
Abstract
The application of arylacetonitrilases from filamentous fungi to the hydrolysis of high concentrations of (R,S)-mandelonitrile (100–500 mM) was demonstrated for the first time. Escherichia coli strains expressing the corresponding genes were used as whole-cell catalysts. Nitrilases from Aspergillus niger, Neurospora crassa, Nectria haematococca, and Arthroderma benhamiae (enzymes NitAn, NitNc, NitNh, and NitAb, respectively) exhibited different degrees of enantio- and chemoselectivity (amide formation). Their enantio- and chemoselectivity was increased by increasing pH (from 8 to 9–10) and adding 4–10 % (v/v) toluene as the cosolvent. NitAn and NitNc were able to convert an up to 500 mM substrate in batch mode. NitAn formed a very low amount of the by-product, amide (<1% of the total product). This enzyme produced up to >70 g/L of (R)-mandelic acid (e.e. 94.5–95.6 %) in batch or fed-batch mode. Its volumetric productivities were the highest in batch mode [571 ± 32 g/(L d)] and its catalyst productivities in fed-batch mode (39.9 ± 2.5 g/g of dcw). NitAb hydrolyzed both enantiomers of 100 mM (R,S)-mandelonitrile at pH 5.0 and is therefore promising for the enantioretentive transformation of (S)-mandelonitrile. Sequence analysis suggested that fungal arylacetonitrilases with similar properties (enantioselectivity, chemoselectivity) were clustered together.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
(R)-Mandelic acid is an important pharmaceutical intermediate used in the synthesis of antibiotics, antiobesity drugs, and cancerostatics, and is also widely used as a chiral resolving agent to obtain optically pure alcohols and amines [1, 2]. Various biocatalytic processes have been proposed for its manufacture, some of them based on the enantioselective hydrolysis of (R,S)-mandelonitrile (Fig. 1a), among them multi-ton processes operated by BASF and Mitsubishi Rayon [3]. The enzymes used to catalyze this reaction have been exclusively bacterial in origin. The first of them was found in wild-type strains of Alcaligenes sp. in the early 1990s (see [4] for a review). A number of nitrilases suitable for the production of (R)-mandelic acid were also found by screening a metagenomic library [5, 6]. The process was recently improved using recombinant strains [1, 2, 7–10] or enzyme mutants [2]. A similar process was also developed for the production of (R)-o-chloromandelic acid, a precursor of the anti-coagulant drug Clopidogrel® [11].

Production of (R)-mandelic acid from (R,S)-mandelonitrile (a) and (S)-mandelic acid from (S)-mandelonitrile (b) by nitrilase 1 (highly enantioselective) and nitrilase 2 (moderately enantioselective). The formation of the side product (S)-mandelamide depends on the enzyme source and the reaction conditions
Nitrilases suitable for the S-selective hydrolysis of (R,S)-mandelonitrile have not been found so far. The majority of characterized nitrilases were R-selective and only a few of them hydrolyzed (S)-mandelonitrile preferentially, but with low enantioselectivities [5, 12]. Therefore, (S)-mandelic acid was synthesized by a stereoretentive hydrolysis of (S)-mandelonitrile using a nitrilase from Pseudomonas fluorescens which exhibited a low enantioselectivity (Fig. 1b) [13–15]. Similar nitrilases have been also found in Bradyrhizobium japonicum [12, 16, 17] and Burkholderia xenovorans [16].
Industrial practice requires running the biocatalytic processes toward high product concentrations. Recently, the arylacetonitrilases from Alcaligenes sp., Alcaligenes faecalis, and Burkholderia cepacia were found to be promising for this purpose, being able to hydrolyze up to 0.5–1 M of mandelonitrile into (R)-mandelic acid with high yields [1, 2, 8, 9].
In contrast to bacterial nitrilases, the biocatalytic potential of nitrilases from filamentous fungi remained unexplored until recently. Until a few years ago, all the fungal nitrilases reported were aromatic nitrilases [18]. It is only since 2011 that arylacetonitrilases have been found in filamentous fungi by genome mining and expression of the artificially synthesized genes [19–22]. These fungal nitrilases are evolutionarily distant from the bacterial ones (see phylogenetic tree; Fig. S1), sharing at most ca. 40 % aa sequence identity. As a result, one could expect their catalytic properties to be different. Six of these enzymes were previously examined for their substrate specificities and were shown to possess high specific activities for (R,S)-mandelonitrile, which, however, was used at a low concentration (25 mM) [20]. The enzymes from Aspergillus niger (NitAn), Neurospora crassa (NitNc), Nectria haematococca (NitNh), and Arthroderma benhamiae (NitAb) were purified and partly characterized [21, 23]. All these enzymes exhibited selectivity for (R)-mandelonitrile, albeit to differing degrees [23, 24].
These findings prompted us to examine the enzymes´ potential for (R)-mandelic acid manufacture from high substrate concentrations. To this end, whole cells of Escherichia coli strains which overproduced these enzymes were used. First, the ability of the enzymes to act on high mandelonitrile concentrations was demonstrated. Second, the effects of the medium properties (pH, organic cosolvent) on the enzyme enantio- and chemoselectivity were examined, and selected enzymes were used for the enantioselective or enantioretentive hydrolysis of (R,S)-mandelonitrile under the optimized conditions. Third, relationships between the aa sequences and catalytic properties of the enzymes were analyzed. The results of this part of the study indicated the possibility of predicting the catalytic properties of further arylacetonitrilases from filamentous fungi.
Materials and Methods
Sequence Analysis
The sequence analysis of fungal arylacetonitrilases was performed using the program BLASTP (http://www.ncbi.nlm.nih.gov/Blast.cgi) with the sequence of NitAn (ref|XP_001397369.1|) as the template. The phylogenetic tree of fungal arylacetonitrilases was calculated and visualized using http://www.ncbi.nlm.nih.gov/blast/treeview/treeView.cgi. Multiple sequence alignment of selected fungal and bacterial arylacetonitrilases was constructed using the Constraint-based Multiple Alignment Tool COBALT (http://www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi).
Nitrilase Production
Fungal nitrilases (NitAb, NitAn, NitNc, NitNh) were produced in E. coli without N-terminal His-tags as described previously [19–23]. Briefly, the genes were designed according to database entries and prepared synthetically by GeneArt (Regensburg, Germany) with their codon frequency optimized for the expression in E. coli. Each gene was cloned into pET 30a(+) plasmid (Novagen), and the resulting plasmids were transformed into E. coli BL21-Gold (DE3) cells. Each strain was cultivated in LB medium (3.5–4 h at 37 °C; induction with 0.5 mM IPTG; 17 h at 25 °C). The cells were harvested by centrifugation, washed with Tris/HCl buffer (100 mM, pH 8), and used for the biotransformation experiments as described below.
Biotransformation of (R,S)-mandelonitrile
Biotransformations of (R,S)-mandelonitrile were carried out with appropriate amounts of whole cells of E. coli in various buffers (120 mM acetic acid/boric acid/phosphoric acid/NaOH buffer, pH 5; 50 or 100 mM Tris/HCl buffer with 150 mM NaCl, pH 8 or 9; 100 mM glycine/NaOH buffer, pH 10). Cells were washed with the corresponding buffer and used without any further pretreatment. The concentrations of whole cells in the suspensions were calculated from the optical densities measured at 600 nm (OD600 = 1 corresponds to ca. 0.3 g dcw/L).
In batch mode, (R,S)-mandelonitrile was added to a final concentration of 100, 250, or 500 mM. Optionally, toluene was added to a final concentration of 4 or 10 % (v/v). The reactions proceeded in 50-mL Falcon tubes (closed with screw caps) with a 10-mL working volume with shaking (300–350 rpm; Eppendorf Thermomixer) and 30 °C. The concentration and the e.e. of the product were determined as described below.
In fed-batch mode, the reactions proceeded in 250-mL Erlenmeyer flasks with a 100-mL working volume with shaking (rotary shaker, 220 rpm) at 28–30 °C. At 1-h intervals, (R,S)-mandelonitrile was added in five or twelve feeds of 2.5 or 5 mmol each, respectively. The total amount of the substrate was added within 4 or 11 h, respectively. The product concentration was monitored for 4.5 and 11.5 h, respectively. The reaction mixtures were then left to stir overnight, and the final concentration and e.e. of the product were determined (see below).
Analytical HPLC
After terminating the reactions with HCl (final concentration 0.2 M), the cells were removed by centrifugation and the substrate and product concentrations determined using a Chromolith Flash RP-18 column (Merck; 25 × 4.6 mm) and mobile phase consisting of water/methanol (9/1) and H3PO4 (0.1 %). The analysis was performed at a flow rate of 2 mL/min and 35 °C. Chiral HPLC analysis of mandelic acid and mandelamide was carried out as described previously [15]. The means and standard deviations were calculated from the results of two to three independent experiments.
Results and Discussion
Production of (R)-mandelic Acid in Fed-Batch Mode
The nitrilases NitAn, NitNc, NitNh, and NitAb were selected for this work as their activities for mandelonitrile were the highest from the set of 13 nitrilases examined [20]. The first three enzymes were highly (R)-selective for this substrate [20, 23, 24] and were therefore promising for the (R)-mandelic acid manufacture. NitAb was able to transform both enantiomers of mandelonitrile efficiently [23] and thus seemed to be rather promising for the enantioretentive hydrolysis of (S)-mandelonitrile.
The catalysts used were whole cells of E. coli. As demonstrated previously, the enzymes were produced in this host at similar levels, forming the major proteins of the soluble protein fractions [21, 23].
First, the catalysts were examined in fed-batch mode with a total of 12.5 mmol (123 mM) of substrate added in five equal feeds within 4 h. To support substrate racemization, the reactions were performed at pH 10. A high racemization rate, which should exceed that of the enzymatic hydrolysis, is a condition for efficient dynamic kinetic resolution. This was demonstrated, for instance, in the enzymatic resolution of racemic amino nitriles [25]. Conversions of ca. 90–98 % were achieved with NitAb, NitAn, and NitNc, but the reaction catalyzed by NitNh ended up with only 42.5 % conversion (Table 1). This was probably caused by the higher sensitivity of NitNh to alkaline pH [23]. The time profile of the reactions was monitored for 4.5 h (Fig. 2a), during which 14.5–16.5 g/L of the product was obtained using NitAn, NitNc, and NitAb. After subsequent overnight incubation, the product concentrations increased to ca. 15.5–18 g/L. The highest e.e. of the product was obtained in NitAn and NitNc (ca. 97.6 and 96.7 %, respectively), while the product of NitAb was only moderately enriched in its (R)-isomer, as expected (Table 1).

Time profile of mandelic acid production by fed-batch transformation of a 12.5 mmol of (R,S)-mandelonitrile using nitrilases NitAn (diamond), NitNc (square), and NitAb (triangle) or b 60 mmol of (R,S)-mandelonitrile using nitrilase NitAn (diamond). The product concentration was determined by achiral HPLC. Arrows indicate the time of addition of a 2.5 mmol and b 5 mmol of substrate. The first feed was added at the start of the reaction (not shown). The reactions were performed at the 100-mL scale in shaken Erlenmeyer flasks
NitAn also exhibited the lowest amide production (0.80 % of the total product), along with a ca. 90 % conversion (Table 1). It was therefore chosen for the following experiment aiming to convert a total of 60 mmol (ca. 560 mM) of (R,S)-mandelonitrile. Here, twelve feeds of 5 mmol were added during the first 11 h, followed by a final incubation of 19.5 h. The product concentration reached ca. 75 g/L after 11.5 h (Fig. 2b) and almost 77 g/L after the total reaction time (Table 1). The e.e. of the product (95.6 %) was slightly lower than in the previous experiment. This indicated the utility of using lower substrate concentrations (25 vs. 50 mM) in the feeds.
Compared to the fed-batch processes catalyzed by nitrilase from Alcaligenes sp. [8], the volumetric productivities of NitAn acting on 560 mM (R,S)-mandelonitrile were almost twice as low [ca. 60 vs. 108 g/(L d)] but the catalyst productivity more than tenfold higher (ca. 40 vs. 3.2 g/g dcw). Recently, a genetically improved variant of an Alcaligenes faecalis nitrilase was prepared and used in (R)-mandelic acid fed-batch production with volumetric and catalyst productivities of ca. 337 g/(L d) and 8.4 g/g dcw, respectively [2]. Thus the latter value was still lower than for NitAn.
Here, the volumetric productivities were calculated for the total reaction times (Table 1). When the reaction was stopped shortly after the last feed, i.e., after 4.5 or 11.5 h in the transformations of 123 and 560 mM substrate, respectively (Fig. 2), the volumetric productivities increased to ca. 77–88 and 156 g/(L d), respectively, albeit at the expense of lower yields (77–88 vs. ca. 90 %).
Effect of pH and Cosolvent on Enzyme Enantio- and Chemoselectivity
The production of high concentrations of (R)-mandelic acid in batch mode will require using the substrate at concentrations which may inhibit the enzymes. For instance, whole cells producing the nitrilase from Alcaligenes sp. was unable to transform (R,S)-mandelonitrile at concentrations of 300 mM or higher. However, this problem was solved by adding toluene (10 %, v/v) which decreased the substrate concentration in the aqueous phase [1]. Alternatively, ethylacetate (30 % v/v) was used as a cosolvent in a process catalyzed by the whole-cell catalyst based on the nitrilase from B. cepacia [10].
The hydrolysis of 500 mM (R,S)-mandelonitrile by E. coli whole-cell catalysts producing the fungal enzymes was first examined at pH 8.0 (near the activity optimum of the enzymes). The enzymes NitAn, NitAb, and NitNc were able to hydrolyze this substrate concentration with high conversions (75–93 %) in the absence of toluene but NitNh only achieved a 25 % conversion. The addition of toluene (10 %, v/v) did not affect the conversions with NitAb or NitNh but decreased conversions with NitAn and NitNc below 60 %. NitNh was only able to transform lower substrate concentrations (250 mM) but it did so almost completely with or without toluene (data not shown).
Thus the majority of the fungal enzymes examined (NitAn, NitAb, NitNc) were more resistant to high concentrations of (R,S)-mandelonitrile than the nitrilase from Alcaligenes sp. [1], but some of them (NitAn, NitNc) were more sensitive to toluene. Higher concentrations of toluene were often observed to decrease nitrilase reaction rates (e.g., [1, 26]). Although toluene is sparingly soluble in water, enzyme deactivation may occur at the water: toluene interface. Apart from this, the effect of high amounts of a non-polar cosolvent may also be mediated by decreasing the substrate concentrations in the aqueous phase, the probability of the enzyme-substrate contact being lowered [1].
NitAn, NitNc, and NitNh were previously found to produce (R)-mandelic acid with high e.e. (89–99 %) from 10 to 25 mM (R,S)-mandelonitrile [20, 23, 24] at pH 8. In contrast, the e.e. of the product obtained from 250 to 500 mM substrate concentrations at pH 8 were only about 70 % (Fig. 3). This effect was probably at least partly due to decreasing pH and, hence, lowering the racemization rate. During the reactions, the pH of the medium (50 mM Tris/HCl, pH 8.0) decreased to ca. 7.5 and 6.0 after the full conversion of 250 and 500 mM substrate, respectively. Increasing the ionic strength of the buffer to 100 mM did not change the enantioselectivity of the enzymes significantly. The decrease in enantioselectivity at high substrate concentrations could also be caused by the different K m-values of each enantiomer of the substrate.

Effect of pH and toluene on e.e. of (R)-mandelic acid (a) and amide production (b) by NitAn, NitNc, and NitNh: The reactions were performed at the 10-mL scale (Falcon tubes; 300–350 rpm; 30 °C). The concentration of (R,S)-mandelonitrile was 500 mM for NitAn and NitNc and 250 mM for NitNh. The concentration of toluene was 4 % (v/v) for NitAn and NitNc and 10 % (v/v) for NitNh. e.e. and % of amide were determined at 70–90 % conversions except for NitNh, which only achieved 50 % conversion at pH 10. Mandelamide was produced by NitAn at less than 2 % of the total product (not shown)
To improve the product e.e., the effects of alkaline pH and toluene were examined (Fig. 3). It could be expected that substrate racemization would be faster at a higher pH, and toluene would decrease the concentration of substrate in the aqueous phase. Toluene was used at 4 % (v/v) for NitAn and NitNc and 10 % (v/v) for NitNh (see above). By adding toluene, the product e.e. increased to 94.5 % in the transformation of 250 mM substrate by NitNh, and to ≥80 % in transformations of 500 mM substrate by NitAn and NitNc at pH 8. By increasing pH to 9, a further increase in e.e. up to ca. 94–96 % was achieved with all these enzymes, and similar values were obtained at pH 10 (Fig. 3a). The enantioselectivity of NitAb remained low, although a similar trend was observed in this enzyme, i.e., an increase in e.e. from 43 % at pH 8 to 70 % in the presence of toluene (10 %, v/v) and pH 9–10 (data not shown).
NitNc and NitNh produced significant amounts of mandelamide as a side product at pH 8. The addition of toluene and the increase in pH to 9–10 lowered its production from ca. 12 to 4 % with NitNh. The percentage of amide formed by NitNc decreased from 32 to ca. 5.5–7 % under these conditions (Fig. 3b). A slightly higher amount of the amide (8.4 %) was produced by NitNc in fed-batch mode without toluene at pH 10 (see above). The production of amide in NitAn and NitAb was below 2 % irrespective of toluene addition or pH.
The decrease in amide production was probably a consequence of the increased enantioselectivity. It was previously observed that the products of the hydrolysis of (R)- and (S)-mandelonitrile exhibited different ratios of acid: amide, i.e., 89:11 and 45:55, respectively, in nitrilase from P. fluorescens [27]. A preferential conversion of (R)-nitrile into acid and (S)-nitrile into amide was also previously observed in NitNc, when using a racemic substrate at pH 5; in this case, the (S)-amide was formed later than the (R)-acid during the reaction [24].
Mandelamide produced by NitNc and NitNh was enriched in its (S)-isomer as expected (e.e. 93 and 81 %, respectively, at pH 8), and its e.e. decreased with increasing pH and toluene addition (data not shown). At pH 10, the stereochemistry of the reaction catalyzed by NitNh was reversed, (R)-mandelamide being obtained in 10 % e.e.
Batch Production of (R)-mandelic Acid in Biphasic Systems
The conditions suitable for maximum enantio- and chemoselectivity of NitAn and NitNc (pH 9.0, 4 % (v/v) toluene) were used to produce (R)-mandelic acid in batch biphasic systems (Table 2). NitAn transformed 500 mM (R,S)-mandelonitrile with a ca. 94.5 % yield to produce over 470 mM (ca. 71 g/L) (R)-mandelic acid (Fig. 4). Hence, the product concentration was similar to that in the fed-batch process with a 560 mM substrate (see above); however, the product e.e. was slightly lower (ca. 94.5 vs. 95.6 %). Nevertheless, this system was found to exhibit the highest volumetric productivity of 571 g/(L d) compared to those reported in this study or in studies of the Alcaligenes nitrilase [1, 2, 8]. Only the nitrilase from B. cepacia gave higher volumetric productivities [up to 982 g/(L d)]; this value was achieved after recycling the catalyst [10] (Table 3).

Time profile of (R)-mandelic acid production by batch transformation of (R, S)-mandelonitrile at concentrations of 100 mM (by NitAb, cross symbol), 250 mM (by NitNh, triangle), and 500 mM (by NitAn, diamond; or NitNc, square). The reactions were performed at the 10-mL scale in shaken Falcon tubes
NitNc also produced (R)-mandelic acid at acceptable concentrations and enantiopurities (ca. 56 g/L, 95.2 % e.e.), but the product still contained a significant percentage of amide (ca. 7 %). NitNh only hydrolyzed up to 250 mM (R,S)-mandelonitrile, providing ca. 32.5 g/L of (R)-mandelic acid which corresponded to a volumetric productivity of ca. 195 g/(L d) (Table 2).
The catalyst productivities in the batch reactions of 500 mM substrate (ca. 9–15 g/g) were lower than in the fed-batch transformation of 560 mM substrate (ca. 40 g/g) but still much higher than in E. coli cells with the Alcaligenes sp. nitrilase when used in a single cycle (1.8–3.7 g/g) [1]. The productivity of the latter catalyst was increased to 55 g/g by cell recycling [1]. The highest catalyst productivity (156 g/g) was achieved with the nitrilase from B. cepacia when the catalyst (immobilized whole cells of E. coli) was recycled [10] (Table 3).
Non-selective Conversion of (R,S)-mandelonitrile at Low pH
Enantioretentive conversion of (S)-mandelonitrile will require running the reactions at acidic pH to suppress substrate racemization. Previously, a moderately selective nitrilase from P. fluorescens was used under these conditions. This enzyme hydrolyzed significant concentrations of (S)-mandelonitrile which was produced in situ by an S-selective oxynitrilase from up to 100 mM benzaldehyde and 300 mM HCN; however, a mixture of acid and amide (at a ratio of 1.2–1.7) was formed [28]. The chemoselectivity for the formation of either (S)-mandelamide or (S)-mandelic acid was increased by the mutations of this enzyme [15, 28, 29], which resulted in obtaining an amide/acid ratio of 19:1 [29]. If (S)-acid is the product required, the by-product amide could be also eliminated by including an amidase in the reaction [14]. In an analogous way, (R)-o-chloromandelic acid may be also prepared via a cascade reaction catalyzed by (1) oxynitrilase and (2) nitrilase (both R-selective). The nitrilase from Alcaligenes faecalis was engineered using directed evolution to enhance its ability to hydrolyze this substrate under enantioretentive conditions, i.e., pH 4.5 [30].
Here, the strain of E. coli producing NitAb—the enzyme with the lowest enantioselectivity—was examined for its activity on high (R,S)-mandelonitrile concentrations at pH 5. Under these conditions, 100 mM (R,S)-mandelonitrile was transformed into ca. 98 mM (ca. 15 g/L) mandelic acid (Table 2). The product was moderately enriched in its R-enantiomer (37 % e.e.) at 30 % conversion but was almost racemic at the end of the reaction. Higher concentrations of (R,S)-mandelonitrile (250 or 500 mM) could not be efficiently converted under these conditions (data not shown). NitAb seems to be promising for the production of (S)-mandelic acid due to its low tendency to produce mandelamide (ca. 1.6 % of the total product).
Sequence Analysis of Fungal Nitrilases Acting on (R,S)-mandelonitrile
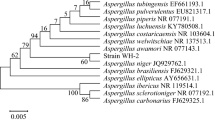
The aa sequence of NitAn was used as a template to search for its closest homologs. Most of the homologous sequences originated from ascomycetes. The nitrilases examined in this study belonged to different subtrees except for NitNh and NitNc which clustered in the same one (Fig. 5). This was in accordance with the similar catalytic properties of these two enzymes (high enantioselectivity and high amide formation). This subtree contained a few hypothetical nitrilases, mainly from the Fusarium genus, with 64–90 % identities to NitNc or NitNh.

Phylogenetic analysis of nitrilases NitAn (ref|XP_001397369.1|), NitNc (emb|CAD70472.1|), NitNh (ref|XP_003050920.1|), NitAb (ref|XP_003011330.1|), and related enzymes. The phylogram was constructed with the Blast Tree View Widget in the program BLASTP (http://blast.ncbi.nlm.nih.gov/Blast.cgi) using NitAn (highlighted) as the template. The biochemically characterized enzymes and their catalytic properties ([19–23]; this study) are highlighted
NitAb was the only characterized member of a different subtree. Sequences in this subtree originated from the Arthroderma and Trichophyton genera and shared ≥93 % identities. These proteins are candidates for nitrilases acting on both enantiomers of mandelonitrile.
NitAn was evolutionarily distant from other fungal arylacetonitrilases and belonged to a subtree consisting of four sequences from Aspergillus sharing 87–96 % identities. NitAn was also previously found to differ from other fungal arylacetonitrilases in the region proximal to the catalytic Cys. Specifically, a hexapeptide upstream of the Cys residue was present in NitNc, NitAb, and NitNh but not in NitAn [21]. The potential function of this structural motif has not been elucidated so far.
The majority of potential nitrilases clustered in a subtree in which two enzymes—from A. niger and A. oryzae—were examined in whole cells and proved to operate on (R,S)-mandelonitrile but with medium activities and moderate R-selectivities [20].
Conclusions
Fungal nitrilases, which are evolutionarily distant from their bacterial counterparts, proved to be promising catalysts of mandelic acid synthesis. Their operational parameters were compared to those of bacterial enzymes (Table 3). Similar to the bacterial enzymes, the fungal nitrilases enabled high concentrations (>70 g/L) of (R)-mandelic acid to be obtained. The volumetric and catalyst productivities of fungal nitrilases were comparable or, in some cases, higher than in bacterial nitrilases used under similar conditions. Higher values for these parameters were only found in a few previous experiments with bacterial nitrilases, mainly when recycling the catalyst. There is room to improve the productivity and enantioselectivity of the fungal enzymes further by modifying the process conditions (cell recycling; substrate, cosolvent and catalyst concentration, reaction time, pH) or by screening for new fungal enzymes. Phylogenetic analysis of fungal arylacetonitrilases indicated that it may be possible to obtain further nitrilases of this type by genome mining and to partially predict their catalytic properties. Fungal nitrilases may also be a source of non-selective catalysts operating on both enantiomers of mandelonitrile and thus promising for (S)-mandelic acid manufacture from (S)-mandelonitrile. The enzymes examined, their homologues and artificial variants are also potentially useful in the production of mandelamide or other chiral amides and acids.
Abbreviations
- aa:
-
Amino acid
- ca.:
-
Circa, approximately
- dcw:
-
Dry cell weight
- IPTG:
-
Isopropyl-β-d-thiogalactopyranoside
- NitAb:
-
Nitrilase from Arthroderma benhamiae CBS 112371 (ref|XP_003011330.1|)
- NitAn:
-
Nitrilase from Aspergillus niger CBS 513.88 (ref|XP_001397369.1|)
- NitNc:
-
Nitrilase from Neurospora crassa OR74A (emb|CAD70472.1|)
- NitNh:
-
Nitrilase from Nectria haematococca mpVI 77-13-4 (ref|XP_003050920.1|)
- vs.:
-
Versus, against
References
Zhang, Z.-J., Pan, J., Liu, J.-F., Xu, J.-H., He, Y.-C., & Liu, Y.-Y. (2011). Significant enhancement of (R)-mandelic acid production by relieving substrate inhibition of recombinant nitrilase in toluene–water biphasic system. Journal of Biotechnology, 152, 24–29.
Liu, Z.-Q., Zhang, X.-H., Xue, Y.-P., Xu, M., & Zheng, Y.-G. (2014). Improvement of Alcaligenes faecalis nitrilase by gene site saturation mutagenesis and its application in stereospecific biosynthesis of (R)-(−)-mandelic acid. Agricultural and Food Chemistry, 6, 4685–4694.
Breuer, M., Ditrich, K., Habicher, T., Hauer, B., Kesseler, M., Stürmer, R., & Zelinski, T. (2004). Industrial methods for the production of optically active intermediates. Angewandte Chemie International Edition, 43, 788–824.
O’Reilly, C., & Turner, P. D. (2003). The nitrilase family of CN hydrolysing enzymes—a comparative study. Journal of Applied Microbiology, 5, 1161–1174.
Robertson, D. E., Chaplin, J. A., DeSantis, G., Podar, M., Madden, M., Chi, E., et al. (2004). Exploring nitrilase sequence space for enantioselective catalysis. Applied and Environmental Microbiology, 70, 2429–2436.
DeSantis, G., Zhu, Z., Greenberg, W. A., Wong, K., Chaplin, J., Hanson, S. R., et al. (2002). An enzyme library approach to biocatalysis: Development of nitrilases for enantioselective production of carboxylic acid derivatives. Journal of the American Chemical Society, 124, 9024–9025.
Banerjee, A., Dubey, S., Kaul, P., Barse, B., Piotrowski, M., & Banerjee, U. C. (2009). Enantioselective nitrilase from Pseudomonas putida: cloning, heterologous expression, and bioreactor studies. Molecular Biotechnology, 41, 35–41.
Zhang, Z.-J., Xu, J.-H., He, Y.-C., Ouyang, L.-M., Liu, Y.-Y., & Imanaka, T. (2010). Efficient production of (R)-(−)-mandelic acid with highly substrate/product tolerant and enantioselective nitrilase of recombinant Alcaligenes sp. Process Biochemistry, 45, 887–891.
Wang, H., Sun, H., & Wei, D. (2013). Discovery and characterization of a highly efficient enantioselective mandelonitrile hydrolase from Burkholderia cenocepacia J2315 by phylogeny-based enzymatic substrate specificity prediction. BMC Biotechnology, 13, 14.
Ni, K., Wang, H., Zhao, L., Zhang, M., Zhang, S., Ren, Y., & Wei, D. (2013). Efficient production or R-(−)-mandelic acid in biphasic system by immobilized recombinant E. coli. Journal of Biotechnology, 167, 433–440.
Zhang, C.-S., Zhang, Z.-J., Li, C.-X., Yu, H.-L., Zheng, G.-W., & Xu, J.-H. (2012). Efficient production of (R)-o-chloromandelic acid by deracemization of o-chloromandelonitrile with a new nitrilase mined from Labrenzia aggregata. Applied Microbiology and Biotechnology, 95, 91–99.
Zhu, D., Mukherjee, C., Yang, Y., Rios, B. E., Gallagher, D. T., Smith, N. N., et al. (2008). A new nitrilase from Bradyrhizobium japonicum USDA 110. Gene cloning, biochemical characterization and substrate specificity. Journal of Biotechnology, 133, 327–333.
Rustler, S., Motejadded, H., Altenbuchner, J., & Stolz, A. (2008). Simultaneous expression of an arylacetonitrilase from Pseudomonas fluorescens and a (S)-oxynitrilase from Manihot esculenta in Pichia pastoris for the synthesis of (S)-mandelic acid. Applied Microbiology and Biotechnology, 80, 87–97.
Chmura, A., Rustler, S., Paravidino, M., van Rantwijk, F., Stolz, A., & Sheldon, R. (2013). The combi-CLEA approach: enzymatic cascade synthesis of enantiomerically pure (S)-mandelic acid. Tetrahedron Asymmetry, 24, 1225–1232.
Sosedov, O., Baum, S., Bürger, S., Matzer, K., Kiziak, C., & Stolz, A. (2010). Construction and application of variants of the Pseudomonas fluorescens EBC191 arylacetonitrilase for increased production of acids and amides. Applied and Environmental Microbiology, 76, 3668–3674.
Seffernick, J. L., Samanta, S. K., Louie, T. M., Wackett, L. P., & Subramanian, M. (2009). Investigative mining of sequence data for novel enzymes: A case study with nitrilases. Journal of Biotechnology, 143, 17–26.
Zhu, D., Mukherjee, C., Biehl, E. R., & Hua, L. (2007). Discovery of a mandelonitrile hydrolase from Bradyrhizobium japonicum USDA 110 by rational genome mining. Journal of Biotechnology, 129, 645–650.
Martínková, L., Vejvoda, V., Kaplan, O., Kubáč, D., Malandra, A., Cantarella, M., et al. (2009). Fungal nitrilases as biocatalysts: recent developments. Biotechnology Advances, 27, 661–670.
Kaplan, O., Bezouška, K., Malandra, A., Veselá, A. B., Petříčková, A., Felsberg, J., et al. (2011). Genome mining for the discovery of new nitrilases in filamentous fungi. Biotechnology Letters, 33, 309–312.
Kaplan, O., Veselá, A. B., Petříčková, A., Pasquarelli, F., Pičmanová, M., Rinágelová, A., et al. (2013). A comparative study of nitrilases identified by genome mining. Molecular Biotechnology, 54, 996–1003.
Petříčková, A., Veselá, A. B., Kaplan, O., Kubáč, D., Uhnáková, B., Malandra, A., et al. (2012). Purification and characterization of heterologously expressed nitrilases from filamentous fungi. Applied Microbiology and Biotechnology, 93, 1553–1561.
Petříčková, A., Veselá, A. B., Kaplan, O., Kubáč, D., Uhnáková, B., Malandra, A., et al. (2012). Erratum to: Purification and characterization of heterologously expressed nitrilases from filamentous fungi. Applied Microbiology and Biotechnology, 97, 9263–9264.
Veselá, A. B., Petříčková, A., Weyrauch, P., & Martínková, L. (2013). Heterologous expression, purification and characterization of arylacetonitrilases from Nectria haematococca and Arthroderma benhamiae. Biocatalysis and Biotransformation, 31, 49–56.
Petříčková, A., Sosedov, O., Baum, S., Stolz, A., & Martínková, L. (2012). Influence of point mutations near the active site on catalytic properties of fungal arylacetonitrilases from Aspergillus niger and Neurospora crassa. Journal of Molecular Catalysis B-Enzymatic, 77, 74–80.
Chaplin, J. A., Levin, M. D., Morgan, B., Farid, N., Li, J., Zhu, Z., et al. (2004). Chemoenzymatic approaches to the dynamic kinetic asymmetric synthesis of aromatic amino acids. Tetrahedron Asymmetry, 15, 2793–2796.
Heinemann, U., Engels, D., Bürger, S., Kiziak, C., Mattes, R., & Stolz, A. (2003). Cloning of a nitrilase gene from the cyanobacterium Synechocystis sp. strain PCC6803 and heterologous expression and characterization of the encoded protein. Applied and Environmental Microbiology, 69, 4359–4366.
Fernandes, B. C. M., Mateo, C., Kiziak, C., Chmura, A., Wacker, J., van Rantwijk, F., et al. (2006). Nitrile hydratase activity of a recombinant nitrilase. Advanced Synthesis & Catalysis, 348, 2597–2603.
Sosedov, O., Baum, S., Bürger, S., Matzer, K., Kiziak, C., & Stolz, A. (2009). Construction and application of variants of the Pseudomonas fluorescens EBC191 arylacetonitrilase for increased production of acids or amides. Applied and Environmental Microbiology, 76, 3668–3674.
Sosedov, O., & Stolz, A. (2014). Random mutagenesis of the arylacetonitrilase from Pseudomonas fluorescens EBC191 and identification of variants, which form increased amounts of mandeloamide from mandelonitrile. Applied Microbiology and Biotechnology, 98, 1595–1607.
Schreiner, U., Hecher, B., Obrowsky, S., Waich, K., Klempier, N., Steinkellner, G., et al. (2010). Directed evolution of Alcaligenes faecalis nitrilase. Enzyme and Microbial Technology, 47, 140–146.
Acknowledgments
Financial support via Project P504/11/0394 and Internal Project RVO61388971 of the Institute of Microbiology of the Academy of Sciences of the Czech Republic is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Veselá, A.B., Křenková, A. & Martínková, L. Exploring the Potential of Fungal Arylacetonitrilases in Mandelic Acid Synthesis. Mol Biotechnol 57, 466–474 (2015). https://doi.org/10.1007/s12033-015-9840-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-015-9840-y