
Abstract
In this study, a chitinase gene (DrChit) that plays a role in the carnivorous processes of Drosera rotundifolia L. was isolated from genomic DNA, linked to a double CaMV35S promoter and nos terminator in a pBinPlus plant binary vector, and used for Agrobacterium-mediated transformation of tobacco. RT-qPCR revealed that within 14 transgenic lines analysed in detail, 57% had DrChit transcript abundance comparable to or lower than level of a reference actin gene transcript. In contrast, the transgenic lines 9 and 14 exhibited 72 and 152 times higher expression level than actin. The protein extracts of these two lines exhibited five and eight times higher chitinolytic activity than non-transgenic controls when measured in a fluorimetric assay with FITC-chitin. Finally, the growth of Trichoderma viride was obviously suppressed when the pathogen was exposed to 100 μg of crude protein extract isolated from line 9 and line 14, with the area of mycelium growth reaching only 56.4% and 45.2%, of non-transgenic control, respectively. This is the first time a chitinase from a carnivorous plant with substrate specificity for long chitin polymers was tested in a transgenic plant with the aim of exploring its antifungal potential.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chitin is the second most important biopolymer consisting of β-(1-4)-N-acetyl-d-glucosamine (GlcNAc) units and is found in the exoskeleton of arthropods and in the cell walls of fungi. In individual species of fungi, chitin accounts for a 3–60% of the total cell wall mass, and it is responsible for the rigidity, physical strength and specific shape of cell wall [1].
Chitinases (EC 3.2.1.14) are enzymes that hydrolyse the β-1,4-linkages in chitin. Based on the sequence similarity and different catalytic mechanisms, chitinases are grouped into glycosyl hydrolase (GH) families 18, 19, and 20 [2]. Plant chitinase genes are classified into seven classes (I–VII) [3, 4]. Class I, II, IV, VI and VII chitinases belong to the GH19 family, while class III and V chitinases are members of the GH18 family [5].
Chitinases play a role during plant growth and developmental processes, such as during pollination, senescence, germination and somatic embryogenesis [6, 7]. Although there is no conclusive information about the endogenous substrates of plant chitinases, arabinogalactan proteins and GlcNAc-containing glycoproteins in cell walls are the presumed substrates of hydrolysis [8]. Another function of chitinases involves abiotic stress responses. Some chitinase genes are induced by a range of abiotic stresses, including osmotic-, cold-, heavy- metal stress, and wounding [9,10,11,12]. Abiotic elicitors stimulate pathogenesis-like responses, which are vital for their survival under varied environmental condition via development of induced systematic resistance [13, 14]. Most attention has been paid to the role of chitinases in defence against invading fungal pathogens [15]. The early stage of pathogenesis is accompanied by the expression of apoplastic class II chitinases that are responsible for releasing elicitor molecules from invading fungal pathogens [16]. A range of defence compounds, including vacuolar class I chitinases, are then stimulated, and they slow the infection from spreading in the affected plant tissue. Antifungal in vitro tests have revealed higher hydrolytic and antifungal potentials in class I chitinases compared with their class II counterparts [17]. The N-terminal chitin-binding domain (CBD) that occurs in addition to the catalytic domain in plant chitinases of class I and IV are responsible for their greater hydrolytic activity. Broglie et al. [18] first generated transgenic tobacco and canola plants utilizing constitutive expression of the bean CH5B gene. The plants with obviously increased chitinolytic activity (44-fold) showed a delay of disease symptoms caused by Rhizoctonia solani infection. Many other studies also showed that mainly the genes for vacuolar class I chitinase genes [19, 20] or their microbial apoplastic chitinase orthologs [10, 21, 22] have the potential to enhance the resistance to fungal pathogens in a range of transgenic crops such as tomato [23], potato [24], cotton [25], wheat [26], rice [27], Indian mustard [28], peanut [29], etc. A comprehensive list of publications dealing with transgenics carrying chitinase genes is provided by review papers [14, 21, 30]. However, disease control has never been complete, while a decrease of symptoms varied with the chitinase transgene and strategy employed as well as with the characteristics of fungal pathogen [30, 31]. The experiments of Dana et al. [10] indicated that two factors were crucial to improving the tolerance of transgenic plants to fungal pathogens when overexpressing chitinase genes—the strength of chitinolytic activity of the transgenic chitinase and its targeting into the apoplast. Such transgenic chitinase(s) fulfil dual roles: inhibiting the growth of invading fungi as well as inducing other defence-related mechanisms. In most plant species, class I chitinases have vacuolar localization; however, carnivorous taxa release chitinases of various classes, including class I, into the apoplast [32, 33].
Our aim here was to investigate the contribution of a novel apoplastic sundew class I chitinase (DrChit) [34] as a enhancement to antifungal potential in transgenic tobacco plants. DrChit expression in the carnivorous species Drosera rotundifolia was detected in tentacles during the digestive processes. Chitinases of several classes have been found in other carnivorous species where they are involved in disrupting the outer chitinous barrier of their captured prey [35, 36].
In this study, the DrChit gene under the control of the CaMV35S promoter, as well as the gene for neomycin phosphotransferase (nptII) driven by the nopaline transferase promoter (nos-P), was introduced into the Nicotiana tabacum L. via Agrobacterium-mediated transformation. Molecular analyses confirmed successful integration and expression of sundew chitinase in regenerated transgenic plants. Subsequently, the DrChit expression level among transformants, their chitonolytic activity and the antifungal potential of crude protein extracts from them were evaluated.
Materials and Methods
Plant and Fungal Material
The Nicotiana tabacum L. (cv. Petit Havana SR1) plants used for genetic transformation were cultured on MS medium (Duchefa, Netherlands) supplemented with 2% (w/v) sucrose and 0.8% agar at 20 ± 2 °C, with a 16 h photoperiod and light intensity of 50 μmol/m2/s.
The plant pathogen Trichoderma viride CCM F486 (obtained from Czech Collection of Microorganisms, Brno; http://www.sci.muni.cz/ccm/) was used for in vitro antifungal activity experiments. The fungal culture was maintained on potato dextrose agar (Sigma-Aldrich) and incubated in the dark at an ambient temperature of 27 °C. Colonies were subcultured to Sabouraud agar medium (40 g/l glucose, 10 g/l peptone, 20 g/l agar, pH 5.6) before the hyphal extension assay.
Expression of rDrchit in Escherichia coli, its purification and detection of enzyme and antifungal activity
For the expression of DrChit gene in bacteria, the vector pET32a-Trx [modified version of pET32a (Millipore) with removed sequence for the 109aa Trx•Tag™ thioredoxin protein] was used. Vector pET32a-Trx (5447 bp) was prepared by ligation of 340 bp SphI–NcoI, 1134 bp NcoI–ScaI and 4003 bp SphI–ScaI fragments; while SphI–NcoI fragment was amplified with FISphI(FOR)–FINcoI(REV) and NcoI–ScaI fragment with FIINcoI(FOR)–FIIScaI(REV) primers on pET32a vector template. SphI–ScaI fragment was isolated from pET32a vector following the digestion with corresponding restriction endonucleases.
Open reading frame of DrChit gene lacking the sequence of signal peptide was isolated NcoI and EcoRI fragment from pGEM-T Easy vector [34] and cloned into the pET32a-Trx vector digested with the same restriction enzymes and used for transformation of E. coli DH5α. After sequencing of the T7/DrChit expression region of pET32a-TrxDrChit, this plasmid was introduced into E. coli BL21-CodonPlus (DE3) RIL expression strain (Agilent). The expression of rDrChit protein was induced by adding 1 mM IPTG to the bacterial culture at OD600 of 0.6 and followed by incubation for 3 h at 37 °C. Upon induction of DrChit protein expression by 1 mM IPTG and subsequent purification on Ni–NTA agarose as it was described by Jopcik et al. [34]. Detection of crude protein extracts and purified DrChit protein were analysed on 12% (w/v) SDS-PAGE (Fig. 1). The same protein samples, but without the heat treatment were separated on 12% SDS-PAGE containing 0.01% glycol chitin. After electrophoresis and re-naturation of separated proteins in the solution containing 50 mmol/l sodium acetate (pH 5.2), and 1% Triton, the bands with chitinase activity were detected as dark zones after staining the gel with 0.01% (w/v) Fluoresecent Brightner 28 for 15 min and UV illumination [34]. Subsequently 50 µg of purified rDrChit protein was tested to suppress the growth of Trichoderma viride compared to 50 μg BSA and clear buffer used using hyphal extension assay described below. The experiment was performed as a technical triplicate and fungal growth values were expressed as the average area in cm2.

a SDS-PAGE analysis of recombinant DrChit protein expressed in E. coli and isolated using His-Tag-based purification. b Detection of endochitinase activity in the gel containing glycol chitin as a substrate. After re-naturation and staining the gel with Fluorescent Brightener 28, the bands with chitinase activity appeared as dark zones after UV illumination. 1, total cell proteins from uninduced E. coli BL21-CodonPlus (DE3) RIL/pET32a-TrxDrChit; 2, total cell proteins from induced E. coli BL21-CodonPlus (DE3) RIL/pET32a-TrxDrChit; 3, Ni–NTA agarose-purified rDrChit protein; c Antifungal activity assays. Fifty μg of purified DrChit protein obviously suppressed Trichoderma viride growth compared to water and 0.1 M sodium acetate buffer (pH 5.1) (BLANK)
Construction of the Plant Expression Vector and Agrobacterium Transformation
The 1665-bp-long sundew (Drosera rotundifolia L.) gene encoding the extracellular chitinase (Drchit), which we previously isolated and characterized [34], was amplified from genomic DNA with the gene-specific P1–P2 primers (Table S1 Online resource 1). The PCR reaction mixture of 50 µl contained 200 ng of DNA template, 20 pmol of each primer, 0.2 mM dNTPs, 1 × PCR buffer, 2.5 mM MgCl2, and 2 U DyNAzymeEXT polymerase (Finnzyme, Finland). The first PCR step was performed at 94 °C for 3 min and was followed by 35 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 40 s. The last step was performed at 72 °C for 10 min. The PCR product was cloned into pCR4-TOPO vector (Invitrogen, Carslad, CA) and subjected to Sanger sequencing (Microsynth, Austria), (pDD1 construct).
The DNA fragment of the 35S terminator was amplified using the P3–P4 primers (Table S1 Online resource 1). The PCR reaction mixture of 25 µl contained 50 ng of DNA template, 20 pmol of each primer, 0.2 mM, dNTPs, 1 × PCR buffer, 2.5 mM MgCl2, and 2 U DyNAzymeEXT polymerase (Finnzyme, Finland). The first PCR step was performed at 94 °C for 3 min and was followed by 35 cycles at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. The final extension was performed at 72 °C for 10 min. The PCR product containing 35S terminator was digested with XbaI–HindIII and the DrChit gene was isolated from the pDD1 construct as a NcoI–XbaI fragment. These were ligated into pSL301 cloning vectors (Invitrogen, Carslad, CA) that were digested with HindIII–NcoI restriction enzymes (pDD2 construct).
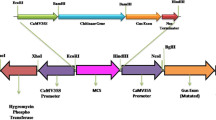
Finally, dCaMV35S promoter was isolated from pCAMBIA1304 vector [37] as a HindIII–NcoI fragment and DrChit gene fused to 35S terminator was isolated as a NcoI–PacI fragment from pDD2 construct. These were ligated into pBinPLUS [38] digested with HindIII–PacI restriction enzymes (pDD3 construct). Subsequently, the pDD3 construct (Fig. 2) was introduced into Agrobacterium tumefaciens strain LBA 4404 and its stability was verified by restriction analysis after re-transformation into E. coli.

The T-DNA region of the vector construct pDD3. The gene for D. rotundifolia chitinase (DrChit gene) was under the control of the double CaMV35S promoter (d35S-P) and terminated by the CaMV35S terminator (35S-T). The gene for neomycin phosphotransferase (nptII gene) was driven by the nopaline transferase promoter (nos-P) and nos terminator (nos-T). Grey arrows indicate the position of the primers used for PCR analyses. EcoRV denotes the restriction enzyme used for Southern blot analysis as a well as predicted fragment size
Plant Transformation Experiments
Agrobacterium tumefaciens strain LBA4404 containing the pDD3 construct was used to transform leaf discs of tobacco (Nicotiana tabacum cv. Petit Havana SR1) following the protocol described by Mlynarova et al. [39]. Regenerated shoots were selected on selective shooting medium containing 50 mg/l kanamycin and 500 mg/l cefotaxime (Duchefa, Netherlands). Six weeks after transformation, the shoots were transferred onto solid MS medium supplemented with 20 g/l sucrose, 50 mg/l kanamycin, and 500 mg/l cefotaxime and cultured until roots developed. The transformation efficiency (%) was defined as the total number of transgenic shoots that roosted on MS-20 Km50/total number of explants used × 100. Individual transgenic plants/lines were subcultured onto fresh medium every 6 weeks.
PCR Analysis
To verify the presence of transgenes in the putative DD3 transgenic plants, genomic DNAs were isolated from leaf tissue of individual transgenic lines and non-transgenic controls using the protocol of Chen et al. [40] and were subjected to PCR. Primers P5–P6 and P7–P8 were used to confirm the presence of nptII and DrChit expression units, respectively (Table S1 Online resource 1, Fig. 2). The 25 µl PCR reaction mixture contained 100–200 ng of DNA template, 10 pmol of each primer, 0.2 mM dNTPs, 1 × PCR buffer, 2.5 mM MgCl2, and 1 U FIREPol Taq DNA polymerase (Solis BioDyne, Estonia). The first PCR step was performed at 94 °C for 2 min, followed by 35 cycles at 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 40 s. The last step was performed at 72 °C for 10 min. Finally, the PCR products were separated on a 1% (w/v) agarose gel and visualized with ethidium bromide staining.
Southern Blot Analysis
Genomic DNA weighing 10 microgrammes was digested with the restriction enzyme EcoRV, separated on a 1% (w/v) agarose gel, and blotted by capillary transfer with 20 × SSC on a positively charged nylon membrane (Roche Applied Science, Germany). A DrChit-specific probe was prepared using PCR with the P9–P10 primer set (Table S1 Online resource 1) and non-radioactively labelled using the DIG Probe Synthesis Kit (Roche, Roche Applied Science, Germany). Hybridization was performed on DIG easy Hyb hybridization solution (Roche, Applied Science, Germany) at 42 °C according to the manufacturer’s instructions. Hybridization signals were visualized by DIG Nucleotic Detection Kit (Roche Applied Science, Germany).
RT-PCR
Total RNA was isolated from leaves of in vitro cultivated transgenic plants and non-transgenic controls using the RNeasy kit, (Qiagen, Germany) and was digested with DNase (Life Technologies, USA) according to manufacturer´s instructions. The integrity of RNA was checked on a 1% (w/v) agarose gel and RNA quantification was performed using a BioSpec-nano spectrophotometer (Shimadzu, Japan). First-strand cDNA was synthesized using the Maxima First strand cDNA Synthesis kit for RT-qPCR (Thermo Fischer Scientific, USA) and the DNA removal step was included. RT-PCR was performed with the primer pairs P11–P12 (sundew chitinase, KU516826) and P13–P14 (tobacco actin, XM_016618658.1) (Table S1 Online resource 1), while the programme involved one cycle at 95 °C 3 min, 35 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 40 s, and a final step at 72 °C for 7 min.
RT-qPCR
RT-qPCR was performed using the Luminaris HiGreen qPCR Master Mix (Thermo Fischer Scientific, USA) according to manufacturer´s recommendation with the primers P15–P16 (tobacco actin, XM_016618658.1) and P17–P18 (sundew chitinase, KU516826) (Table S1 Online resource 1). The reaction was initiated by a uracil-DNA glycosylase step at 50 °C for 2 min followed by one cycle at 95 °C for 10 min. The reaction proceeded for 40 cycles at 95 °C for 15 s and 60 °C for 60 s, and was completed with a melting curve analysis step to confirm the specificity of amplified products. A LigthCycler Nano (Roche Applied Science, Germany) was used for qPCR. Experiments were performed in biological triplicate (three independent isolations, reverse transcriptions, qPCR reactions for each plant) and technical duplicate, and the threshold Ct value was set according automatic calling methods and melting curve analysis of the associated software. The Pfaffl method was employed to process the relative gene expression data [41]. Briefly, values were expressed as a ratio between the target (transgenic chitinase) and a housekeeping gene (actin). The correct primer efficiency (E) was calculated using a standard curve which was obtained from qPCR reactions using 5 point tenfold diluted cDNA as a template. The target gene expression ratio was then calculated with the Pfaffl equation: ratio = (Etarget)Cttarget/(Eactin)Ctactin.
The Endochitinolytic Activity Assay
The endochitinolytic activity of crude protein extracts isolated from individual transgenic plants and non-transgenic controls was measured by fluorometric assay with N-fluorescein-labelled chitin (FITC-chitin) [42]. Briefly, leaves of individual plants were homogenized with the aid of liquid nitrogen and extracted using 0.1 M sodium acetate buffer (pH 5.2). Following a 10 min centrifugation at 4 °C, the proteins present in the cleared protein lysates were quantified according to Bradford [43] against a BSA calibration curve. The enzymatic reaction mixtures were 200 μl and consisted of 10 mg of FITC-chitin dissolved in the 0.1 M sodium acetate buffer (pH 5.2) and 10 μg of the individual protein extracts. The reaction mixtures were incubated at 37 °C and continuously shaken for 90 min. After centrifugation for 1 min, 100 μl of supernatant (without the sedimented pellet) was transferred into a new microcentrifuge tube with 400 μl 0.5 M Tris–HCl (pH 8.9) mixed and centrifuged for 15 min at 15,000 rpm. Next, three 10 μl aliquots of the upper phase—as technical replicates—were tenfold diluted in 0.5 M Tris–HCl (pH 8.9) and transferred into 96-well black-sided assay plates and measured on a Synergy™ H1 microplate reader (BioTek, Winooski, VT, USA) using 490/520 nm excitation and emission filters, gain sensitivity of 120, and probe distance of 4.75 mm. As in the RT-qPCR, three biological replicates of each sample were analysed. Chitinolytic activity values were expressed as an average of the relative fluorescent units normalized to the blank (reaction without enzyme).
Hyphal Extension Assay
Using a hole drill, a 9.8-mm-diameter hole was pierced in the centre of Petri dishes that were filled with 20 ml Sabouraud agar medium. The bottoms of the holes were sealed with melted medium to create central cistern. A 2.8-mm disc of 4 day-old Trichoderma viride mycelium (previously grown on the same medium) and 0.1 M sodium acetate buffer (pH 5.1) containing 100 μg of analysed protein extract were added to the cisterns. Clear buffer or 100 μg BSA, respectively, were added as controls. The Petri dishes were incubated for 36 h at 27 °C. Fungal growth was documented, and subsequently, photographs were analysed with ImageJ [44] by manually marking the mycelial area. The experiment was performed as a technical triplicate, and fungal growth values were expressed as the average area in cm2.
Statistical Analysis
For the statistical comparison of RT-qPCR, chitinolytic activity and fungal growth area among the individually analysed plants, the online Kruskal–Wallis rank sum test was used (http://astatsa.com/KruskalWallisTest/). For post hoc pairwise comparison of individual’s pairs, the Connover method was used with p values adjustment by family-wide error rate (FWER) method of Holm and the superior false discovery rate (FDR) method of Benjamini-Hochberg.
Results
Expression of rDrChit in E. coli Protein Against Trichoderma viride Detected in In Vitro Test
To detect the antifungal activity of the DrChit protein, the open reading frame of DrChit gene without the putative signal peptid was PCR-amplified, cloned into a pET32a-Trx vector and introduced into E. coli BL21-CodonPlus (DD3) RIL strain. SDS-PAGE analysis of protein extract from IPTG-induced bacterial culture revealed the presence of a predominant band of approximately ~ 32 kDa corresponding to recombinant DrChit protein (Fig. 1a, lane 2). Following the purification on Ni–NTA agarose and the purified fraction was detected at the position corresponding to the overexpressed protein (Fig. 1a, lane 3). Moreover, the samples of total E. coli protein extracts containing rDrChit protein as well as purified rDrChit protein alone showed chitinolytic activity in PAGE containing glycol chitin as a substrate (Fig. 1b lanes 2, 3).
In hyphal growth inhibition assay, 50 μg of purified rDrChit protein effectively inhibited the growth T. viride compared to control used (Fig. 1c). These results showed that purified sundew chitinase protein of ~ 32 kDa with detected chitinolytic activity exhibits obvious antifungal potential.
Generation of Transgenic Tobacco Plants
The vector construct pDD3 was prepared by inserting of the CaMV 35S promoter with a double enhancer fused to the 1.6 kb genomic clone of sundew chitinase and the CaMV35S terminator into the binary vector pBinPLUS [38]. To facilitate selection of plant transformants the T-DNA contains kanamycin resistance gene (nptII) (Fig. 2).
Tobacco leaf explants were successfully transformed using A. tumefaciens strain LBA 4404 harbouring the pDD3 construct, producing kanamycin resistant shoots 4–6 weeks after cocultivation. All putative transgenic shoots developed roots in the presence of 50 mg/l kanamycin, while the morphological appearance did not differ from non-transformed control plants. In total, we generated 55 transgenic plants in two sets of transformation experiments, separated by 18 months’ time. Transformation efficiency was similar and ranged from 59 to 61%. In total, 14 plants, 1–7 and 8–14 from first and second transformation experiments, respectively, were subjected to detailed molecular, biochemical and antifungal analyses.
Molecular Characterization of Transgenic Plants
The transgenic character of all regenerated plants was verified on genomic DNA template by PCR. The expected PCR products of 500 bp and 630 bp length were identified with the P5–P6 and P7–P8 primers and corresponded to the nptII and DrChit–35S-T sequences, respectively (Figs. 2, 3).

PCR verification of transgenes in DD3 plants. PCR reaction was carried out on genomic DNA with P5–P6 and P7–P8 primers and yielded the 500 bp nptII and the 630 bp DrChit–35S-T amplicons, respectively. Lane M—100 bp GeneRuler (Thermo Fisher Scientific, USA); NT non-transformed plant; lanes 1–14 represent the PCR products of individual transgenic plants
Following transgene verification, the transgenic plants (1–14) and non-transgenic controls were analysed for copy number determination. EcoRV digestion of plant DNA and probing with the Drchit probe resulted in the hybridization of the RB junction fragments longer than 2.4 kb (Figs. 2, 4). Except for transgenic plant 8, all analysed plants contained one or two copies of the DrChit gene (Fig. 3). However, in case of the transgenic plant 7, one incomplete T-DNA copy presented problems with DrChit transgene expression.

Southern blot analysis with EcoRV-digested DNA from transgenic DD3 tobacco plants. The blot was hybridized with a 1.2 kb DIG-labelled fragment containing the DrChit gene as a probe. All bands (> 2.4 kb correspond to right border fragments and indicate number of independent transgene copies. Lanes labelled 1–7 and 8–14 represented transgenic plants from the first (A) and second (B) transformation experiments, respectively. Lane NT was the non-transgenic control
The expression of the DrChit transgene was investigated using RT-PCR. The P11–P12 primers enabled amplification of a 725 bp stretch of cDNA that ensured detection of nearly the full chitinase transcript, while the same primers amplified 1413 bp fragment on the genomic DNA template. The cDNA quality of individual samples was assessed using the P13–P14 primers that yielded 510 bp fragment of the actin reference gene. Similarly, DrChit RT-PCR products of the expected size were amplified from all of the analysed lines, except for line 7 (Fig. 5). The lack of a RT-PCR product from transgenic line 7 was a consequence of this line lacking a complete copy of the DrChit gene. Sequencing revealed that line 7 contained a deletion at the 5´end of DrChit gene, including the sequence annealing location of the P11 primer (data not shown).

RT-PCR product analysis from tobacco lines (1–14) carrying DrChit transgene. Expression of the DrChit transgene was assessed with the P11–P12 primers yielding 725 bp fragments from cDNA and 1413 bp from genomic DNA (lane 14*). Tobacco actin was used as an endogenous control, and it exhibited the expected size of 510 bp with the P13–P14 primers. Lane M was the 100 bp Gene Ruler (Thermo Fisher Scientific, USA) and lane NT was the non-transgenic control
Quantification of Transgene Gene Expression Level
The expression level of the DrChit transgene driven by the double CaMV35S promoter was conducted using RT-qPCR analysis on mRNA isolated from leaves of fourteen transgenic lines (coming from both sets of transformants). DrChit expression varied substantially among individual transgenic lines. A large group of transgenic lines (1, 3, 4, 5, 6, 7, 10, 13) had DrChit transcript levels comparable or lower than the levels of the actin transcript (Fig. 6). A relatively low expression of the DrChit gene, but higher than actin expression, was observed in transgenic lines 2, 8, 11, and 12. In contrast, transgenic tobacco lines 9 and 14 had high DrChit transcript abundance, equalling 72 and 152 times higher than their actin transcript abundance, respectively. Lines 9 and 14 were suitable candidates for analysing the effect of DrChit gene expression in tobacco with respect to its antifungal potential.

Relative expression levels of the DrChit gene in transgenic tobacco lines (1–14). The relative expression values were calculated with the Pfaffl method as the ratio between transcript abundance of the target DrChit transgene and the endogenous actin gene control. Normalization was performed-based on amplification efficiency results from a 5 point 10 fold dilution standard curve (actin: E-1.9300, R2 = 0.9993; chitinase: E-1.8870, R2 = 0.9984). Error bars are standard deviation calculated from three independent biological samples, each with technical duplicates (Table S2 Online resource 1). Similarity matrix between the individual plants is available in Table S5 Online resource 1
Chitinolytic Activity of Crude Protein Extracts
Chitinolytic activity assays were used to investigate the contribution of the DrChit enzyme to the chitinolytic activity of crude protein extracts from transgenic lines and non-transgenic controls. Crude protein extracts of each tested plant (10 μg) were incubated with FITC-chitin, and the fluorescence of soluble FITC-labelled chitooligosaccharides was detected at 90 min. Transgenic lines (1, 3, 4, 5, 6, 7, 10, 13) with extremely low DrChit expression exhibited chitinolytic activity comparable with non-transgenic controls (CP1–CP3) or only slightly higher than the controls (line 11) (Fig. 7). Lines 2, 8 and 13 had chitinolytic activity of 2.5–2.9 times higher than that of the control. Transgenic line 9 and line 14 had chitinolytic activities nearly five and eight times greater, respectively, than that of the non-transgenic controls. Statistical analysis with Kruskal–Wallis test confirmed differences of lines 2, 8, 9, 12 and 14 from each of non-controls at significance of p < 0.01 (Table S3, Table S6 Online resource 1).

The chitinolytic activity assays of crude protein extracts from transgenic tobacco lines (1–14) expressing the DrChit gene, and of non-transgenic controls (CP1–CP3) against FITC chitin. The measurements were performed with three biological and the small bars represent standard deviation (Table S3 Online resource 1). The tobacco lines 2, 8, 9, 12 and 14 significantly differed from each of non-transgenic controls at p < 0.01. Similarity matrix between the individual plants is available in Table S6 Online resource 1
In Vitro Antifungal Activity Assay
Antifungal activity assays were used to determine whether the overexpressed DrChit gene in transgenic tobacco plants had the ability to increase their antifungal potential. Crude protein extracts (100 μg) from transgenic tobacco plants were dissolved in sodium acetate buffer and tested against Trichoderma viride. Protein extracts from non-transgenic plants (CP1–CP3) in sodium acetate buffer, and 100 μg of BSA in sodium acetate buffer were used as controls. Trichoderma viride had the greatest growth in the presence of BSA and protein extracts from transgenic and non-transgenic control plants retarded fungal growth compared to BSA. Mycelium size varied when we evaluated the influence of protein extracts from individual transgenic plants and non-transgenic controls on the growth of T. viride. Chitinolytic activity was not the only factor influencing antifungal potential of crude protein extracts.
Protein extracts from transgenic plants (2, 8, 9, 12, 14) with the greatest DrChit gene expression had the greatest inhibitory potential among the transgenic plants and significantly differed (p < 0.01) from non-transgenic controls. The greatest ability to suppress T. viride growth was with the protein extract from line 14 (Figs. 8, 9) and mycelium growth with this extract only reached 45.2% of the area of non-transgenic control. Transgenic lines 9, 8, and 2 followed, with 56.4%, 63.8%, and 65.5% of the area of the control, respectively. All of these lines (2, 8, 9, and 14) exhibited obvious DrChit gene expression and increased chitinolytic activity. Extracts of remaining transgenic lines did not exhibit statistically different growth inhibition relative to the non-transgenic controls (p < 0.01).

The effects of 100 μg of crude protein extracts isolated from individual transgenic tobacco lines (1–14) and non-transformed controls (CP1–CP3) on the growth Trichoderma viride grown on Sabouraud agar in Petri dishes. As controls, sodium acetate buffer (blank) and 100 μg BSA (blank BSA) were tested. Standard deviations indicated by bars were calculated from technical triplicates (Table S4 Online resource 1). Similarity matrix between the individual plants is available in Table S7 Online resource 1

Antifungal activity assays. The growth Trichoderma viride in the presence of 100 μg of crude protein extract from transgenic line 14, which had the highest chitinolytic activity (14 a–c), 100 μg crude protein extract from non-transgenic control plant 3 (CP3 a–c), 0.1 M sodium acetate buffer (pH 5.1) used for protein extraction (BLANK a–c), and 100 μg BSA dissolved in acetate buffer (BSA a–c). The fungal growth values were expressed as the average area size in cm2 after 36 h-incubation at 27 °C
Discussion
Increasing plant tolerance to fungal pathogens through the expression of heterologous genes, whose products have antifungal activity, has been reported previously [21, 45]. However, there is a constant need to achieve broad-spectrum resistance to several phytopathogens, necessitating continued testing of new genes from different genetic sources.
Here, we tested a chitinase isolated from the carnivorous plant species Drosera rotundifolia. Our previous work found that a class I chitinase from D. rotundifolia with substrate specificity for long chitin polymers actively participates in cell wall degradation of captured insect prey [34]. In addition to a catalytic domain, class I chitinases contain a chitin-binding domain that enhances their antifungal potential. When gene for an antimicrobial peptide (an alfalfa defensin) was fused to the rice chitinase CBD using a helix-forming linker and introduced in tobacco, transgenic plants exhibited enhanced resistance to Fusarium solani [46]. Our experiments showed that DrChit protein overexpressed in E. coli expression system and purified on Ni–NTA agarose has obvious potential to suppress the growth of T. viride tested in hyphal extension assay. Following the generation of intact transgenic tobacco plants, fourteen randomly selected individuals were subjected to detailed molecular and biochemical analyses. Southern blot analysis indicated that most of these plants contained one or two copies of the sundew chitinase transgene. Except of one transgenic line, all tested plants yielded RT-PCR products with the P11–P12 primers. Although T-DNA insertion into the plant genome is often accompanied by small or large rearrangements at junction sites [47], deletions in transgenes or regulatory sequences were also reported [48, 49]. Except for one line, all contained at least one complete T-DNA copy since RT-PCR confirmed the expression of a functional DrChit gene.
The expression level of DrChit was analysed using a RT-qPCR approach. Only six out of 13 RT-PCR positive plants exhibited DrChit expression higher than the reference gene (actin). The considerable variability of transgene expression that is often observed within a population of transgenic plants transformed with the same transgene construct [29, 50, 51] is attributable to the position effect, transgene copy number, and various epigenetic silencing phenomena [52]. In addition, CaMV 35S promoter was reported to frequently yield a bimodal expression pattern in populations of transformants that is characterized by a limited number of plants with high transgene expression, but the majority of transformants with very low expression levels [50, 53].
Nevertheless, the transgenic lines 9 and 14 exhibited 72 and 152 times higher DrChit transgene expression than that of the internal control. Both high-expression transgenic lines had high chitinolytic activity, as measured 90 min after incubating their crude protein extracts with FITC-chitin. The suitability of FITC-chitin as a substrate for the DrChit enzyme was tested in a previous study [34] and the purified protein exhibited long oligomer-specific endochitinase activity on glycol chitin and FITC-chitin, but not short oligomer-specific endochitinase [substrate 4MU-(GLCNAC)3] or chitobiosidase [substrate 4MU-(GLCNAC)2] activities.
Enzymatic activity difference among plants with significant DrChit expression and non-transgenic control plants was observed only after protein extracts and substrate were incubated for 90 min. It is unclear why increase in fluorescence was not detectable after 30 or 60 min. In this fluorometric assay, the fluorescence signal is a function of soluble FITC-chitooligomers resulting from tobacco as well as sundew chitinolytic activities. Since we do not know the length of FITC-chitooligomers resulting from DrChit activity, we suppose that it was a mixture of long soluble and insoluble FITC-chitooligomers. However, increased fluorescence after 90 min of incubation may result from subsequent conversion of insoluble long FITC-chitooligomers (product of sundew chitinase) into soluble FITC-chitooligomers by tobacco endochitinases. A second explanation takes into account the different kinetic properties of sundew chitinase and tobacco chitinases. The fluorimetric method using FITC-chitin as a substrate for determining endochitinase activity [34] in plant extracts is a very rapid and more sensitive method relative to the turbidimetric and viscosimetric methods [54, 55].
Finally, crude protein extracts from transgenic and non-transgenic plants were tested for their ability to inhibit the growth of Trichoderma viride hyphae in in vitro assays. To evaluate correctly the contribution of DrChit endochitinase in the protein extracts, several controls were also included. As expected, T. viride exhibited the highest growth rate in the presence of BSA. The buffer (nutrient-free control) had a comparable influence on fungal growth than crude protein extracts from non-transgenic plants, suggesting that crude protein extracts from non-transgenic plants contained proteins of nutritional character and antifungal proteins, including chitinolytic enzymes in balance, resulting in no obvious effect on fungal growth. Transgenic plants had variable sundew chitinase expression resulting in variable fungal growth retardation. Unlike the clear association between DrChit expression and chitinolytic activity, mycelium size did not always correspond to high or low DrChit expression or chitonolytic activity. For example, one line (11) had only slightly increased DrChit transgene expression compared to the reference gene, slightly increased chitinolytic activity, but no increased antifungal activity, suggesting that it was insufficient for suppression of fungal growth. In contrast, a clear association between hyphal growth inhibition and DrChit expression, as well as chitinolytic activity, was reported for two transgenic lines (9 and 14). These lines exhibited 72 and 152 times higher DrChit transcript abundance than actin transcript level, and their protein extracts significantly inhibited fungal growth relative to the control by 44 and 55%, respectively.
This study demonstrates that an endochitinase enzyme that is natively involved in the digestive processes of the carnivorous plant, Drosera rotundifolia has the potential to inhibit growth of plant phytopathogens with chitin in their cell walls. Optimal evaluation of antifungal effect of transgene protein in plants includes both, in vitro as well as in vivo assays. The former responds the question if tested hydrolytic enzyme suitably complements modes of actions of endogenous plant hydrolytic enzymes required for maximum efficacy [56], while quantification of obtained data represents the advantage of this approach. As in vitro assay, this study also confirmed the strong correlation between the fungal growth inhibition level and endochitinase activity of crude protein extracts from transgenic tobacco plants; in vivo testing of other transgenic valuable crops against significant pathogens can follow. The determination of disease symptoms in in vivo tests is performed on ordinal scale, while (hidden) factors of the environment can complicate the final evaluation. Therefore, confirming of antifungal activity in in vitro test can be helpful, when the plant–pathogen interaction is evaluated in in vivo tests.
Conclusions
This is the first time an endochitinase natively involved in the digestive processes of the carnivorous plant, Drosera rotundifolia, was tested in transgenic plants with the aim of exploring its antifungal potential. Our results showed that the constitutive expression of sundew extracellular class I chitinase in tobacco resulted in enhanced antifungal potential of crude protein extracts against Trichoderma viride. This work and further research will probably add the sundew chitinase gene to the list of chitinases that are useful in genetic manipulation strategies.
References
Lipke, P. N., & Ovalle, R. (1998). Cell wall architecture in yeast: New structure and new challenges. Journal of Bacteriology, 180(15), 3735–3740.
Henrissat, B., & Bairoch, A. (1993). New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochemical Journal, 293, 781–788. https://doi.org/10.1042/bj2930781.
Neuhaus, J. M. (1999). Plant chitinases (PR-3, PR-4, PR-8, PR-11). In S. K. Datta & S. Muthukrishnan (Eds.), Pathogenesis-related proteins in plants (pp. 77–105). Boca Raton, FL: CRC Press.
Sarma, K., Dehury, B., Sahu, J., Sarmah, R., Sahoo, S., Sahu, M., et al. (2012). A comparative proteomic approach to analyse structure, function and evolution of rice chitinases: A step towards increasing plant fungal resistance. Journal of Molecular Modeling, 18(11), 4761–4780. https://doi.org/10.1007/s00894-012-1470-8.
Ohnuma, T., Numata, T., Osawa, T., Inanaga, H., Okazaki, Y., Shinya, S., et al. (2012). Crystal structure and chitin oligosaccharide-binding mode of a loopful’ family GH19 chitinase from rye, Secale cereale, seeds. FEBS Journal, 279(19), 3639–3651. https://doi.org/10.1111/j.1742-4658.2012.08723.x.
van Hengel, A. J., Tadesse, Z., Immerzeel, P., Schols, H., van Kammen, A., & de Vries, S. C. (2001). N-acetylglucosamine and glucosamine-containing arabinogalactan proteins control somatic embryogenesis. Plant Physiology, 125(4), 1880–1890. https://doi.org/10.1104/pp.125.4.1880.
Fraterova, L., Salaj, T., Matusikova, I., & Salaj, J. (2013). The role of chitinases and glucanases in somatic embryogenesis of black pine and hybrid firs. Central European Journal of Biology, 8(12), 1172–1182. https://doi.org/10.2478/s11535-013-0234-5.
Kasprzewska, A. (2003). Plant chitinases—Regulation and function. Cellular & Molecular Biology Letters, 8(3), 809–824.
van Loon, L. C., Rep, M., & Pieterse, C. M. J. (2006). Significance of inducible defense-related proteins in infected plants. Annual Review of Phytopathology, 44, 135–162. https://doi.org/10.1146/annurev.phyto.44.070505.143425.
Dana, M. D., Pintor-Toro, J. A., & Cubero, B. (2006). Transgenic tobacco plants overexpressing chitinases of fungal origin show enhanced resistance to biotic and abiotic stress agents. Plant Physiology, 142(2), 722–730. https://doi.org/10.1104/pp.106.086140.
Meszaros, P., Rybansky, L., Hauptvogel, P., Kuna, R., Libantova, J., Moravcikova, J., et al. (2013). Cultivar-specific kinetics of chitinase induction in soybean roots during exposure to arsenic. Molecular Biology Reports, 40(3), 2127–2138. https://doi.org/10.1007/s11033-012-2271-y.
Guleria, P., Kumar, V., & Guleria, S. (2017). Genetic engineering: A possible strategy for protein-energy malnutrition regulation. Molecular Biotechnology, 59(11–12), 499–517. https://doi.org/10.1007/s12033-017-0033-8.
Acharya, K., Chakraborty, N., Dutta, A. K., Sarkar, S., & Acharya, R. (2011). Signaling role of nitric oxide in the induction of plant defense by exogenous application of abiotic inducers. Archives of Phytopathology and Plant Protection, 44(15), 1501–1511. https://doi.org/10.1080/03235408.2010.507943.
Cletus, J., Balasubramanian, V., Vashisht, D., & Sakthivel, N. (2013). Transgenic expression of plant chitinases to enhance disease resistance. Biotechnology Letters, 35(11), 1719–1732. https://doi.org/10.1007/s10529-013-1269-4.
Graham, L. S., & Sticklen, M. B. (1994). Plant chitinases. Canadian Journal of Botany-Revue Canadienne De Botanique, 72(8), 1057–1083. https://doi.org/10.1139/b94-132.
Fink, W., Liefland, M., & Mendgen, K. (1988). Chitinases and beta-1,3-glucanases in the apoplastic compartment of oat leaves (Avena sativa L). Plant Physiology, 88(2), 270–275. https://doi.org/10.1104/pp.88.2.270.
Sela-Buurlage, M. B., Ponstein, A. S., Bresvloemans, S. A., Melchers, L. S., Vandenelzen, P. J. M., & Cornelissen, B. J. C. (1993). Only specific tobacco (Nicotiana tabacum) chitinases and beta-1,3-glucanases exhibit antifungal activity. Plant Physiology, 101(3), 857–863. https://doi.org/10.1104/pp.101.3.857.
Broglie, K., Chet, I., Holliday, M., Cressman, R., Biddle, P., Knowlton, S., et al. (1991). Transgenic plants with enhanced resistance to the fungal pathogen Rhizoctonia solani. Science, 254(5035), 1194–1197. https://doi.org/10.1126/science.254.5035.1194.
Maximova, S. N., Marelli, J. P., Young, A., Pishak, S., Verica, J. A., & Guiltinan, M. J. (2006). Over-expression of a cacao class I chitinase gene in Theobroma cacao L. enhances resistance against the pathogen, Colletotrichum gloeosporioides. Planta, 224(4), 740–749. https://doi.org/10.1007/s00425-005-0188-6.
Zeng, X. F., Li, L., Li, J. R., & Zhao, D. G. (2016). Constitutive expression of McCHIT1-PAT enhances resistance to rice blast and herbicide, but does not affect grain yield in transgenic glutinous rice. Biotechnology and Applied Biochemistry, 63(1), 77–85. https://doi.org/10.1002/bab.1342.
Lorito, M., Woo, S. L., Fernandez, I. G., Colucci, G., Harman, G. E., Pintor-Toro, J. A., et al. (1998). Genes from mycoparasitic fungi as a source for improving plant resistance to fungal pathogens. Proceedings of the National academy of Sciences of the United States of America, 95(14), 7860–7865. https://doi.org/10.1073/pnas.95.14.7860.
Ziaei, M., Motallebi, M., Zamani, M. R., & Panjeh, N. Z. (2016). Co-expression of chimeric chitinase and a polygalacturonase-inhibiting protein in transgenic canola (Brassica napus) confers enhanced resistance to Sclerotinia sclerotiorum. Biotechnology Letters, 38(6), 1021–1032. https://doi.org/10.1007/s10529-016-2058-7.
Jabeen, N., Chaudhary, Z., Guffraz, M., Rashid, H., & Mirza, B. (2015). Expression of rice chitinase gene in genetically engineered tomato confers enhanced resistance to Fusarium Wilt and Early Blight. Plant Pathology Journal, 31(3), 252–258. https://doi.org/10.5423/PPJ.OA.03.2015.0026.
Moravcikova, J., Libantova, J., Heldak, J., Salaj, J., Bauer, M., Matusikova, I., et al. (2007). Stress-induced expression of cucumber chitinase and Nicotiana plumbaginifolia beta-1,3-glucanase genes in transgenic potato plants. Acta Physiologiae Plantarum, 29(2), 133–141. https://doi.org/10.1007/s11738-006-0017-y.
Emani, C., Garcia, J. M., Lopata-Finch, E., Pozo, M. J., Uribe, P., Kim, D. J., et al. (2003). Enhanced fungal resistance in transgenic cotton expressing an endochitinase gene from Trichoderma virens. Plant Biotechnology Journal, 1(5), 321–336. https://doi.org/10.1046/j.1467-7652.2003.00029.x.
Cheng, W., Li, H.-P., Zhang, J.-B., Du, H.-J., Wei, Q.-Y., Huang, T., et al. (2015). Tissue-specific and pathogen-inducible expression of a fusion protein containing a Fusarium-specific antibody and a fungal chitinase protects wheat against Fusarium pathogens and mycotoxins. Plant Biotechnology Journal, 13(5), 664–674. https://doi.org/10.1111/pbi.12289.
Karmakar, S., Molla, K. A., Chanda, P. K., Sarkar, S. N., Datta, S. K., & Datta, K. (2016). Green tissue-specific co-expression of chitinase and oxalate oxidase 4 genes in rice for enhanced resistance against sheath blight. Planta, 243(1), 115–130. https://doi.org/10.1007/s00425-015-2398-x.
Kamble, S., Mukherjee, P. K., & Eapen, S. (2016). Expression of an endochitinase gene from Trichoderma virens confers enhanced tolerance to Alternaria blight in transgenic Brassica juncea (L.) czern and coss lines. Physiology and Molecular Biology of Plants, 22(1), 69–76. https://doi.org/10.1007/s12298-016-0340-8.
Iqbal, M. M., Nazir, F., Ali, S., Asif, M. A., Zafar, Y., Iqbal, J., et al. (2012). Over expression of rice chitinase gene in transgenic peanut (Arachis hypogaea L.) improves resistance against leaf spot. Molecular Biotechnology, 50(2), 129–136. https://doi.org/10.1007/s12033-011-9426-2.
Ceasar, S. A., & Ignacimuthu, S. (2012). Genetic engineering of crop plants for fungal resistance: Role of antifungal genes. Biotechnology Letters, 34(6), 995–1002. https://doi.org/10.1007/s10529-012-0871-1.
Punja, Z. K. (2001). Genetic engineering of plants to enhance resistance to fungal pathogens—A review of progress and future prospects. Canadian Journal of Plant Pathology-Revue Canadienne De Phytopathologie, 23(3), 216–235.
Matusikova, I., Salaj, J., Moravcikova, J., Mlynarova, L., Nap, J. P., & Libantova, J. (2005). Tentacles of in vitro-grown round-leaf sundew (Drosera rotundifolia L.) show induction of chitinase activity upon mimicking the presence of prey. Planta, 222(6), 1020–1027. https://doi.org/10.1007/s00425-005-0047-5.
Ishisaki, K., Arai, S., Hamada, T., & Honda, Y. (2012). Biochemical characterization of a recombinant plant class III chitinase from the pitcher of the carnivorous plant Nepenthes alata. Carbohydrate Research, 361, 170–174. https://doi.org/10.1016/j.carres.2012.09.001.
Jopcik, M., Moravcikova, J., Matusikova, I., Bauer, M., Rajninec, M., & Libantova, J. (2017). Structural and functional characterisation of a class I endochitinase of the carnivorous sundew (Drosera rotundifolia L.). Planta, 245(2), 313–327. https://doi.org/10.1007/s00425-016-2608-1.
Eilenberg, H., Pnini-Cohen, S., Schuster, S., Movtchan, A., & Zilberstein, A. (2006). Isolation and characterization of chitinase genes from pitchers of the carnivorous plant Nepenthes khasiana. Journal of Experimental Botany, 57(11), 2775–2784. https://doi.org/10.1093/jxb/erl048.
Hatano, N., & Hamada, T. (2012). Proteomic analysis of secreted protein induced by a component of prey in pitcher fluid of the carnivorous plant Nepenthes alata. Journal of Proteomics, 75(15), 4844–4852. https://doi.org/10.1016/j.jprot.2012.05.048.
Roberts, C. S., Rajagopal, S., Smith, L. A., Nguyen, T. A., Yang, W., Nugroho, S., et al. (2002). A comprehensive set of modular vectors for advanced manipulations and efficient transformation of plants by both Agrobacterium and direct DNA uptake methods. CAMBIA, Canberra, Australia. http://www.cambia.org.
van Engelen, F. A., Molthoff, J. W., Conner, A. J., Nap, J. P., Pereira, A., & Stiekema, W. J. (1995). pBINPLUS—An improved plant transformation vector based on pBIN19. Transgenic Research, 4(4), 288–290. https://doi.org/10.1007/BF01969123.
Mlynarova, L., Loonen, A., Heldens, J., Jansen, R. C., Keizer, P., Stiekema, W. J., et al. (1994). Reduced position effect in mature plants conferred by the chicken lysozyme matrix-associated region. Plant Cell, 6(3), 417–426.
Chen, J., Greenblatt, I. M., & Dellaporta, S. L. (1992). Molecular analysis of Ac transposition and DNA replication. Genetics, 130(3), 665–676.
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research, 29(9), e45. https://doi.org/10.1093/nar/29.9.e45.
Tikhonov, V. E., Lopez-Llorca, L. V., Salinas, J. S., & Monfort, E. (2004). Endochitinase activity determination using N-fluorescein-labeled chitin. Journal of Biochemical and Biophysical Methods, 60(1), 29–38. https://doi.org/10.1016/j.jbbm.2004.04.013.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72(1–2), 248–254. https://doi.org/10.1006/abio.1976.9999.
Schneider, C. A., Rasband, W. S., & Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods, 9(7), 671–675. https://doi.org/10.1038/nmeth.2089.
Vellicce, G. R., Ricci, J. C. D., Hernandez, L., & Castagnaro, A. P. (2006). Enhanced resistance to Botrytis cinerea mediated by the transgenic expression of the chitinase gene ch5B in strawberry. Transgenic Research, 15(1), 57–68. https://doi.org/10.1007/s11248-005-2543-6.
Badrhadad, A., Nazarian-Firouzabadi, F., & Ismaili, A. (2018). Fusion of a chitin-binding domain to an antibacterial peptide to enhance resistance to Fusarium solani in tobacco (Nicotiana tabacum). 3 Biotech, 8(9), 391. https://doi.org/10.1007/s13205-018-1416-7.
Forsbach, A., Schubert, D., Lechtenberg, B., Gils, M., & Schmidt, R. (2003). A comprehensive characterization of single-copy T-DNA insertions in the Arabidopsis thaliana genome. Plant Molecular Biology, 52(1), 161–176. https://doi.org/10.1023/A:1023929630687.
Wu, Y. H., Zhang, L., Wu, G., Nie, S. J., & Lu, C. M. (2014). Characterization of genomic integration and transgene organization in six transgenic rapeseed events. Journal of Integrative Agriculture, 13(9), 1865–1876. https://doi.org/10.1016/S2095-3119(13)60628-0.
Boszoradova, E., Libantova, J., Matusikova, I., & Moravcikova, J. (2014). Application of Arabidopsis tissue-specific CRUC promoter in the Cre/loxP self-excision strategy for generation of marker-free oilseed rape: potential advantages and drawbacks. Acta Physiologiae Plantarum, 36(6), 1399–1409. https://doi.org/10.1007/s11738-014-1518-8.
Elmayan, T., & Vaucheret, H. (1996). Expression of single copies of a strongly expressed 35S transgene can be silenced post-transcriptionally. Plant Journal, 9(6), 787–797. https://doi.org/10.1046/j.1365-313X.1996.9060787.x.
Ribeiro, T. P., Arraes, F. B. M., Lourenco-Tessutti, I. T., Silva, M. S., Lisei-de-Sa, M. E., Lucena, W. A., et al. (2017). Transgenic cotton expressing Cry10Aa toxin confers high resistance to the cotton boll weevil. Plant Biotechnology Journal, 15(8), 997–1009. https://doi.org/10.1111/pbi.12694.
Butaye, K. M. J., Cammue, B. P. A., Delaure, S. L., & De Bolle, M. F. C. (2005). Approaches to minimize variation of transgene expression in plants. Molecular Breeding, 16(1), 79–91. https://doi.org/10.1007/s11032-005-4929-9.
Butaye, K. M. J., Goderis, I., Wouters, P. F. J., Pues, J., Delaure, S. L., Broekaert, W. F., et al. (2004). Stable high-level transgene expression in Arabidopsis thaliana using gene silencing mutants and matrix attachment regions. Plant Journal, 39(3), 440–449. https://doi.org/10.1111/j.1365-313X.2004.02144.x.
Ohtakara, A. (1988). Viscosimetric assay for chitinase. Methods in Enzymology, 161, 426–430.
Tronsmo, A., & Harman, G. E. (1993). Detection and quantification of N-acetyl-beta-D-glucosamidase, chitobiosidase, and endochitinase in solutions and on gels. Analytical Biochemistry, 208(1), 74–79. https://doi.org/10.1006/abio.1993.1010.
Haran, S., Schickler, H., & Chet, I. (1996). Molecular mechanisms of lytic enzymes involved in the biocontrol activity of Trichoderma harzianum. Microbiology-UK, 142, 2321–2331. https://doi.org/10.1099/00221287-142-9-2321.
Acknowledgements
The authors would like to thank Anna Fabelova for in vitro plant care. This work was co-funded by a grant from the Slovak Grant Agency VEGA 2/0075/17 and Research Centre AgroBioTech built in the framework of European Community project Building Research Centre “AgroBioTech” ITMS 26220220180.
Author information
Authors and Affiliations
Contributions
JL and MJ and designed research. DD, MJ, MR and JM conducted the experiments. MJ analysed data. JL wrote the manuscript.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Durechova, D., Jopcik, M., Rajninec, M. et al. Expression of Drosera rotundifolia Chitinase in Transgenic Tobacco Plants Enhanced Their Antifungal Potential. Mol Biotechnol 61, 916–928 (2019). https://doi.org/10.1007/s12033-019-00214-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-019-00214-1