
Abstract
The macrofungus Cordyceps militaris contains many kinds of bioactive ingredients that are regulated by functional genes, but the functions of many genes in C. militaris are still unknown. In this study, to improve the frequency of homologous integration, a genetic transformation system based on a split-marker approach was developed for the first time in C. militaris to knock out a gene encoding a terpenoid synthase (Tns). The linear and split-marker deletion cassettes were constructed and introduced into C. militaris protoplasts by PEG-mediated transformation. The transformation of split-marker fragments resulted in a higher efficiency of targeted gene disruption than the transformation of linear deletion cassettes did. The color phenotype of the Tns gene deletion mutants was different from that of wild-type C. militaris. Moreover, a PEG-mediated protoplast transformation system was established, and stable genetic transformants were obtained. This method of targeted gene deletion represents an important tool for investigating the role of C. militaris genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cordyceps militaris (L.) Fr., a well-known edible and medicinal fungus, is considered valuable as a mushroom used in traditional Chinese medicines and health supplements. C. militaris is very similar to the famous Cordyceps sinensis in medicinal value and some functional ingredients [1]. Therefore, C. militaris is widely used as a substitute for C. sinensis. Many cultivation methods of C. militaris have been developed to reduce market pressure on wild C. militaris and C. sinensis. However, the production of C. militaris still cannot meet the needs of the market due to its low content of bioactive ingredients. Genetic engineering is a promising method for increasing the content of bioactive ingredients in C. militaris. Therefore, it is necessary to develop an efficient gene knockout system for gene function studies.
Phenotypic analysis by gene deletion is one of the most powerful methods for understanding gene function. However, it is difficult to knock out a target gene, because non-homologous end-joining (NHEJ) in most filamentous fungi is the main process. Integration of foreign genes often results in low frequencies of homologous recombination (HR) [2]. The frequency of targeted gene deletion can be improved by a split-marker disruption strategy [3, 4]. However, this method has not yet been employed in C. militaris. In this study, we aimed to establish the split-marker technology by polyethylene glycol (PEG)-mediated protoplast transformation for C. militaris. The frequency of targeted gene disruption was studied by transforming split-marker fragments and linear deletion cassettes. The Tns gene was knocked out by using this method. The correct mutants were identified and the mitotic stability was investigated.
Materials and Methods
Plasmids and Strains
The vector pCAMBIA0390-Bar, containing a phosphinothricin acetyltransferase gene (bar) under the control of Aspergillus nidulans trpC promoter and trpC terminator, was constructed in our laboratory based on pCAMBIA0390 (Supplementary Fig. 1a). A laboratory and commercial strain of C. militaris, CM10, from Ningyang County, Haixin Biological Technology Co., Ltd., was used as the DNA recipient host for targeted gene disruption.
Preparation of Protoplasts
Conidia were harvested from wild-type (WT) C. militaris plates using sterile double-distilled water (ddH2O) and filtered through four-layer lens papers. Conidia were inoculated into 100 mL of potato dextrose broth (PDB) and cultured at 25 °C on a 150 rpm shaker for 48 h. The PDB culture was filtered through cotton wool to remove the mycelia. The filtrate was centrifuged at 8000×g for 10 min to obtain blastospores that were observed under a 1000 × microscope (Leica, Germany). The harvested blastospores were rinsed with osmotic stabilizer (0.8 M KCl) and then digested with lywallzyme (Guangdong Institute of Microbiology, China) solution (25 mg/mL in 0.8 M KCl). After 2.5 h with gentle shaking at 24 °C, undigested large particles were removed by filtration through four-layer lens papers. The filtrate containing the protoplasts was centrifuged at 3000×g for 10 min. The protoplast pellet was resuspended in STC buffer [18.2% sorbitol, 10 mM Tris–HCl (pH 7.5), 25 mM CaCl2] to a final concentration of 1 × 108 protoplasts per milliliter. The concentration of protoplasts was counted with a hemocytometer under a 400 × microscopy.
Generation of Deletion Cassettes
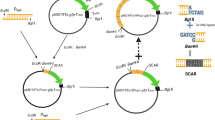
As proof of principle, the Tns gene (GenBank: NW_006271971.1) was used as a target for knockout [5]. To develop an efficient gene replacement strategy, we made deletion cassettes with different forms of the selectable marker (split or linear) using single-joint PCR (SJ-PCR) and double-joint PCR (DJ-PCR) as described previously [6]. The complete procedure for generating the deletion constructs is shown in Supplementary Fig. 1. The split-marker gene fragments flanked with the Tns gene were constructed using a SJ-PCR method that required only two rounds of PCR (Supplementary Fig. 1a). Four gene-specific primers and four selectable marker primers were required for each deletion (Supplementary Table 1). The linear deletion cassette was constructed by using the DJ-PCR approach (Supplementary Fig. 1b). PCR was performed according to the following parameters: 94 °C for 2 min; 30 cycles of 98 °C for 10 s, Tm—5 °C for 30 s, and 68 °C for 1 min for every 1 kb of template. The obtained deletion cassettes were used to transform protoplasts.
PEG-Mediated Transformation of C. militaris Protoplasts
To test the sensitivity of C. militaris to glufosinate ammonium, blastospores were plated onto peptone-added potato dextrose agar (PPDA) plates (20% potato, 0.3% peptone, 2% dextrose, and 1.5% agar, w/v) with different glufosinate ammonium concentrations (100, 150, 200, 250, 300, 350, 400, and 500 μg/mL).
At least 100 μL protoplasts (1 × 108 protoplasts per milliliter) were used for each transformation. Aliquots of 100 μL were added to five 1.5-mL centrifuge tubes and treated separately as follows: (a) the first tube received 10 μg of the split-marker fragments (5 μg of each split-marker fragment); (b) no DNA was added to the second tube as a negative control; (c) the third tube was mixed with 5 μg of the 5′ construct; (d) the fourth tube was mixed with 5 μg of the 3′ construct; (e) the fifth tube was mixed with 10 μg of the linear deletion cassette. These five tubes were incubated on ice for 5 min. Then, 50 μL of 25% PEG 4000 buffer [25% PEG 4000, 10 mM Tris–HCl (pH 7.5), 25 mM CaCl2] was added, and the solution was incubated on ice for 30 min. An additional 0.5 mL of PEG 4000 buffer was added to each tube, and the solution was incubated at 28 °C for 20 min. The mixtures were combined with 1.0 mL of STC buffer and spread onto PPDA plates containing mannitol (0.8 M) and glufosinate ammonium (300 μg/mL). Resistant transformants were grown at 25 °C in the dark for 7–14 days and then were transferred to fresh PPDA selective plates containing glufosinate ammonium (300 μg/mL). All experiments were repeated three times.
Screening of Positive Mutants
Successful deletion of the Tns gene was validated using PCR and Southern blot hybridization. Several combinations of primers were used for this purpose (Supplementary Table 1).
Transformants were screened by PCR using primers JCBar-F/JCBar-R that amplified an 890-bp fragment of the bar cassette to confirm the presence of the bar gene. Then, the JCK4-F/JCK4-R primers were used to amplify a 993-bp fragment of the Tns gene to verify that gene replacement had occurred. In addition, primer pairs F5/R5 and F6/R6 were used to confirm correct site-specific replacement by amplifying from outside the left and right flanking sequences, respectively, to the bar cassette. PCR products were sequenced at BGI Co. to verify the sequences.
Southern hybridization was used to confirm the deletion of the Tns gene and the copy number of the bar gene in the mutants. A 433-bp fragment within the bar gene amplified with primers BF/BR and a 526-bp fragment within the Tns gene amplified with primers TnsF/TnsR were probed by DIG High Prime DNA Labelling and Detection Starter Kit II (Roche Applied Sciences) as a bar probe and a Tns probe, respectively (Supplementary Fig. 2). Six mutants and one mutant that had been identified by PCR were randomly selected from the first group and the fifth group, respectively. Genomic DNA was extracted by using the HP Fungal DNA Kit (Omega, USA). 10 μg of EcoRV-digested DNA was separated by electrophoresis on a 1% (w/v) agarose gel and transferred to a Hybond-N+ nylon membrane (Amersham, USA). The hybridization and detection procedure was carried out following the manufacturer’s protocol.
Phenotypes and Mitotic Stability of Mutants
To observe the phenotypic changes, positive mutants identified by PCR and Southern blot were inoculated on fresh PPDA selective plates at 25 °C for 14 days and then exposed to light with an intensity of 500 lux at 25 °C for 7 days. The color phenotype was observed.
To determine the genetic stability, positive mutants were cultured on PPDA plates without glufosinate ammonium at 25 °C for 14 days. Then the culture was transferred onto fresh PPDA. This procedure was repeated five additional times. Then, the resistance of these mutants to glufosinate ammonium was tested by growing them on PPDA selective plates.
Data Analysis
All trials were performed in triplicate. The values were expressed as the mean ± SD by using SPSS 18.0 (SPSS Inc., Chicago, USA).
Results
Preparation of C. militaris Protoplasts
The harvested blastospores of C. militaris were bar-shaped (Fig. 1a) [5]. C. militaris protoplasts were obtained by digesting blastospores, and the yield of C. militaris protoplasts reached up to 3.12 × 107 protoplasts per milliliter. The protoplasts were identified as round cells that appeared slightly translucent under a microscope (Fig. 1b).

Preparation of C. militaris protoplasts and deletion of the Tns gene. a Blastospores. b Protoplasts (black arrow). c Generation of deletion cassettes. Lanes 1, 7: 5′ flanking region; lane 2: “PB” fragment; lane 3: 5′ split-marker fragment; lane 4: “BT” fragment; lanes 5, 9: 3′ flanking region; lane 6: 3′ split-marker fragment; lane 8: bar cassette; lane 10: linear deletion cassette. d PCR analysis of the Tns gene deletion. Four DNA templates (lanes 1, 4, 8, 11: genome of the mutant; lanes 2, 5, 9, 12: genome of wild-type C. militaris; lane 6: pCAMBIA0390-Bar; lanes 3, 7, 10, 13: ddH2O) were used as DNA templates. M: DNA marker. The 993-bp fragment of the Tns gene was amplified (lanes 1–3). The 890-bp fragment of the bar cassette was amplified (lanes 4–7). The upstream flanking sequence (2108 bp) was amplified (lanes 8–10). The downstream flanking sequence (1762 bp) was amplified (lanes 11–13). e The 5′ and 3′ split-marker fragments were co-transformed into protoplasts. f No DNA was transformed. g The 5′ split-marker fragment was transformed. h The 3′ split-marker fragment was transformed. i The linear deletion cassette was transformed
Generation of Deletion Cassettes
The SJ-PCR results indicated that the 5′ split-marker fragment was conveniently obtained by fusing the 5′ flanking sequence and the “PB” fragment; the 3′ split-marker fragment was also quickly obtained by fusing the 3′ flanking sequence and the “BT” fragment (Fig. 1c). The DJ-PCR results suggested that the linear deletion cassette was successfully constructed by fusing three fragments (5′ flanking region, bar cassette, and 3′ flanking region) in Fig. 1c.
Transformation of C. militaris Protoplasts and Disruption of the Tns Gene
The effect of the minimum inhibitory concentration of glufosinate ammonium on the growth of blastospores was determined. The growth of blastospores was completely inhibited at a glufosinate ammonium concentration of 300 μg/mL, which suggested that this concentration was appropriate for screening C. militaris putative mutants.
The Tns gene was deleted to verify the efficiency of the split-marker method. The results of the five transformation groups were significantly different. The first transformation group yielded 45.33 ± 7.50 putative mutants per 10 μg of the split-marker fragments (5 μg of each split-marker fragment) (Fig. 1e), and 73.67 ± 7.02 putative mutants were obtained in the fifth transformation group per 10 μg of the linear deletion cassette (Fig. 1i). However, no putative mutants were found in the other groups (Fig. 1f–h). The results of the first group showed that the split-marker system successfully took effect in C. militaris. Putative mutants arose only from HR events between the two separate split-marker fragments and the genomic locus of interest (Supplementary Fig. 1a). The results of the second group suggested that C. militaris protoplasts were completely inhibited in growth on PPDA selective plates containing glufosinate ammonium (300 μg/mL). The results of the third and fourth groups indicated that only a single split-marker fragment could not encode resistance to glufosinate ammonium. The results of the fifth group showed clearly that the linear deletion cassette could be integrated into the C. militaris genome. Compared with the first group, more putative mutants appeared in the fifth group.
PCR results suggested that the Tns gene had been knocked out (Fig. 1d). The results of hybridization with the bar probe indicated that there was only a single hybridized band in the PCR-positive mutants and no hybridized band in wild-type C. militaris (Fig. 2a). The results of hybridization with the Tns probe revealed that there was no hybridized band in the mutants and one hybridized band in wild-type C. militaris (Fig. 2b). Based on these results, the Tns gene was successfully knocked out and the copy number of the bar gene in the PCR-positive putative mutants was single copy. Table 1 shows the gene disruption efficiency data. Linear deletion cassettes gave a higher transformation efficiency than split-marker cassettes, but only 1.36% of the putative mutants produced with the linear cassettes were positive mutants. In contrast, the split-marker cassettes gave a lower transformation efficiency but with a higher frequency (13.24%) of targeted gene disruption than that of the linear cassette group.

Southern blot analysis of PCR-positive putative mutants. Lane M, DIG-labeled marker; P1, vector pCAMBIA0390-Bar (11,574 bp); WT, wild-type C. militaris; S1–S6, PCR-positive putative mutants from the first group; L1, PCR-positive putative mutant from the fifth group; P2, vector pMD18T-Tns (3810 bp) containing the Tns gene. a Digested DNA was probed with the bar probe. b Digested DNA was probed with the Tns probe
Phenotypes and Mitotic Stability of C. militaris Mutants
The color of the C. militaris colony is pure white in the absence of light, because the pigment is induced by light [7]. The colony color changed from pure white to orange when wild-type C. militaris was illuminated (Fig. 3a, b). However, the color of the positive mutants changed from pure white to very pale yellow under the same light conditions (Fig. 3c–f). This great change in color phenotype was due to the knockout of the Tns gene.

Color phenotypes of wild-type C. militaris and mutants. a Wild-type C. militaris was cultured on PPDA in the dark for 14 days. b Wild-type C. militaris was cultured on PPDA in the dark for 14 days and illuminated for 7 days. c Positive mutants were cultured on PPDA selective plates in the dark for 14 days. d–f Three different positive mutants were cultured on PPDA selective plates in the dark for 14 days and illuminated for 7 days
The mitotic stability of the mutants was tested, and the results indicated that all the mutants maintained the ability to grow in the presence of 300 μg/mL glufosinate ammonium after five generations on PPDA without selective pressure.
Discussion
Gene replacement is a fundamental method used for the functional characterization of fungal genes. To date, several techniques have been developed for such characterization. However, gene-targeting specificity and accuracy of the target genes in filamentous fungi are very low, which greatly hinders the study of fungal functional genes. With the increased availability of fungal whole-genome sequences, the split-marker approach is becoming more widely adopted. In this study, split-marker technology via PEG-mediated protoplast transformation was successfully applied to C. militaris for the first time. The gene-targeting specificity and accuracy with split-marker fragments was better than that with linear deletion cassettes. The mutants could maintain resistance to glufosinate ammonium. Moreover, the Tns gene was effectively knocked out in C. militaris by using the split-marker approach.
Fungal hyphae and blastospores have been widely used to prepare protoplasts [8, 9]. Blastospores were characterized with thin-walled [10]. It is well known that the amount of chitin was higher in the mycelial form of Candida albicans than in blastospores [9]. It has been reported that C. militaris could produce large numbers of blastospores [11]. Therefore, in this study, blastospores of C. militaris were used for the first time to prepare protoplasts. The results indicated that a large number of protoplasts were obtained, and the bar gene was successfully transformed into protoplasts generated from C. militaris blastospores.
Targeted gene disruption is a tedious operation in filamentous fungi due to the low frequencies of homologous recombination [12]. The split-marker strategy was successfully used in Epichloë festucae to increase the frequency of HR [13]. To efficiently knock out the target gene, the split-marker fragments and linear cassettes prepared by PCR were first used to transform C. militaris protoplasts. In our present work, we obtained more mutants by transforming the linear deletion cassette, but the frequency of targeted gene disruption was low. This result may be because only one double crossover HR event is required to produce resistant mutants. In contrast, a higher frequency of targeted gene disruption was achieved by transforming the split-marker fragments, yet the transformation efficiency was slightly lower. The reason is probably that the split-marker strategy requires three HR events to produce resistant mutants. Considering that the purpose of this study was targeted gene disruption, the split-marker strategy was a good choice for studying the function of C. militaris genes.
The target gene mutant has a significant change in color phenotype compared with that of wild-type C. militaris. This result suggested that the Tns gene was closely related to pigment synthesis in C. militaris. It is well known that C. militaris produces carotenoids as terpenoids induced by light, as previously reported [14]. We believe that the Tns gene may be responsible for the synthesis of C. militaris carotenoids.
Conclusions
The split-marker fragments and linear deletion cassettes were successfully transformed into protoplasts generated from C. militaris blastospores. The frequency of targeted gene disruption by transforming the split-marker fragments was higher than that by transforming the linear cassette. This is the first report in which the split-marker approach was successfully applied in C. militaris, and the Tns gene was disrupted, resulting in color phenotype changes. The bar gene in the mutants can be stably maintained. Furthermore, the split-marker strategy will allow us to identify the functions of C. militaris genes.
References
Shrestha, B., Zhang, W., Zhang, Y., & Liu, X. (2012). The medicinal fungus Cordyceps militaris: Research and development. Mycological Progress, 11, 599–614.
Xu, C., Zhang, X., Qian, Y., Chen, X., Liu, R., Zeng, G., et al. (2014). A high-throughput gene disruption methodology for the entomopathogenic fungus Metarhizium robertsii. PLoS ONE, 9, e107657.
Fu, J., Hettler, E., & Wickes, B. L. (2006). Split marker transformation increases homologous integration frequency in Cryptococcus neoformans. Fungal Genetics and Biology, 43, 200–212.
Lin, C. H., & Chung, K. R. (2010). Specialized and shared functions of the histidine kinase- and HOG1 MAP kinase-mediated signaling pathways in Alternaria alternata, the filamentous fungal pathogen of citrus. Fungal Genetics and Biology, 47, 818–827.
Zheng, P., Xia, Y., Xiao, G., Xiong, C., Hu, X., Zhang, S., et al. (2011). Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biology, 12, R116.
Yu, J. H., Hamari, Z., Han, K. H., Seo, J. A., Reyes-Domínguez, Y., & Scazzocchio, C. (2004). Double-joint PCR: A PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genetics and Biology, 41, 973–981.
Wang, F., Song, X., Dong, X., Zhang, J., & Dong, C. (2017). DASH-type cryptochromes regulate fruiting body development and secondary metabolism differently than CmWC-1 in the fungus Cordyceps militaris. Applied Microbiology and Biotechnology, 101, 4645–4657.
Patil, N. S., & Jadhav, J. P. (2015). Penicillium ochrochloron MTCC 517 chitinase: An effective tool in commercial enzyme cocktail for production and regeneration of protoplasts from various fungi. Saudi Journal of Biological Sciences, 22, 232–236.
Elorza, M. V., Rico, H., Gozalbo, D., & Sentandreu, R. (1983). Cell wall composition and protoplast regeneration in Candida albicans. Antonie van Leeuwenhoek, 49(4–5), 457–469.
Bidochka, M. J., Pfeifer, T. A., & Khachatourians, G. G. (1987). Development of entomopathogenic fungus Beauveria bassiana in liquid. Mycopathologia, 99(2), 77–83.
Xiong, C. H., Xia, Y. L., Zheng, P., Shi, S. H., & Wang, C. S. (2010). Developmental stage-specific gene expression profiling for a medicinal fungus Cordyceps militaris. Mycology, 1(1), 25–66.
Delmas, S., Llanos, A., Parrou, J. L., Kokolski, M., Pullan, S. T., Shunburne, L., et al. (2014). Development of an unmarked gene deletion system for the filamentous fungi Aspergillus niger and Talaromyces versatilis. Applied and Environmental Microbiology, 80, 3484–3487.
Rahnama, M., Forester, N., Ariyawansa, K. G., Voisey, C. R., Johnson, L. J., Johnson, R. D., et al. (2017). Efficient targeted mutagenesis in Epichloë festucae using a split marker system. Journal of Microbiological Methods, 134, 62–65.
Dong, J. Z., Wang, S. H., Ai, X. R., Yao, L., Sun, Z. W., Lei, C., et al. (2013). Composition and characterization of cordyxanthins from Cordyceps militaris fruit bodies. Journal of Functional Foods, 5, 1450–1455.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31572178, 31372116), and the Projects of Science and Technology of Guangdong Province (Grant Nos. 2014B020205003, 2016A030313404). We are grateful to Prof. Gang Liu and Yuanyuan Pan, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China, for providing pAg1-H3 vector.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lou, H., Ye, Z., Yun, F. et al. Targeted Gene Deletion in Cordyceps militaris Using the Split-Marker Approach. Mol Biotechnol 60, 380–385 (2018). https://doi.org/10.1007/s12033-018-0080-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-018-0080-9