p53 in the HEK293 cells. However, overexpression of a USP19 catalytic mutant did not show any significant changes in the protein levels of p53 (Fig. 1B). Next, we demonstrated that the sgRNA targeting USP19 upregulated the p53 protein levels, while the USP19-depleted cells reconstituted with Flag-USP19 reversed the stabilization effect on the p53 protein level (Fig. 1C, lane 7 vs. 5, 6). Furthermore, we cross-confirmed this negative effect of USP19 on endogenous p53 protein levels in HeLa cells. Similar to our previous results, we observed that USP19 destabilized the endogenous p53 protein level in a concentration dependent manner (Fig. 1D), but not with USP19CA (Fig. 1E). Additionally, USP19-mediated destabilization of the endogenous p53 protein level (Fig. 1F, lane 3) was reversed by sgRNA targeting USP19 (Fig. 1F, lane 4). Together, these results suggest that the deubiquitinating activity of USP19 shows negative regulation on p53 protein levels.
We next sought to determine whether USP19 interacts with p53 protein. To this end, we performed co-immunoprecipitation assays using specific antibodies against endogenous USP19 and p53. The results indicated that endogenous USP19 interacts with p53 protein and vice versa under physiological conditions in HeLa cells (Fig. 1G). Similarly, to confirm the interaction between USP19 and p53, we ectopically transfected HEK293 cells with Flag-USP19 and Myc-p53. Our results showed that Flag antibody co-immunoprecipitated Myc-p53 along with USP19 and vice-versa (Fig. 1H). Additionally, we used a Duolink PLA assay to demonstrate the interaction between USP19 and p53 in HeLa cells. In situ USP19–p53 interaction was confirmed by red fluorescence PLA dots when immunostained with both USP19 and p53; however, no PLA dots were observed when the cells were stained with either USP19 or p53 antibody alone (Fig. 1I), indicating that USP19 interacts with p53.
In order to investigate whether the interaction between USP19 and p53 is direct or not, we performed an in vitro pull down assay using purified proteins. To this end, we expressed and purified bacterial recombinant protein Histidine (His)-p53. The binding between p53 and USP19 was determined by incubating purified His-p53 recombinant protein with HEK293 cell lysate expressing USP19. His-alone was used as the control sample in the experiment. The results showed that USP19 protein was pulled down by His-p53 (Fig. 1J, lane 4) but not with His-alone (Fig. 1J, lane 3). Altogether, these results suggest that the deubiquitinase, USP19, directly interacts with p53 protein.

USP19 interacts with and negatively regulates p53 levels. A and B HEK293 cells were transfected with constant amount of Myc-p53 and increasing concentrations of (A) Flag-USP19 and (B) Flag-USP19CA to check the exogenous levels of p53 protein. C The effect of Flag-USP19 and sgRNA1/2 targeting USP19 on exogenous p53 protein. D and E HeLa cells were transfected with increasing amount of (D) Flag-USP19 and (E) Flag-USP19CA to check its effect on endogenous p53 protein levels. F The effect of reconstitution of Flag-USP19 on endogenous p53 protein in USP19-depleted HeLa cells was validated. GAPDH was used as a loading control. G Interaction between endogenous USP19 and p53 proteins were analyzed in HeLa cells. H Interaction between exogenous Flag-USP19 and Myc-p53 proteins were analyzed in HEK293 cells. Cell lysates were immunoprecipitated and immunoblotted with the indicated antibodies. Protein expression was checked using Western blotting. GAPDH was used as a loading control. I HeLa cells were subjected to the Duolink PLA assay to analyze the interaction between USP19 and p53 using specific antibodies. In-situ USP19–p53 interaction was observed as red PLA dots when USP19 and p53 were immunostained together but not when they were stained with individual antibodies. Scale bar: 10 μm. J Pull down assay using His-tagged p53 purified protein and cell lysate expressing USP19, western blotting was performed using specific antibodies
USP19 Promotes Polyubiquitination of p53
To further validate the negative regulation of p53 by USP19, we analyzed the ubiquitination status of p53 in the presence of wild-type USP19, catalytic mutant USP19, and sgRNA targeting USP19. The ubiquitination assay revealed that wild-type USP19 promotes endogenous p53 ubiquitination, as evident by the increased polyubiquitin smear (Fig. 2A, lane 2) whereas the catalytic mutant USP19CA did not promote endogenous p53 ubiquitination (Fig. 2A, lane 3). In contrast, the transient knockdown of USP19 reduced the p53 linked-polyubiquitin smear in HeLa cells when compared with the control (Fig. 2A, lane 4). Similar results were obtained for an ubiquitination assay performed using HEK293 cells transfected with the ectopically expressed p53 construct. A greater polyubiquitin smear of p53 was observed in the presence of USP19 but not with USP19CA (Fig. 2B). Thus, the data suggest that USP19 accelerates the degradation of p53 protein by promoting its polyubiquitination.
USP19 Reduces the Half-Life of p53 Protein
To access the influence of the deubiquitinating activity of USP19 on p53 protein turnover, we next treated cells with the protein synthesis inhibitor cycloheximide (CHX) and analyzed the half-life of p53 protein in the presence and absence of USP19. We first evaluated the half-life of endogenous p53 in HeLa cells and found that under physiological conditions, the half-life of p53 was around 30 min (Fig. 2C). However, the half-life of endogenous p53 was extended in USP19-depleted cells (Fig. 2D). The silencing of USP19 showed a similar stabilization effect on the ectopically expressed p53 protein level in the HEK293 cells (Fig. 2E F). These data suggest that the silencing of USP19 increases half-life of p53 protein.

USP19 promotes ubiquitination and reduces the half-life of p53. A The ubiquitination of endogenous p53 was analyzed by transfecting HeLa cells with Flag-USP19, Flag-USP19CA, or sgRNA targeting USP19 followed by immunoprecipitation with an anti-p53 antibody and immunoblotting with an anti-ubiquitin antibody. The cells were treated with MG132 for 6 h prior to harvest. B HEK293 cells were transfected with Myc-p53, HA-ubiquitin, Flag-USP19, Flag-USP19CA. The ubiquitination of exogenous p53 was confirmed by co-immunoprecipitation with the anti-Myc antibody and immunoblotting with anti-HA antibody. C The half-life of endogenous p53. D The half-life of endogenous p53 along with sgRNA targeting USP19 was analyzed in HeLa cells. E The half-life of exogenous p53. F The half-life of exogenous p53 along with sgRNA targeting USP19 was analyzed by transfecting HEK293 cells with Myc-p53. (C-F) CHX (150 µg/mL) was administered for the indicated time, and the cells were then harvested for Western blotting with the indicated antibodies. The rate of p53 decay was quantified using ImageJ software with reference to GAPDH control and is mentioned at the bottom of the blot
Expression Patterns of USP19 and p53 in Cancer
We used the Cancer Cell Line Encyclopedia (CCLE) database to check the correlation between USP19 and p53. The high scores of USP19 mRNA were inversely correlated with p53 mRNA levels across a wide range of cancer types (Fig. 3A, Table S4). Moreover, a scatterplot of the USP19-p53 expression patterns gave an r value of -0.2119 across different tissues, suggesting a negative correlation between USP19 and p53 (Fig. 3B). Our analysis showed that USP19 and p53 are negatively correlated with each other, suggesting that USP19 might play a critical role in promoting carcinogenesis.

Correlation between USP19 and p53 in different cancer types. A A heat map showing mRNA expression levels of USP19 and p53 derived from the CCLE panel for different cancer types. Representative samples are arranged from low to high mRNA levels of USP19, and corresponding p53 values are sorted. B A scatterplot showing the expression correlation between USP19 and p53 mRNA levels. Pearson correlation (r) quantifying the relationship between USP19 and p53 are presented
Generation of Single Cell-Derived Knockout Clones of USP19 in HeLa Cells
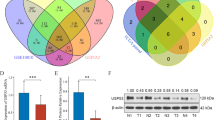
Next, we speculate that the depletion of USP19 might attenuate carcinogenesis in cervical cancer cells by enhancing the tumor suppressive role of p53. To test this hypothesis, we generated a USP19-knockout cell line in cervical cancer cells (HeLa) using the CRISPR/Cas9 system. We designed sgRNA1 and sgRNA2 targeting exon 1 and exon 2 of USP19, respectively (Fig. 4A). The T7E1 assays showed that sgRNA1 and sgRNA2 disrupted USP19 in HeLa cells (Fig. 4B). Next, we used both sgRNA1 and sgRNA2 to generate single-cell-derived USP19-knockout HeLa cell clones. The sgRNA1 and sgRNA2-mediated knockout of USP19 in single-cell-derived clones were analyzed using the T7E1 assay and Western blotting (Fig. S1A-S1B and S2A-S2B). The USP19 gene knockout clones, USP19-KO1 #18 and USP19-KO2 #22, were further analyzed using Sanger sequencing. The results indicated out-of-frame mutations in USP19-KO1 #18 (hereafter referred to as USP19-KO), while USP19-KO2 #22 knockout clones generated in HeLa cells showed out-of-frame mutations along with mixed wild type sequences (Fig. 4C). USP19 gene disruption and its effect on p53 were further investigated by Western blot analysis. We observed a significant reduction in USP19 protein levels in USP19-KO cells when compared with mock controls (Fig. 4D). Consistent with this finding, USP19-KO cells displayed significant upregulation of p53 protein when compared with mock controls (Fig. 4D), suggesting that USP19 may play an important role in regulating p53 protein levels during carcinogenesis.

Generation and validation of USP19-knockout in cervical cancer cells. A Schematic representation showing the sgRNA design strategy targeting USP19 at exon 1 and 2. The position of the designed sgRNA is denoted by the red arrowhead. The target sequence is in red and the PAM sequence is in bold blue font. B The knockout efficiency of the designed sgRNAs in HeLa cells was validated using T7E1 assay. The arrowhead indicates the cleaved bands after T7E1 assay. The band intensity was calculated using ImageJ software and the indel percentage is provided at the bottom of the gel. C The USP19 depletion was confirmed using Sanger sequencing. The data shows the disrupted USP19 gene sequences of two clones (USP19-KO1 #18 and USP19-KO2 #22). The sgRNA recognition site is indicated in red and PAM sequence in bold blue font. The deleted bases are indicated with dashes, whereas the inserted bases are denoted with a green color, and the number of deleted or inserted bases is indicated in parentheses. The number of occurrences of the indicated sequences is shown in parentheses (for example, X4 and X2 indicate the number of each clone sequenced). D The expression of USP19 and p53 in USP19 knock-out cell lines (USP19-KO1 and USP19-KO2) was validated by Western blotting. GAPDH was used as a loading control
Loss of USP19 Suppresses Cervical Cancer Progression
To further assess the consequences of USP19 regulation of p53 in cancer progression, we performed several carcinogenesis-related assays. An anchorage-independent colony formation assay showed that USP19-KO cells formed fewer colonies than mock control cells (Fig. 5A). However, increased carcinogenic activity was observed in USP19-KO cells reconstituted with Flag-USP19 as evidenced by increased colonies but not with Flag-USP19CA (Fig. 5A). Similarly, we evaluated the effect of USP19 on the cellular migration and invasion abilities in HeLa cells. We found that the migration and invasion abilities of USP19-KO cells were reduced when compared to mock controls. In contrast, USP19-KO cells reconstituted with Flag-USP19 showed increased cell migration and invasion (Fig. 5B C). However, USP19-KO cells reconstituted with Flag-USP19CA showed no significant increase in cell migration or invasion (Fig. 5B C). Collectively, our data demonstrate that the loss of USP19 suppresses carcinogenesis in cervical cancer.

Depletion of USP19 supports tumor suppression in a p53-dependent manner. A Mock, USP19-KO1, USP19-KO1 reconstituted with USP19 and USP19-KO1 reconstituted with USP19CA were subjected to anchorage independent colony formation assay. (scale bar = 200 μm). The colony numbers were quantified and are presented graphically as mean and standard deviation of three independent experiments (n=3) (right panel). One-way ANOVA followed by Dunnett’s post-hoc test was used and P values are indicated. B The indicated cell groups were subjected to matrigel invasion assay (scale = 100 μm). The number of invaded cells/field are presented graphically as mean and standard deviation of three independent experiments (n=3) (right panel). One-way ANOVA followed by Dunnett’s post-hoc test was used and P values are indicated. C The same cells were assessed for their migratory activity using a wound-healing assay. Images were captured at the indicated time points (scale = 200 μm). The percentage of wound closure is presented graphically as mean and standard deviation of three independent experiments (n=3) (right panel). One-way ANOVA followed by Dunnett’s post-hoc test was used and P values are indicated
Discussion
Cells are constantly exposed to different types of stressors that introduce DNA damage and gene aberrations such as mutations, deletions, or gene translocations. Accumulation of genetic aberrations results in genetic instability, which can lead to the development of cancers and other diseases [38]. Therefore, cells have evolved protective responses to counteract stress signals and prevent malignant transformation of cells. p53 is a tumor suppressor gene that plays a variety of roles in regulating cellular stress responses. p53 activates several target genes and channels the activation of pro-apoptotic signals and cell-cycle arrest [39,40,41,42]. Previous reports have demonstrated that over 50% of cancers have mutations in the p53 gene [43], indicating that wild-type p53 is critical for maintaining cellular integrity and genomic stability [44,45,46,47]. Moreover, studies on p53-deficient mice have revealed their susceptibility to oncogenic transformation [48]. Therefore, regulation of the optimal level of p53 protein is critical for the maintenance of cellular proteostasis.
DUBs are proteases that reversely modify substrate proteins by removing ubiquitin moieties on target proteins, thus preventing their protein degradation. DUBs regulate diverse biological functions such as DNA damage response, apoptosis, transcriptional regulation, cell cycle control, immune response, and oncogenesis [49,50,51,52,53]. The majority of DUBs are known to stabilize their protein substrate by preventing protein degradation through the 26 S proteasomal pathway [53]. However, in some cases, DUBs can negatively regulate their protein substrate depending on the cellular condition [54, 55]. There are several instances where DUBs negatively regulate their protein substrates and promote for rapid proteolysis.
Under normal conditions, p53 is maintained at low levels by the E3 ligase Mdm2; however, upon DNA damage, p53 levels are elevated by phosphorylation, acetylation, and deubiquitination [22, 56, 57]. DUBs are directly or indirectly involved in the regulation of the basal levels of p53 in normal or stress conditions [22, 58]. In 2002, USP7 was the first DUB candidate identified for p53 stabilization. USP7 has both a direct and indirect influence on p53 stabilization [59]. USP10 is another DUB that stabilizes p53 under physiological conditions as well as in response to DNA damage to maintain a stable level of p53 in cytosol [10]. In response to DNA damage, USP11 is known to interact with and stabilizes p53 protein. Additionally, upon DNA damage, USP11 activates p53 along with p53-targeted genes such as Puma, Bax, and p21 [19].
Certain DUBs, however, negatively regulate and promote the degradation of p53 protein [24, 60, 61]. These DUBs downregulate the p53 protein level by stabilizing E3 ligases such as ARF-BP1, Mdm2, E6AP, CARP, and Pirh2, which are known to regulate ubiquitin-mediated p53 protein degradation [24, 62,63,64,65,66]. For instance, in 2007 Stevenson et al. demonstrated that USP2a interacts with and stabilizes Mdm2 resulting in rapid p53 protein degradation [26]. A direct interaction between USP4 and ARF-BP1 E3 ligase resulted in destabilization of p53 protein level [24]. Overexpression of USP5 stabilized Beclin1 subsequently led to MDM2-mediated p53 protein instability [25, 67]. Importantly, a partial knockdown of USP7 results in destabilization of endogenous p53, while the complete loss of USP7 leads to stabilization of p53 [68]. USP15 showed a similar destabilization effect on p53 protein by stabilizing MDM2 and promoted cancer cell survival [23].
In contrast, some DUBs showed a direct effect on p53 stabilization without regulating E3 ligases. In 2006, Dayal et al. demonstrated that USP5 destabilizes p53 protein. Silencing of USP5 increased the stability of p53 by inhibiting proteasomal degradation of p53 without affecting the proteasomal degradation of Mdm2, suggesting direct negative regulation on p53 [69]. Likewise, in this study, we identified USP19 exhibiting a negative regulatory role in p53 protein stabilization. Overexpression of USP19 showed a decreased p53 level, while depletion of USP19 showed stabilization of p53 (Fig. 1). Moreover, the endogenous USP19 and p53 protein bind each other, which was evident by interaction studies using specific antibodies (Fig. 1). In order to investigate whether the interaction between USP19 and p53 is direct or not, we performed a direct protein-protein binding pull down assay using purified proteins. Interestingly, p53 showed a direct interaction with USP19 (Fig. 1). We further demonstrated that USP19 promotes ubiquitination of p53 and reduces its half-life (Fig. 2). The above observation is in line with our previous report on CYLD showing negative regulation of NoxO1 protein stabilization where CYLD promoted ubiquitination and further reduced the half-life of NoxO1 protein [55].
USP19 plays a critical role in the control of tumorigenesis and cancer dissemination [28]. USP19 is also associated with regulation of several cellular process by stabilizing key proteins that regulate cancer such as HIF1-alpha, c-IAP1, c-IAP2, SOAT1, MMP2/MMP9, and ME1 [31, 70,71,72,73]. In 2022, we reported that USP19 stabilizes survivin protein and regulates mitotic progression and tumorigenesis in colon cancer [74]. In this current study, we applied the CRISPR/Cas9 system to generate USP19 gene disruption in HeLa cells and investigated its role in p53-mediated carcinogenesis (Fig. 4). We demonstrated that the complete knockout of USP19 reduced the oncogenic potential of cervical cancer cells. The USP19-depleted cells reconstituted with USP19 promoted cancer progression in HeLa cells resulting in increased colony formation, migration, and invasion (Fig. 5). The above observation is in line with our previous report on USP32 exhibiting a similar negative regulatory role on its protein substrate SLC35F2 promoting tumorigenesis in breast cancer [54]. In summary, we have identified USP19 as a negative regulator of p53 protein stabilization. Moreover, the loss of USP19 extended p53 protein half-life and suppressed cervical cancer progression.
Data Availability
Data availability is not applicable as no datasets were generated in this study.
References
Hager, K. M., & Gu, W. (2014). Understanding the non-canonical pathways involved in p53-mediated tumor suppression. Carcinogenesis, 35(4), 740–746. https://doi.org/10.1093/carcin/bgt487.
Hu, X., Chandler, J. D., Park, S., Liu, K., Fernandes, J., Orr, M., Smith, M. R., Ma, C., Kang, S. M., Uppal, K., Jones, D. P., & Go, Y. M. (2019). Low-dose cadmium disrupts mitochondrial citric acid cycle and lipid metabolism in mouse lung. Free radical biology & medicine, 131, 209–217. https://doi.org/10.1016/j.freeradbiomed.2018.12.005.
Kruiswijk, F., Labuschagne, C. F., & Vousden, K. H. (2015). p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nature reviews Molecular cell biology, 16(7), 393–405. https://doi.org/10.1038/nrm4007.
Song, H., Hollstein, M., & Xu, Y. (2007). p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nature cell biology, 9(5), 573–580. https://doi.org/10.1038/ncb1571.
Fearon, E. R., & Vogelstein, B. (1990). A genetic model for colorectal tumorigenesis. Cell, 61(5), 759–767. https://doi.org/10.1016/0092-8674(90)90186-i.
Malkin, D., Li, F. P., Strong, L. C., Fraumeni, J. F. Jr., Nelson, C. E., Kim, D. H., Kassel, J., Gryka, M. A., Bischoff, F. Z., Tainsky, M. A., et al. (1990). Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science (New York NY), 250(4985), 1233–1238. https://doi.org/10.1126/science.1978757.
Miller, L. D., Smeds, J., George, J., Vega, V. B., Vergara, L., Ploner, A., Pawitan, Y., Hall, P., Klaar, S., Liu, E. T., & Bergh, J. (2005). An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proceedings of the National Academy of Sciences of the United States of America, 102(38), 13550–13555. https://doi.org/10.1073/pnas.0506230102.
Olivier, M., & Taniere, P. (2011). Somatic mutations in cancer prognosis and prediction: Lessons from TP53 and EGFR genes. Current opinion in oncology, 23(1), 88–92. https://doi.org/10.1097/CCO.0b013e3283412dfa.
Rivlin, N., Brosh, R., Oren, M., & Rotter, V. (2011). Mutations in the p53 tumor suppressor gene: Important milestones at the various steps of Tumorigenesis. Genes & cancer, 2(4), 466–474. https://doi.org/10.1177/1947601911408889.
Yuan, J., Luo, K., Zhang, L., Cheville, J. C., & Lou, Z. (2010). USP10 regulates p53 localization and stability by deubiquitinating p53. Cell, 140(3), 384–396. https://doi.org/10.1016/j.cell.2009.12.032.
Pei, D., Zhang, Y., & Zheng, J. (2012). Regulation of p53: A collaboration between Mdm2 and mdmx. Oncotarget, 3(3), 228–235. https://doi.org/10.18632/oncotarget.443.
Wade, M., Wang, Y. V., & Wahl, G. M. (2010). The p53 orchestra: Mdm2 and Mdmx set the tone. Trends in cell biology, 20(5), 299–309. https://doi.org/10.1016/j.tcb.2010.01.009.
Li, Y., Ma, C., Zhou, T., Liu, Y., Sun, L., & Yu, Z. (2016). TRIM65 negatively regulates p53 through ubiquitination. Biochemical and biophysical research communications, 473(1), 278–282. https://doi.org/10.1016/j.bbrc.2016.03.093.
Freedman, D. A., & Levine, A. J. (1998). Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Molecular and cellular biology 18 (12):7288–7293. doi:https://doi.org/10.1128/mcb.18.12.7288.
Chène, P. (2003). Inhibiting the p53-MDM2 interaction: An important target for cancer therapy. Nature reviews Cancer, 3(2), 102–109. https://doi.org/10.1038/nrc991.
Sane, S., & Rezvani, K. (2017). Essential roles of E3 ubiquitin ligases in p53 regulation. International journal of molecular sciences, 18(2), https://doi.org/10.3390/ijms18020442.
Lim, S. K., Shin, J. M., Kim, Y. S., & Baek, K. H. (2004). Identification and characterization of murine mHAUSP encoding a deubiquitinating enzyme that regulates the status of p53 ubiquitination. International journal of oncology, 24(2), 357–364.
Cummins, J. M., Rago, C., Kohli, M., Kinzler, K. W., Lengauer, C., & Vogelstein, B. (2004). Tumour suppression: Disruption of HAUSP gene stabilizes p53. Nature, 428(6982), 1–pfollowing486. https://doi.org/10.1038/nature02501.
Ke, J. Y., Dai, C. J., Wu, W. L., Gao, J. H., Xia, A. J., Liu, G. P., Lv, K. S., & Wu, C. L. (2014). USP11 regulates p53 stability by deubiquitinating p53. Journal of Zhejiang University Science B, 15(12), 1032–1038. https://doi.org/10.1631/jzus.B1400180.
Liu, J., Chung, H. J., Vogt, M., Jin, Y., Malide, D., He, L., Dundr, M., & Levens, D. (2011). JTV1 co-activates FBP to induce USP29 transcription and stabilize p53 in response to oxidative stress. The EMBO journal, 30(5), 846–858. https://doi.org/10.1038/emboj.2011.11.
Hock, A. K., Vigneron, A. M., Carter, S., Ludwig, R. L., & Vousden, K. H. (2011). Regulation of p53 stability and function by the deubiquitinating enzyme USP42. The EMBO journal, 30(24), 4921–4930. https://doi.org/10.1038/emboj.2011.419.
Kwon, S. K., Saindane, M., & Baek, K. H. (2017). p53 stability is regulated by diverse deubiquitinating enzymes. Biochimica et biophysica acta reviews on cancer 1868 (2):404–411. doi:https://doi.org/10.1016/j.bbcan.2017.08.001.
Zou, Q., Jin, J., Hu, H., Li, H. S., Romano, S., Xiao, Y., Nakaya, M., Zhou, X., Cheng, X., Yang, P., Lozano, G., Zhu, C., Watowich, S. S., Ullrich, S. E., & Sun, S. C. (2014). USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nature immunology, 15(6), 562–570. https://doi.org/10.1038/ni.2885.
Zhang, X., Berger, F. G., Yang, J., & Lu, X. (2011). USP4 inhibits p53 through deubiquitinating and stabilizing ARF-BP1. The EMBO journal, 30(11), 2177–2189. https://doi.org/10.1038/emboj.2011.125.
Li, J., Wang, Y., Luo, Y., Liu, Y., Yi, Y., Li, J., Pan, Y., Li, W., You, W., Hu, Q., Zhao, Z., Zhang, Y., Cao, Y., Zhang, L., Yuan, J., & Xiao, Z. J. (2022). USP5-Beclin 1 axis overrides p53-dependent senescence and drives Kras-induced tumorigenicity. Nature communications, 13(1), 7799. https://doi.org/10.1038/s41467-022-35557-y.
Stevenson, L. F., Sparks, A., Allende-Vega, N., Xirodimas, D. P., Lane, D. P., & Saville, M. K. (2007). The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. The EMBO journal, 26(4), 976–986. https://doi.org/10.1038/sj.emboj.7601567.
Lahav-Baratz, S., Kravtsova-Ivantsiv, Y., Golan, S., & Ciechanover, A. (2017). The testis-specific USP26 is a deubiquitinating enzyme of the ubiquitin ligase Mdm2. Biochemical and biophysical research communications 482 (1):106–111. doi:https://doi.org/10.1016/j.bbrc.2016.10.135.
Rossi, F. A., & Rossi, M. (2022). Emerging role of ubiquitin-specific protease 19 in oncogenesis and Cancer Development. Frontiers in cell and developmental biology, 10, 889166. https://doi.org/10.3389/fcell.2022.889166.
Hu, W., Su, Y., Fei, X., Wang, X., Zhang, G., Su, C., Du, T., Yang, T., Wang, G., Tang, Z., & Zhang, J. (2020). Ubiquitin specific peptidase 19 is a prognostic biomarker and affect the proliferation and migration of clear cell renal cell carcinoma. Oncology reports, 43(6), 1964–1974. https://doi.org/10.3892/or.2020.7565.
Kang, H., Choi, M. C., Kim, S., Jeong, J. Y., Kwon, A. Y., Kim, T. H., Kim, G., Joo, W. D., Park, H., Lee, C., Song, S. H., Jung, S. G., Hwang, S., & An, H. J. (2021). USP19 and RPL23 as candidate prognostic markers for Advanced-Stage High-Grade Serous Ovarian Carcinoma. Cancers, 13(16), https://doi.org/10.3390/cancers13163976.
Mei, Y., Hahn, A. A., Hu, S., & Yang, X. (2011). The USP19 deubiquitinase regulates the stability of c-IAP1 and c-IAP2. The Journal of biological chemistry, 286(41), 35380–35387. https://doi.org/10.1074/jbc.M111.282020.
Mirza, A., McGuirk, M., Hockenberry, T. N., Wu, Q., Ashar, H., Black, S., Wen, S. F., Wang, L., Kirschmeier, P., Bishop, W. R., Nielsen, L. L., Pickett, C. B., & Liu, S. (2002). Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene, 21(17), 2613–2622. https://doi.org/10.1038/sj.onc.1205353.
Wang, Z., Fukuda, S., & Pelus, L. M. (2004). Survivin regulates the p53 tumor suppressor gene family. Oncogene, 23(49), 8146–8153. https://doi.org/10.1038/sj.onc.1207992.
Cheng, L., Zhou, Z., Flesken-Nikitin, A., Toshkov, I. A., Wang, W., Camps, J., Ried, T., & Nikitin, A. Y. (2021). Correction to: Rb inactivation accelerates neoplastic growth and substitutes for recurrent amplification of cIAP1, cIAP2 and Yap1 in sporadic mammary carcinoma associated with p53 deficiency. Oncogene, 40(9), 1754. https://doi.org/10.1038/s41388-021-01647-2.
Ramakrishna, S., Kwaku Dad, A. B., Beloor, J., Gopalappa, R., Lee, S. K., & Kim, H. (2014). Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome research, 24(6), 1020–1027. https://doi.org/10.1101/gr.171264.113.
Suresh, B., Ramakrishna, S., Kim, Y. S., Kim, S. M., Kim, M. S., & Baek, K. H. (2010). Stability and function of mammalian lethal giant larvae-1 oncoprotein are regulated by the scaffolding protein RanBPM. The Journal of biological chemistry, 285(46), 35340–35349. https://doi.org/10.1074/jbc.M110.156836.
Suresh, B., Ramakrishna, S., & Kim, H. (2017). Cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA for genome editing. Methods in molecular biology. (Clifton NJ), 1507, 81–94. https://doi.org/10.1007/978-1-4939-6518-2_7.
Elledge, S. J. (1996). Cell cycle checkpoints: Preventing an identity crisis. Science (New York NY), 274(5293), 1664–1672. https://doi.org/10.1126/science.274.5293.1664.
Sionov, R. V., & Haupt, Y. (1999). The cellular response to p53: The decision between life and death. Oncogene, 18(45), 6145–6157. https://doi.org/10.1038/sj.onc.1203130.
McLean, D. E., Kearney, J., & Cawley, M. F. (1999). Environmentally responsive temperature instability in pediatric spinal cord injury. Spinal cord, 37(10), 705–709. https://doi.org/10.1038/sj.sc.3100888.
Vousden, K. H., & Lu, X. (2002). Live or let die: The cell’s response to p53. Nature reviews Cancer, 2(8), 594–604. https://doi.org/10.1038/nrc864.
Prives, C., & Hall, P. A. (1999). The p53 pathway. The Journal of pathology, 187(1), 112–126. https://doi.org/10.1002/(sici)1096-9896(199901)187:1<112::Aid-path250>3.0.Co;2-3.
Feki, A., & Irminger-Finger, I. (2004). Mutational spectrum of p53 mutations in primary breast and ovarian tumors. Critical reviews in oncology/hematology, 52(2), 103–116. https://doi.org/10.1016/j.critrevonc.2004.07.002.
Baker, S. J., Fearon, E. R., Nigro, J. M., Hamilton, S. R., Preisinger, A. C., Jessup, J. M., vanTuinen, P., Ledbetter, D. H., Barker, D. F., Nakamura, Y., White, R., & Vogelstein, B. (1989). Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science (New York NY), 244(4901), 217–221. https://doi.org/10.1126/science.2649981.
Nigro, J. M., Baker, S. J., Preisinger, A. C., Jessup, J. M., Hostetter, R., Cleary, K., Bigner, S. H., Davidson, N., Baylin, S., Devilee, P., et al. (1989). Mutations in the p53 gene occur in diverse human tumour types. Nature, 342(6250), 705–708. https://doi.org/10.1038/342705a0.
Takahashi, T., Nau, M. M., Chiba, I., Birrer, M. J., Rosenberg, R. K., Vinocour, M., Levitt, M., Pass, H., Gazdar, A. F., & Minna, J. D. (1989). p53: A frequent target for genetic abnormalities in lung cancer. Science (New York NY), 246(4929), 491–494. https://doi.org/10.1126/science.2554494.
Hollstein, M. C., Metcalf, R. A., Welsh, J. A., Montesano, R., & Harris, C. C. (1990). Frequent mutation of the p53 gene in human esophageal cancer. Proceedings of the National Academy of Sciences of the United States of America, 87(24), 9958–9961. https://doi.org/10.1073/pnas.87.24.9958.
Donehower, L. A., Harvey, M., Slagle, B. L., McArthur, M. J., Montgomery, C. A. Jr., Butel, J. S., & Bradley, A. (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature, 356(6366), 215–221. https://doi.org/10.1038/356215a0.
Clague, M. J., Coulson, J. M., & Urbé, S. (2012). Cellular functions of the DUBs. Journal of cell science, 125(Pt 2), 277–286. https://doi.org/10.1242/jcs.090985.
Amerik, A. Y., & Hochstrasser, M. (2004). Mechanism and function of deubiquitinating enzymes. Biochimica et biophysica acta, 1695(1–3), 189–207. https://doi.org/10.1016/j.bbamcr.2004.10.003.
Sun, S. C. (2008). Deubiquitylation and regulation of the immune response. Nature reviews Immunology, 8(7), 501–511. https://doi.org/10.1038/nri2337.
Sarodaya, N., Karapurkar, J., Kim, K. S., Hong, S. H., & Ramakrishna, S. (2020). The role of deubiquitinating enzymes in hematopoiesis and hematological malignancies. Cancers, 12(5), https://doi.org/10.3390/cancers12051103.
Ramakrishna, S., Suresh, B., & Baek, K. H. (2011). The role of deubiquitinating enzymes in apoptosis. Cellular and molecular life sciences: CMLS, 68(1), 15–26. https://doi.org/10.1007/s00018-010-0504-6.
Chandrasekaran, A. P., Kaushal, K., Park, C. H., Kim, K. S., & Ramakrishna, S. (2021). USP32 confers cancer cell resistance to YM155 via promoting ER-associated degradation of solute carrier protein SLC35F2. Theranostics, 11(20), 9752–9771. https://doi.org/10.7150/thno.63806.
Haq, S., Sarodaya, N., Karapurkar, J. K., Suresh, B., Jo, J. K., Singh, V., Bae, Y. S., Kim, K. S., & Ramakrishna, S. (2022). CYLD destabilizes NoxO1 protein by promoting ubiquitination and regulates prostate cancer progression. Cancer letters, 525, 146–157. https://doi.org/10.1016/j.canlet.2021.10.032.
Ashcroft, M., Kubbutat, M. H., & Vousden, K. H. (1999). Regulation of p53 function and stability by phosphorylation. Molecular and cellular biology, 19(3), 1751–1758. https://doi.org/10.1128/mcb.19.3.1751.
Tang, Y., Zhao, W., Chen, Y., Zhao, Y., & Gu, W. (2008). Acetylation is indispensable for p53 activation. Cell, 133(4), 612–626. https://doi.org/10.1016/j.cell.2008.03.025.
He, M., Zhou, Z., Shah, A. A., Zou, H., Tao, J., Chen, Q., & Wan, Y. (2016). The emerging role of deubiquitinating enzymes in genomic integrity, diseases, and therapeutics. Cell & bioscience, 6, 62. https://doi.org/10.1186/s13578-016-0127-1.
Li, M., Chen, D., Shiloh, A., Luo, J., Nikolaev, A. Y., Qin, J., & Gu, W. (2002). Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature, 416(6881), 648–653. https://doi.org/10.1038/nature737.
Cho, J., Park, J., Shin, S. C., Jang, M., Kim, J. H., Kim, E. E., & Song, E. J. (2020). USP47 promotes tumorigenesis by negative regulation of p53 through Deubiquitinating Ribosomal protein S2. Cancers, 12(5), https://doi.org/10.3390/cancers12051137.
Qi, S. M., Cheng, G., Cheng, X. D., Xu, Z., Xu, B., Zhang, W. D., & Qin, J. J. (2020). Targeting USP7-Mediated deubiquitination of MDM2/MDMX-p53 pathway for Cancer Therapy: Are we there yet? Frontiers in cell and developmental biology, 8, 233. https://doi.org/10.3389/fcell.2020.00233.
Sheng, Y., Saridakis, V., Sarkari, F., Duan, S., Wu, T., Arrowsmith, C. H., & Frappier, L. (2006). Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nature structural & molecular biology, 13(3), 285–291. https://doi.org/10.1038/nsmb1067.
Vos, R. M., Altreuter, J., White, E. A., & Howley, P. M. (2009). The ubiquitin-specific peptidase USP15 regulates human papillomavirus type 16 E6 protein stability. Journal of virology, 83(17), 8885–8892. https://doi.org/10.1128/jvi.00605-09.
Yang, W., Rozan, L. M., McDonald, E. R. 3rd, Navaraj, A., Liu, J. J., Matthew, E. M., Wang, W., Dicker, D. T., & El-Deiry, W. S. (2007). CARPs are ubiquitin ligases that promote MDM2-independent p53 and phospho-p53ser20 degradation. The Journal of biological chemistry, 282(5), 3273–3281. https://doi.org/10.1074/jbc.M610793200.
Leng, R. P., Lin, Y., Ma, W., Wu, H., Lemmers, B., Chung, S., Parant, J. M., Lozano, G., Hakem, R., & Benchimol, S. (2003). Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell, 112(6), 779–791. https://doi.org/10.1016/s0092-8674(03)00193-4.
Lee, J. T., & Gu, W. (2010). The multiple levels of regulation by p53 ubiquitination. Cell death and differentiation, 17(1), 86–92. https://doi.org/10.1038/cdd.2009.77.
Tavana, O., & Gu, W. (2017). Modulation of the p53/MDM2 interplay by HAUSP inhibitors. Journal of molecular cell biology, 9(1), 45–52. https://doi.org/10.1093/jmcb/mjw049.
Li, M., Brooks, C. L., Kon, N., & Gu, W. (2004). A dynamic role of HAUSP in the p53-Mdm2 pathway. Molecular cell, 13(6), 879–886. https://doi.org/10.1016/s1097-2765(04)00157-1.
Dayal, S., Sparks, A., Jacob, J., Allende-Vega, N., Lane, D. P., & Saville, M. K. (2009). Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53. The Journal of biological chemistry, 284(8), 5030–5041. https://doi.org/10.1074/jbc.M805871200.
Altun, M., Zhao, B., Velasco, K., Liu, H., Hassink, G., Paschke, J., Pereira, T., & Lindsten, K. (2012). Ubiquitin-specific protease 19 (USP19) regulates hypoxia-inducible factor 1α (HIF-1α) during hypoxia. The Journal of biological chemistry, 287(3), 1962–1969. https://doi.org/10.1074/jbc.M111.305615.
Zhu, Y., Gu, L., Lin, X., Zhou, X., Lu, B., Liu, C., Li, Y., Prochownik, E. V., Karin, M., Wang, F., & Li, Y. (2023). P53 deficiency affects cholesterol esterification to exacerbate hepatocarcinogenesis. Hepatology (Baltimore Md), 77(5), 1499–1511. https://doi.org/10.1002/hep.32518.
Dong, Z., Guo, S., Wang, Y., Zhang, J., Luo, H., Zheng, G., Yang, D., Zhang, T., Yan, L., Song, L., Liu, K., Sun, Z., Meng, X., Zheng, Z., Zhang, J., & Zhao, Y. (2020). USP19 enhances MMP2/MMP9-Mediated tumorigenesis in gastric Cancer. OncoTargets and therapy, 13, 8495–8510. https://doi.org/10.2147/ott.S240543.
Zhu, Y., Gu, L., Lin, X., Zhou, X., Lu, B., Liu, C., Lei, C., Zhou, F., Zhao, Q., Prochownik, E. V., & Li, Y. (2021). USP19 exacerbates lipogenesis and colorectal carcinogenesis by stabilizing ME1. Cell reports, 37(13), 110174. https://doi.org/10.1016/j.celrep.2021.110174.
Chandrasekaran, A. P., Tyagi, A., Poondla, N., Sarodaya, N., Karapurkar, J. K., Kaushal, K., Park, C. H., Hong, S. H., Kim, K. S., & Ramakrishna, S. (2022). Dual role of deubiquitinating enzyme USP19 regulates mitotic progression and tumorigenesis by stabilizing survivin. Molecular therapy: the journal of the American Society of Gene Therapy, 30(11), 3414–3429. https://doi.org/10.1016/j.ymthe.2022.07.019.
Acknowledgements
We would like to thank all the members of the Suri Lab and KSK Lab. We would also like to thank Professor Ryu Seong Eon, Hanyang University, Seoul, South Korea for his help in recombinant protein purification.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grants (2021M3A9H3015389 and 2021R1I1A1A01052637), a Korean Fund for Regenerative Medicine (KFRM) grant funded by the Korea government (Ministry of Science and ICT, Ministry of Health & Welfare) (22A0304L1-01) and Medical Research Center (2017R1A5A2015395), funded by the National Research Foundation of Korea (NRF) of the Ministry of Science, ICT and Future Planning, Korea.
Author information
Authors and Affiliations
Contributions
A.T., J.K.K, and S.R.K. designed the study. A.T, J.K.K, and J.C.C conducted experiment and analyzed and interpreted the data. A.T. and S.R.K. co-wrote the manuscript. J.C.C. conducted all revision experiment. B.S., N.S., A.M.A., K.K., S.H., A.P.C., S.D., and V.S. assisted A.T with the experiments. B.S., K.S.K., and S.R. procured financial support and reviewed the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors have no competing interests regarding the content of this manuscript to declare.
Ethics Approval
This study was approved by the Hanyang University Institutional Review Board (approval number HYI-17-137-10).
Consent to Participate
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tyagi, A., Karapurkar, J.K., Colaco, J.C. et al. USP19 Negatively Regulates p53 and Promotes Cervical Cancer Progression. Mol Biotechnol 66, 2032–2045 (2024). https://doi.org/10.1007/s12033-023-00814-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-023-00814-y