
Abstract
The evolution of the complex immune system is equipped to defend against perilous intruders and concurrently negatively regulate the deleterious effect of immune-mediated inflammation caused by self and nonself antigens. Regulatory T-cells (Tregs) are specialized cells that minimize immune-mediated inflammation, but in malignancies, this feature has been exploited toward cancer progression by keeping the antitumor immune response in check. The modulation of Treg cell infiltration and their induction in the TME (tumor microenvironment) alongside associated inhibitory molecules, both soluble or membranes tethered in the TME, have proven clinically beneficial in boosting the tumoricidal activity of the immune system. Moreover, Treg-associated immune checkpoints pose a greater obstruction in cancer immunotherapy. Inhibiting or blocking active immune checkpoint signaling in combination with other therapies has proven clinically beneficial. This review summarizes the ontogeny of Treg cells and their migration, stability, and function in the TME. We also elucidate the Treg-associated checkpoint moieties that impede effective antitumor activity and harness these molecules for effective and targeted immunotherapy against cancer nuisance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The integration of cancerous cells with immune-associated cells and the availability of various metabolites produced in the tumor milieu pose immunosuppression that derail antitumor activity and have been linked with the failure of immunotherapies [1]. The tumor mass construction comprises various other cell populations that fabricate the immunosuppressive microenvironment, eventually leading to the tumor’s systematic dissemination [2]. Regulatory T-cells (Tregs) are among the most potent immunosuppressive cells, belong to the CD4+ T-cell lineage, and characteristically express the master switch gene FOXP3, which is crucial for the maintenance and development of Treg cells [3]. These cells are equipped to maintain immune homeostasis between inflammatory and anti-inflammatory responses and tolerance by controlling various facets of immune responses [4]. Any sort of perturbance in the Treg cell pool is often associated with various diseases, such as autoimmunity and cancer [5,6,7]. Any deleterious mutations in the FOXP3 gene are consequent in impairment and dysfunctional Treg cells that further account for multiple autoimmune disorders, and this condition in humans is known as IPEX syndrome (immune dysregulation polyendocrinopathy enteropathy X linked). Indeed, a similar FOXP3 mutation was observed, where mice (Scurfy) deficient in Treg cells succumb to systematic autoimmunity [8].
FOXP3 acts as a master transcription regulator of which expression determines phenotypic fate and immunosuppressive functionality (Fig. 1) [5, 9]. Treg cells migrate to the inflamed site and mitigate inflammation by inhibiting other immune cells, such as cytotoxic T-cells (CTLs) and helper T-cells (TH cells) [10, 11]. In cancer, Tregs infiltrate inflamed tumors along with other cells and contribute to the development and progression of various types of malignancies [12]. The only exception is colon cancer, where high Treg cell infiltration during the early phase favors a better prognosis [13]. A high Treg to CD8+ population ratio among TILs has a poor prognosis [14, 15]. The harsh milieu of the TME and the occurrence of various immune checkpoints aid in de-escalation of antitumor activity and promote tumor growth and decimations to secondary sites [16, 17]. Treg cell-associated immune checkpoint molecules, such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein-1 (PD-1), T-cell immunoglobulin, and mucin-domain-containing 3 (TIM-3), and band T lymphocyte attenuator (BTLA), obstruct the antitumor response. Targeting these molecules ameliorates the cancer condition and good therapeutic outcomes in various cancers [18]. Targeting ICIs (immune checkpoint inhibitors) have shown profound therapeutic significance in cancer, suggesting that targeting tumor-infiltrated Tregs or their localized depletion may be clinically beneficial, preventing systematic autoimmunity, and affirming central tolerance. Here, we reviewed Treg cell ontogeny, the mechanism of suppression, and accumulation in the tumor and the strategies to target predominantly the Treg-specific membrane surface molecules that are expressed by Treg cells upon activation, infiltration, and survival in the tumor milieu. We have also documented Treg-associated checkpoint molecules and their role in immunosuppression.

Phonotypical markers of Tregs for the development of immune suppression
Regulatory T-cells: subtypes, development, and location
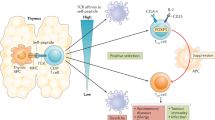
The Treg cell population holds 5–10% of the T-cell pool share in humans and mice in the peripheral compartment, consisting of natural/thymic and peripherally generated Tregs [19]. The ontogeny of Tregs involves nTregs, which spontaneously matures in the thymus with higher avidity and interacts with MHC-II-bound self-antigens and the IL-2 receptor. Induced Tregs constitute conventional CD4+ T-cells in the periphery and are tolerogenic to self-antigens named pTreg cells [20]. TCR activation with TGF-β or retinoic acid converts naive T-cells into iTreg cells [21, 22]. In addition, suboptimal costimulation and high levels of TGF-β, retinoic acid, and IL-2 favor FOXP3 induction [23, 24]. In humans, such interactions generate inflammatory cytokines rather than suppressing the immune system [25]. CD25+CD4+ Treg cells with inhibitory function display low amounts of CD127, the α-chain of the IL-7 receptor [26]. Naive T-cells, driven by TCR-mediated signaling, transiently upregulate FOXP3 and downregulate CD127, suggesting that the CD4+CD25+CD127low T-cell fraction may include activated non-Treg cells. Naive Treg cells multiply and mature into highly suppressive and terminally differentiated eTreg cells following TCR activation, which may hinder antigen-specific maturation of dendritic cells (DCs). eTreg cells hamper other effector cells via high affinity IL-2 receptor signaling, secreting inhibitory cytokines such IL-10, TGF-β, and IL-35, and cleaving ATP into adenosine. Antigen-specific Treg cells outperformed antigen-nonspecific Treg cells in a TCR transgenic animal model [27]. Antigen-presenting cells (APCs) in the lymph nodes prime naive T-cells (CD8+ T and CD4+ FOXP3 T-cells) and detect cognate antigens for activation in the TME [28]. Treg cells in the TME contain a TCR repertoire that is different from that of other conventional CD4+ T lymphocytes and is selective for tumor-specific antigens, indicating recognition of tumor-specific antigens by Treg cells [29].
Effector functions of Tregs in various pathologies
Tregs control the immune response in both antigen-dependent and antigen-independent ways via the immunosuppressive cytokines IL-10 and TGF-β [30]. IL-35, composed of Ebi3-IL-12 alpha heterodimer immunosuppressive cytokines, is constitutively expressed on Treg cells and hampers the effector T-cell immune response [31]. Treg cells with higher expression of IL-2R alpha (CD25) increase their affinity for IL-2, which deprives activated T-effector cells of IL-2 and prevents them from proliferating, leading to metabolic disruption and apoptosis [32]. IL-2-dependent downstream STAT-5 activation is essential for homeostasis and function in Treg cells. In addition, cAMP assists in Treg-mediated immunosuppression by injecting cAMp through gap junctions or catabolizing extracellular nucleotides into adenosine through ectonucleases. CD39 and CD73 present on Treg cells mediate the immunosuppression of T-effector cells [33, 34]. Contact-dependent production of the serine protease granzyme B by Tregs allows them to induce apoptosis in T-effector cells [35]. CTLA-4 expressed on Tregs competes with other effector T-cells for molecular interactions with CD80/86 on DCs. This interaction triggers the release of IFN-γ, which induces the production of indoleamine 2,3-dioxygenase (IDO), a potent enzyme that catabolizes the tryptophan to kynurenine metabolite that suppresses T-cell effector activity [36]. The expression of lymphocyte activation gene (LAG) on the Treg cell surface is structurally homologous to the CD4 receptor and has significantly higher affinity for MHC-II [37]. The LAG:MHC-II interaction involves the downstream recruitment of ERK and SHP-1, which inhibits the maturation and immune stimulatory capacity of DCs mediated through ITAMs [38]. Notably, the high expression of the PD-1 molecule (CD279) on exhausted T-cells and tumor-infiltrating immunosuppressive Treg cells quenches antitumor activity [39]. PD-L1 expression on tumor and Treg cells sends inhibitory signals that help the tumor escapes the immune system [40].
Regulatory T-cells and TME
Tregs are located throughout the TME and act as metabolic and trafficking ‘barriers’ to keep effector cells out of the TME or reduce the activity of effector cells already present in the TME, resulting in a decrease in CD8+ T-cells in the proximity of tumor cells, which is linked to poor outcomes [41]. Poor peripheral T activation, disorganized vasculature, lymphatic structures in the TME, and the occurrence of a stroma prevent cell migration into and in the vicinity of the tumor bed, constituting primary barriers constructed by Tregs that hinder the infiltration of proinflammatory cells (Fig. 2) [42, 43].

Overview of the tumor microenvironment and associated cell population of Tregs
Cancer cells have an inherent genetic instability to produce abnormal proteins called neoantigens that spontaneously trigger CD8+ T-cell responses. Immune surveillance eradicates cancer cells that display these highly immunogenic neoantigens [44]. The prevalence of a large number of Tregs and a lower ratio of CD8+ T lymphocytes to Tregs in the TME are both related to a poor prognosis in a variety of cancer types [45]. There is a correlation between good prognosis and the increase in FOXP3+ non-Treg cells in the TME of a type of colorectal tumor, including high levels of TGF-β and IL-12. In contrast, a significant number of eTreg cells in the TME are linked to poor prognosis in other forms of cancer in which FOXP3+ non-Treg cells are virtually never found [46]. The increase in Treg cell number in the TME occurs mainly through chemoattraction [47]. Treg cells tend to cross talk with infiltrated immune cells, stromal cells, and tumor cells [48, 49]. IL-10 also plays a central role in the induction of Tregs in the TME area and in the periphery, as validated by isolation of IL-10 mRNA transcripts from various tumor types, and the major contributors are tumor cells and infiltrated leukocytes [50]. TGF-β is another cytokine that induces naive T-cells into Treg cells that promote cancer progression and dampen effective immune cytokines [51]. The elevated level of TGF-β in the TME correlates with advanced cancer progression and with poor prognosis [52]. TGF-β, IL-2, and prostaglandin E2 (PGE2) are abundant in the TME area and are crucial in converting Th17 cells into Tregs. Various cytokine gradients established by both tumor and immune cells serve as driving forces for nTreg cell entry into the TME. The use of a systemic CCR5 antagonist slows tumor development, improves survival, and reduces infiltrating Tregs in the TME [53]. Treg cells are migrated to the TME area in response to CXC and CC chemokines, and the chemokine receptors CXCR3, CCR4, CCR8, and CCR10 are responsible for this migration. Treg cells, as opposed to effector and memory T lymphocytes, are better able to detect the vast majority of tumor-associated self-antigens that are released by dying tumor cells in the TME, resulting in activation and proliferation of Tregs, and creating an immunosuppressive TME [54]. In addition, the inducible T-cell costimulatory ICOS, which belongs to the CTLA-4/PD-1/CD28 family, is also expressed in regulatory T-cells. It plays a role in the activation and proliferation of Tregs by binding to an ICOS ligand that is expressed on plasmacytoid DCs [55, 56]. Treg cells inhibit the activation of various other immune cells directly by engaging with immunosuppressive receptors or indirectly by various soluble molecules (Fig. 3).

Treg suppression. Tregs repress many cell types directly and indirectly. Treg cytokines influence T-cells (IL-10, IL-35, and TGF-ß). Perforin and granzyme damage target cell membranes and trigger apoptosis. Strong CD25 expression sequesters IL-2 from the microenvironment, reducing effector T-cell growth. IL-2 deficiency impairs NK cell proliferation and effector function. TGF-ß from Tregs directly affects NK cells. Tregs directly affect B-cells and DCs through PD-L1/PD-1 and CTLA-4/LAG-3. CTLA-4 blocks costimulation to downregulate CD80/CD86 and upregulate IDO. CD39 expression. CD39 on Tregs converts ATP to adenosine and AMP to suppress T-effector proliferation. Tregs may turn monocytes into M2 macrophages instead of proinflammatory M1 macrophages
14th The TME plays an important role in anticancer immunity including effect in immunotherapy and other treatments [57, 58]. Cancer cells and its interaction with the extracellular matrix and stromal cells influence TME, forming a heterogeneous environment which fosters chronic inflammation, immune suppression, and angiogenesis [59, 60]. Avoidance of immune-suppressive networks in the TME is a major factor for the elimination of tumor cells which can be achieved by targeting and reprogramming the TME via enhancing the T-cell activity which likely reduces the immunosuppression [61, 62]. Studies on TME composition and its role on immune surveillance attenuation may also guide the development of strategies to manipulate the TME and benefit cancer patients [63]. The role of immune cell involvement and major transcription factors is also critical for developing therapies that target inefficient T-cells within the TME. TME-derived cannabinoid receptors (CB1 and CB2) in immune cells residing in TME aggravate the non-small cell lung cancer and compositions of the immune cell population. Inhibition of these receptors favors the occurrence of tumor-killing lymphocytes. The absence of CB1 and CB2 significantly improved the outcome of immunotherapy indicating the importance of TME [64]. Ni et al. reported that signal pathways and immunosuppressive subtypes of cells in TME may provide new avenues for improvement in personalized immunotherapy against glioblastoma progression [65]. Aru et al. showed that dual inhibition of the CXCL12-CXCR4 and PD-1-PD-L1 axes alleviates the immunosuppressive TME in acute myeloid leukemia (AML). This also includes the role of checkpoint blockade in the bone marrow microenvironment of the bone marrow of patients [66]. Aberrant metabolic processes like dysregulated amino acid metabolism impaired the function of cytotoxic CD8+ T-cells in the immunosuppressive microenvironment in multiple myeloma [67]. Immunomodulatory drugs could alter TME and may improve the treatment outcome in hematological malignancies including AML, CML, MM, and B-cell neoplasias. This could be achieved via immune checkpoint inhibitors or metabolic interventions [68].
Treg migration in TME
Treg cells have been shown to infiltrate the tumor microenvironment (TME) in a number of murine and human cancers (Fig. 4) [45]. iTregs are generated from naive CD4+ T-cells in the periphery in response to tumor cues that induce differentiation toward a regulatory phenotype. The production of iTregs in the TME is largely dependent on IL-10 activity. The TME produces IL-10 from a number of different sources, including the tumor cells themselves, as well as invading leukocytes such as T- and B-cells, macrophages, and NK cells [69]. In an autocrine fashion, greater amounts of TGF-ß are secreted by tumors as their growth progresses [70]. In addition, the thick stromal network that surrounds the tumor is also a source of this cytokine [71]. The synergistic effect of TGF-ß and IL-10 on the development of human iTregs is shown by the upregulated expression of both FOXP3 and CTLA-4 [72]. The differentiation of iTregs in the TME is related to overall worse patient survival in cancer subtypes. Induction of iTregs in the TME is responsible for driving tumor development.

Induction of naive T-cells in the TME area into Treg cells and chemokine-mediated recruitment of Treg cells in the TdLN and TME
Targeting Tregs: relevance in tumor immunotherapy
The tumors are immunologically divided into inflamed and noninflamed types based on the activation status of infiltrated immune cells. In general, immunotherapies work better against inflamed tumors than noninflamed tumors. FOXP3 in Tregs is critical for immune homeostasis and immunological self-tolerance. These cells are targeted either by depleting or weakening their suppressive abilities to generate an effective antitumor immune response [73]. Various immunotherapies have been designed to target intratumoral Treg cells, including depleting and targeting key surface molecules that are highly expressed and impart immunosuppressive functions. Targeting the molecules PD-1, Tim-3, CD25, CTLA-4, OX40, TIGIT, 4-1BB, CCR4, CCR8, etc., has had some clinical success. Recently, our laboratory showed that galunisertib (inhibitor for TGF-βR1) improves the effectiveness of immunotherapy using IL-15-activated dendritic cells by reducing the regulatory T-cell population in addition to ablation of p-SMAD2 and neuropilin-1, leading to significantly better survival in a murine model of lymphoma [74]. Importantly, a first study showed that eliminating Treg cells caused tumors to shrink in some tumor cell lines (CD25 positive), such as Meth A (methylcholanthrene-induced fibrosarcoma) and RLmale 1 (radiation-induced leukemia with CD25 marker), but not in others (CD25 negative), such as AKSL2 and RLfemale 8 (radiation-induced leukemia lacking CD25) [75]. As a result, treatment aimed against Treg cells is unlikely to be successful against all cancers. A phase I clinical study of Treg depletion in patients with solid tumors by the infusion of anti-CCR4 monoclonal antibody (mAb) resulted in enhanced antitumor immune responses in many patients, although clinical responses were not detected in the majority of patients [76]. Systemic Treg cell reduction may enhance immune-related adverse events (irAEs), such as autoimmunity-related toxicities. Instead of systemic depletion of Treg cells, specific TME eTreg cell depletion may boost anticancer effects without irAEs. Thus, we must define the TME immune-suppressive network to develop biomarkers that discriminate tumors in which Treg cells are critical for tumor progression. By changing the cytokine and/or chemokine axis, cell-intrinsic signaling, or metabolites generated in the TME area, Treg cells might alter their activity and invasion. IDO and adenosine in the TME strongly alter Treg cell activity and lineage stability. Fatty acid metabolism boosts Treg cell growth. Cancer cells use glucose and increase TME lactic and fatty acids via glycolytic metabolism. FOXP3 decreases glycolysis and increases nicotinamide adenine dinucleotide oxidation by reducing Myc [71]. Treg cells use oxidative phosphorylation and β-oxidation of fatty acids to absorb lactic acid and fatty acids for survival and immunological suppression in the TME. PI3K modulation also affects FOXP3 [77]. These signaling pathways and the metabolites associated with Treg cells may be therapeutic targets in cancer patients, although further research is needed for better understanding.
2nd The control of Treg stability, plasticity, and functions is important to guide the development of novel therapies to treat human disease. Regulatory T-cells (CD4+CD25highCD127−FOXP3+, Tregs) are fundamentally critical for maintaining immune homeostasis by modulating the immune response against self-antigens, allergens, pathogens, and tumors. Tregs are dynamic population with a higher degree of plasticity than previously thought and have a far broader role in disease mediated by their interaction with several immune and non-immune cells. Treg program is a fairly stable homeostatic condition that includes Foxp3 expression and an occurrence of Treg-specific epigenetic signature, acquired during Treg development in the thymus [78]. Stable and long-term Foxp3 expression in Tregs is critical for Treg function and is partly controlled by the demethylation of Treg-specific epigenetic signature genes, including Foxp3 [79]. The plasticity of Tregs triggers diverse changes including the generation of inflammatory cytokines and instability including FOXP3 expression and may associate with diseases including autoimmune, allergic, and infectious diseases [80]. Treg plasticity and instability episodes are controlled by intrinsic signaling pathways such as the PI3K/AKT pathways [4, 5]. The TME is responsible for the phenotypes and functionality of specific infiltrating immune cells [81] Toor SM et al. characterize Tregs, infiltrating the tumor tissue in colorectal cancer patients, and compare them to normal colon tissues and peripheral blood. There is an enrichment of highly suppressive Tregs (Foxp3+Helios+) in the tumor microenvironment, which upregulate a number of inhibitory receptors including CTLA-4, TIM-3, PD-1, and LAG-3[82].
Immune checkpoint inhibitors targeting Treg cells
Targeting immune checkpoint (IC) molecules in Tregs, including CTLA-4, TIGIT, PD-1, and GITR, may offer a cure response toward cancer [83]. CTLA-4 maintains Treg suppression by binding to its ligands CD80 and CD86 on APCs with great affinity. Thus, anti-CTLA-4 antibodies may inhibit Tregs and activate effector cells [84]. Ipilimumab and tremelimumab (TremeIgG1), which inhibit CTLA-4, have shown persistent therapeutic efficacy in a group of advanced solid cancer patients by enhancing effector T-cell-mediated immune responses. In mice, Fc-dependent anti-CTLA-4 mAbs preferentially deplete intratumoral FOXP3+ Tregs. Ipilimumab and tremelimumab boost intratumoral CD4+ and CD8+ cells without decreasing TME FOXP3 + cells. Anti-CTLA-4 immunotherapy does not deplete FOXP3+ cells in human tumors, suggesting that altering the Fc sections of the mAbs to promote Fc-mediated intratumoral Treg depletion might improve effectiveness [85]. In vivo, ipilimumab and tremelimumab-like antibodies deplete intratumoral Tregs, raising the CD8/Treg ratio and encouraging tumor rejection [86, 87]. Clinically effective anti-CTLA-4 mAb rejects tumors through host Fc receptor-dependent pathways [88]. Anti-CTLA-4 binding to FcRs depleted intratumoral Tregs and activated and degranulated intratumoral NK cells. Anti-CTLA-4 and IL15/IL15Rα complexes together improved tumor suppression [89]. Fc-region-modified anti-CTLA-4 mAb with significant ADCC and ADCP activity selectively depleted CTLA-4+ FOXP3 + Tregs and increased tumor antigen-specific CD8+ T-cells. ADCC may not work for colorectal, liver, prostate, and ovarian cancer [90, 91]. The first FDA-approved immune checkpoint blocker (ICB) for metastatic melanomas was ipilimumab [92]. Anti-CTLA-4 antibody treatment may generate severe irAEs, limiting its therapeutic advantages despite patient survival. A lower dose of anti-CTLA-4 antibodies and anti-PD-1 combination therapy may produce significant irAEs in many individuals. If irAEs can be managed, targeting CTLA-4 provides long-term benefits for cancer immunotherapy [93, 94]. Zhang et al. observed that while irAE-prone ipilimumab and tremelimumab quickly guide cell surface CTLA-4 toward lysosomal destruction, their nonirAE-prone antibodies, HL12 or HL32, detach from CTLA-4 after endocytosis and enable LRBA-dependent CTLA-4 recycling to the cell surface. Increasing tremelimumab pH sensitivity with planned tyrosine-to-histidine mutations inhibits antibody-triggered lysosomal CTLA-4 downregulation and significantly reduces irAEs.
6th Targeting immune check point molecule like PD-1, PD-L1, and CTLA-4 in cancer also increased immune-related adverse events (irAEs). This typically displayed delayed with prolonged duration and frequently involves the occurrence of low grade and treatable; however, adverse effects can be severe leading to permanent disorders. irAE involved and correlated with treatment outcome with ICI in certain malignancies. However, only limited data or consensus on guidelines for the management of irAE are available [95]. We have already included a discussion on the topic in the original submission. There are no specific irAEs that are involved in our approach of therapy and expect it to be similar to others proposed including its management [96].
pH-sensitive anti-CTLA-4 antibodies, which avoid CTLA-4 downregulation and have higher bioavailability, are more effective in depleting intratumoral Tregs and rejecting large established tumors [97]. Intratumoral injections of Pam3CSK4 and anti-CTLA-4 mAb improved antitumor immune responses compared to anti-CTLA-4 alone, and its effectiveness relied on CD4 and CD8 T-cells, FcR IV, and macrophages. The TLR1/2 ligand boosted macrophage FcR IV expression, which depleted Tregs in melanoma and improved the effectiveness of anti-CTLA-4 mAbs in combination therapy [98]. In preclinical HPV+ oral cancer models, targeting interferon signaling and CTLA-4 improves anti-PD-1 immunotherapy [99]. ATOR-1015 targets CTLA-4 and OX40 concurrently to increase immune activation in tumors, making it a possible next-generation CTLA-4 targeting object. ATOR-1015 may synergize with anti-PD-1/PD-L1 treatment [100]. Pharmacologically altering T-cell EZH2 expression may enhance anti-CTLA-4-induced antitumor responses [101]. TIGIT signaling controls the Treg phenotype and reduces antitumor immunity through Tregs but not CD8+ T-cells. TIM-3 and TIGIT also suppress antitumor immune responses [102]. Androgen deprivation treatment (ADT) and TI-Treg depletion using an anti-CTLA-4 antibody delayed castration resistance and extended the longevity of certain tumor-bearing animals [103]. In advanced HCC, checkpoint inhibitor-positive Tregs were negatively linked with age and produced more IL-10 and IL-35. Tregs suppressed CD8+ T-cell IFN-γ and cytotoxicity in HCC patients, partly attenuated by neutralizing PD-1 and PD-L1 antibodies [104]. In HPD patients, anti-PD-1 mAb therapy significantly boosted tumor-infiltrating proliferative eTregs in GC tissue samples. Compared to PD-1 eTregs, circulating and tumor-infiltrating PD-1 + eTregs are highly active and express CTLA-4 [105]. Anti-GITR mAbs deplete and destabilize TI-Tregs; residual Tregs have less suppressive behavior, and a highly active fraction of CD44hi ICOShi TI-Tregs is selectively eliminated. Intratumoral CD8+ T-cells became more functional by downregulating PD-1 and LAG-3 due to Treg alterations. Agonistic GITR signaling and Treg depletion reversed CD8+ T-cell exhaustion [106]. In vitro, a STAT3 inhibitor reduced T-cell IL-10 production, increased suppressive activity, and decreased Tregs [107]. STAT3-FOXP3 complexes expand FOXP3c transcriptional activity to additional STAT3 target genes without FOXP3-binding sites [108]. In glioblastoma, upregulated PD-L1 expands Tregs, which may sustain immunosuppressive Tregs that shorten patient survival. PD-L1/PD-1 inhibition may diminish Treg growth and increase T-cell activity [109]. Antitumor effectiveness requires strict Treg depletion and checkpoint inhibition [110]. Anti-PD-1 immunotherapy downregulates TIM-3 on HNSCC-isolated Tregs in rats and mice, reversing their suppressive activity [111]. In mice treated with radiation (RT) and dual-ICB, targeted Treg depletion improved antitumor immunity, tumor rejection, and immunologic memory [112]. Anti-TIM-3 mAb reduced Tregs in transgenic HNSCC mice with increased IFN-γ+ CD8+ T lymphocytes and augmented antitumor immunity [113]. Anti-ICOS/ICOSL antibodies eliminate Tregs in follicular B-cell lymphoma [114, 115].
By inhibiting CD28 and TCR signals and making effector T-cells useless, PD-1 prevents excessive activation of T lymphocytes that are responsible for immune responses (so-called exhausted T-cells) [116]. Because the expression of PD-1 on Treg cells is comparable to that on effector T-cells, specifically in the TME area, and because Treg cells are dependent on TCR and costimulatory CD28 signals for both their survival and their function, blocking PD-1 may activate the suppressive functions of Tregs. PD-1-deficient Treg cells exhibit powerful immunosuppressive activity and are able to rescue autoimmune abnormalities [117]. In certain patients with gastric cancer, treatment with anti-PD-1 enhances the immune-suppressive activity mediated by Treg cells, which leads to hyperprogression [105]. However, another study found that PD-1-blocked Treg cells have limited immune-suppressive function, suggesting that the roles of PD-1 in effector T lymphocytes and Treg cells depend on the type of cancer [118]. An overview of various target receptors and enzymes expressed on Tregs that can be harnessed to induce effective antitumor immunity is shown in Fig. 5.

Key approaches and target enzymes and receptors expressed on Tregs: Potential application in the induction of antitumor immunity
Depletion of Treg cells: prospects in cancer immunotherapy
Numerous cell surface markers, including CD25, CTLA-4, PD-1, ICOS, OX40, GITR, CD15s [119], CCR4, and CCR8, are expressed by Tregs in the TME zone and may be exploited to eliminate Treg cells.
CD25
A number of studies have investigated the possibility of depleting Treg cells by targeting CD25 with antibodies or a recombinant protein that is composed of IL-2, and the active domain belongs to diphtheria toxin [120]. In a clinical trial, Treg cell depletion following anti-CD25 mAb treatment was evaluated with a median progression-free survival of 4.8 months in breast cancer patients treated with an anti-CD25-depleting mAb called daclizumab [121]. In another study, daclizumab eliminated both effector T lymphocytes and Treg cells, but there was no evidence of an anticancer immune response or antibody generation. Since CD25 expression is upregulated after the activation of effector T lymphocytes, CD25-targeted Treg cell depletion could be accompanied by a reduction in effector cells, indicating that there is a limited scope for CD25-targeted Treg cell depletion for antitumor immunity.
Cytotoxic T lymphocyte antigen-4
After activation, CTLA-4 is expressed more strongly by CD4+ and CD8+ effector T-cells; then, it is by FOXP3+CD4+ Treg cells, which express it constitutively. Initially, it was believed that anti-CTLA-4 mAb counteracted an inhibitory signal present on activated CD4+ and CD8+ effector T-cells, reviving those cells and allowing them to restore their ability to fight tumors [122]. However, a recent study indicated that the tumoricidal effects of anti-CTLA-4 mAb are dependent on the depletion of CTLA-4-expressing Treg cells in the TME through ADCC. The tumoricidal effect of anti-CTLA-4 mAb is completely abrogated when Fc function is depleted [123, 124]. Therefore, increased antitumor effects caused by anti-CTLA-4 mAb primarily caused suppression of the function of Tregs and their elimination in the TME. Further study on the roles of CTLA-4 in effector T lymphocytes and Treg cells in cancer patients is essential.
OX40 and GITR
OX40 [125], GITR [126], and LAG-3 [127], which are mainly expressed by T-regulatory cells, are also possible choices for the depletion and functional modification of Treg cells. GITR is a costimulatory molecule that is produced at low levels by resting CD4+ and CD8+ T lymphocytes and at high levels constitutively by FOXP3+CD4+ Treg cells. Upon activation of GITR either with an agonistic anti-GITR mAb or GITR ligands, the suppressive action of FOXP3+CD4+ Treg cells is inhibited, and effector T-cells become resistant to the suppression mediated by FOXP3+CD4+ Treg cells [128]. OX40 (a TNF superfamily member) expression occurs momentarily but persistently by the activation of effector T-cells, while FOXP3+CD4+ Tregs express OX40 in a constitutive manner. Agonistic anti-OX40 mAb attenuates the immune suppression caused by FOXP3+ CD4+ Treg cells and activates effector T-cells [125]. Clinical studies are now being conducted to investigate the effectiveness of potential treatments that target GITR and OX40 [129]. Folate receptor 4 (FR4) is more common in rodent Tregs, and folate influx depends on it. FR4 expression increases many times in FOXP3+ Tregs [130], indicating that FR4 could be a good target for Treg depletion. Folate is needed for other cells to grow and divide, which may limit the use of antibodies against FR4.
Chemokines and chemokine receptors
Inhibiting the CCL28-CCR10, CCL1-CCR8, and CCL22-CCR4 pathways may reduce the formation of Treg cells in the TME area, which in turn boosts immune responses against the tumor. Hypoxia in the TME promotes the production of CCL28, which in turn chemoattracts CCR10+ Treg cells. Intratumoral delivery of an anti-CCR10 immunotoxin elevates the immune response in a mouse model of cancer by preventing the interaction between CCL28 and CCR10 and decreases the number of Treg cells in the TME [131]. After elimination of CCR4 + eTreg cells in melanoma patients, enhanced tumor antigen (NY-ESO-1)-specific CD4+ and CD8+ T-cell responses were found [132]. Anti-CCR4 mAb is essential in activating and boosting antitumor immunity in cancer patients, and a phase Ia/Ib multicenter clinical study has been carried out [76]. In patients with advanced disease or recurring solid tumors, the administration of an anti-CCR4 mAb (mogamulizumab) results in a considerable reduction in the number of eTreg cells in the peripheral blood. In addition, humoral immunity against NY-ESO-1 and XAGE1 antigens was detected in 3 of 5 and 5 of 5 patients, respectively. These findings were obtained in patients who had previously been diagnosed with NY-ESO-1 or XAGE1 antigens [76]. Myeloid cells that express CD11b and CD14 and produce CCL1 are engaged in the process of Treg cell invasion [133]. Treg and NKT cells, especially in cancer patients, have been shown to have elevated expression of CCR8, which is a receptor for CCL1. CCL1 and CCR8 mutual interaction via STAT3 increases the expression of FOXP3, and activated CCR8+ Treg cells effectively decrease antitumor immunity by boosting ATP-adenosine metabolism by CD39 and the production of granzyme B and IL-10 [134]. Patients with higher infiltration of CCR8+ FOXP3+ Treg cells have a considerably worse overall survival rate than those who have low infiltration [135].
Targeting checkpoint receptors on Treg cells
Treg cells regulate the immunosuppressive response by secreting cytokines such as TGF-β, IL-6, IL-10, and IL-35 or coinhibitory receptors such as CTLA-4, PD-1, LAG-3, Tim-3, and Nrp-1 upon contact with pathogenic immunotypes at the site of inflammation [118]. Many patients do not react to immunotherapies targeting CTLA-4 and PD-L1/PD-1 because additional coinhibitory receptors (Tim-3, LAG-3, and TIGIT) expressed on tumor-infiltrating lymphocytes (TILs) hamper effective immunotherapy [136]. The expression of Tim-3 in T-regulatory cells was also promoted by PD-1 blockade with mAbs, resulting in the failure of anti-PD-1/PD-L1 immunological treatment [136]. Targeting the inhibitory receptors CLTA-4 [137], TIGIT [138], and LAG-3 [139] compromises immunosuppressive Treg cell functions or populations. In clinical trials, inhibitors targeting the coinhibitory receptors had only minimal anticancer benefits when delivered alone, but in combination, they dramatically increased the antitumor impact [140].
Protein 1 associated with Programmed Cell Death (PD-1 Inhibitors)
The immunoinhibitory transmembrane protein PD-1, which is a member of the CD28 Ig superfamily, is an essential component for immune evasion by tumors. PD-1 is expressed on activated T- and B-cells, NKT cells, tumor-infiltrating lymphocytes, and subsets of dendritic cells. Tumor-infiltrating T lymphocytes in cancer patients have high quantities of PD-1. FDA-approved pembrolizumab and nivolumab can limit the activity of Treg cells by disrupting the interaction between PD-1 and PD-L1 and boost antitumor activity. Ribas et al. [135] reported that there was no change in the percentage of circulating Treg cells in metastatic melanoma patients who were treated with pembrolizumab. A recent preclinical osteosarcoma animal model study noted a decrease in the number of tumor-infiltrating Treg cells following anti-PD-1 therapy [141]. A counterintuitive rise in the number of circulating Treg cells was observed in patients with resected high-risk melanoma who were treated with nivolumab with a reduction in Treg-suppressive ability [107]. In hyperprogressive advanced gastric cancer patients, a considerable rise in the circulating and intratumoral levels of PD-1+ Treg cells inhibits antitumor immunity in such individuals [105]. Anti-PD-1-mediated stimulation of the TGF-β eta/Smad3 pathway might enhance the induction of Treg cells and immunosuppression [142]. Therefore, limiting TGF-β in conjunction with blocking PD-1 is a potential strategy to reduce tumor-infiltrating Treg cells to enhance infiltration of effector T lymphocytes, causing cytotoxic death of the tumor [143]. In addition, a combination therapy consisting of a PD-1 inhibitor and a CTLA-4 inhibitor has been shown to be more effective than monotherapy against melanoma and non-small cell lung cancer, with the ability to decrease Tregs and increase CD8+ effector T-cells in the TME [144,145,146]. Combining cell therapy with doxorubicin successfully downregulates the Treg cell population and PD-1 expression in treated animals [147]. Combining a PD-1 inhibitor with an FC-optimized CD25 inhibitor successfully suppressed existing tumors in preclinical models of sarcoma, colon cancer, and melanoma [148]. Studies are now being conducted to make headway toward the discovery of prospective biomarkers and the clinical response of PD-1 blockade in relation to Treg cells [149].
T-cell immunoglobulin and mucin-domain-containing 3
Tim-3 (CD366) is linked to T-cell exhaustion in the TME [150]. Tim-3+ FOXP3+ Treg cells have a higher expression of IL-10, CD39, CD73, and TGF-β, as well as higher levels of other checkpoint receptors, such as CTLA-4, LAG-3, and PD-1 [151]. High expression of Tim-3 in Treg cells and CD8+ T-cells is related to a worse prognosis in individuals with colon cancer [152]. In a mouse model of transgenic head and neck squamous cell cancer, treatment with anti-Tim-3 monoclonal antibodies led to a reduction in Tregs and an increase in the production of interferon alpha by CD8+ T-cells [113]. However, anti-Tim-3 mAb monotherapy had very little or no anticancer efficacy in another mouse tumor model [153]. Tim-3 signaling plays a supporting or amplifying role in the regulation of the immune response. Patients with non-small cell lung cancer (NSCLC) who receive PD-1 inhibition had increased expression of Tim-3 and TIGIT on their CD8+ T-cells [136]. In mice carrying CT26 tumors, inhibiting pathways associated with Tim-3, TIGIT, and PD-1 had a greater antitumor impact than any monotherapy. The combination of an anti-Tim-3 antibody (TSR-022) and an anti-PD-1 monoclonal antibody is now being evaluated in clinical studies in patients with advanced solid malignancies.
5th Several clinical studies indicates the role of Tregs in immune check point inhibitor (ICI)-based immunotherapy against various types of cancer. In patients with non-small-scale lung cancer, gastric cancer, and malignant, melanoma effect of anti-PD-1 antibodies (nivolumab or pembrolizumab) or anti-PD-L1 antibody atezolizumab was evaluated [154]. Nonresponsive patients showed higher PD-1 on Tregs with elevated tumor-infiltrating PD-1+CD8+ T-cells, indicating potential biomarker potential of PD-1 + Tregs and its importance in the clinical efficacy of ICI in lung cancer therapy [155]. In another study, pembrolizumab or nivolumab treatment in NSCLC patients downregulate the tumor-infiltrating PD-L1hi Tregs following PD-1 inhibition with improved clinical outcome compared to patients with low frequency of tumor-infiltrating PD-L1hi Tregs [156].
Koh J et al. reported that following ICI-based immunotherapy (anti-PD-1 administration) in NSCLC patients results in improved response including longer progression-free and overall survival with reduced peripheral Tregs and inhibition in elevated levels of TGF-β [157]. These results also indicate that increased peripheral Tregs and elevated TGF-β may work as a biomarker for the clinical outcome of the disease.
Tregs and checkpoint inhibitors: role of reactive oxygen species (ROS)
ROS play a crucial role in many cellular activities, but excessive levels can cause cellular damage and oxidative stress [158]. To preserve optimum health, it is critical to maintain a balance between ROS generation and neutralization [159]. Extreme apoptosis occurs in Treg cells when they are exposed to the high levels of oxidative stress seen in the tumor microenvironment compared to the comparatively lower levels seen in effector T-cells [160]. In addition, increased synthesis of the antioxidant thioredoxin-1 (Trx-1) in naturally occurring Tregs is associated with increased resistance to oxidative stress [161]. There is evidence that NOX2-derived ROS contribute to the regulation of Treg formation, as NOX2-/- mice were shown to have fewer peripheral CD4+CD25+ Tregs and lower FOXP3 expression [162].
The direct suppression of CD4+ effector T lymphocytes by Tregs was found to be inhibited or decreased when ROS were abolished by antioxidants or NADPH oxidase inhibitors [163]. In addition, ROS have the potential to stimulate the generation and development of Treg cells [164]. Metformin reduces mitochondrial ROS, prevents the maturation of immature CD4+ T-cells into Tregs, and suppresses FOXP3 expression, resulting in a lower number of tumor-infiltrating Treg cells [165]. TCR signaling may create ROS that can specifically block the degradation of SENP3 (a positive regulator of Tregs) and, thus, help Tregs maintain immunosuppressive functions [166]. SENP3 expression may be inhibited by treatment with N-acetyl cysteine (NAC), which alters the stability of Treg cells, resulting in an improved immune response to the tumor. In comparison to effector CD4+ T-cells, Tregs are less likely to perish as a result of oxidative stress, likely due to greater expression levels of antioxidative enzymes in Tregs (Fig. 6) [167].

Overview of ROS activity and Treg functions in cancer immunotherapy
Following T-cell receptor activation, Ca2+ is released, which increases mitochondrial functions, such as the tricarboxylic acid (TCA) cycle and the production of ROS [168]. Inhibition of PD-1 signaling showed mitochondrial activities, including ROS production [169]. Blocking PD-1 also activates AMPK and mTOR, which in turn upregulates the expression of peroxisome proliferator-activated receptor (PPAR)-gamma coactivator 1 (PGC-1), resulting in increased mitochondrial activity and inhibiting tumor progression [169]. ROS have a considerable impact on PD-1 and PD-L1 expression. However, the mechanism of communication and crosstalk between ROS and PD-L1 is not completely understood. It has also been suggested that an elevated amount of ROS in tumor cells is involved in EMT, which plays an essential role in the production of PD-L1 in neoplastic cells [170]. With regulated ROS production and optimum therapeutic advantages, a combination regimen of anti-PD-L1 plus photodynamic or sonodynamic therapy could be an appealing choice for the treatment of extremely resistant and advanced neoplasias, such as pancreatic tumors. We have recently shown that a tiny biomolecular drug combination between 5-FU and bilirubin induced extremely significant ROS production in highly metastatic Dalton's lymphoma (DL) [171]. Enhanced Tim-3 expression in a subpopulation of hepatic macrophages of mice with a diet deficient in methionine and choline alleviates liver injury by regulating ROS generation and secretion of proinflammatory cytokines from the macrophages. This indicates that Tim-3 has a protective function against liver damage by controlling the activation of macrophages. The activation of the Nod-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) inflammasome was caused by the conditional deletion of Tim-3 in dendritic cells, which led to an enhanced build-up of ROS.
Implications of Treg cell heterogeneity in the TME for the effective Immunotherapy
10th Tregs are a heterogeneous group of population and the biology underlying specific subpopulations is important for selectively regulating the immune response while maintaining autotolerance. This is critical to understand different roles of Tregs in various diseases. The contradiction exists with reference to the general autoimmune disease and tumor pathogenesis theories. While cancers are triggered by excessive Treg activity, the autoimmune processes are due to low Tregs activity. Both cancers and autoimmune pathologies may well be concurrent. Tregs are selective to various types of immune responses which depends on antigenic stimuli and the spatial localization of the process. Thus, it may be assumed that pathology is caused by the failure of a specific Treg cluster that features a common TCR repertoire of similar antigenic specificity. TCR plays role in maintaining the homeostasis, or activation of Tregs, and also in the phenotypic selection due to the antigenic specificity of TCR that determines Tregs regulated immune response. This was documented by the differences in location and transcription profiles of Tregs that differ in TCR specificity [172, 173]. This also indicates a clonal organization of the Treg pool based on the antigenic specificity of TCR in the same clone. Experimental results indirectly support this hypothesis with reference to the in vivo application of Tregs with a chimeric antigen receptor (CAR) for the treatment of autoimmune diseases and in transplantation. These CAR Tregs are specifically migrated to the target sites and demonstrated significant antigen-specific suppressive activity indicating the occurrence of Treg clones with specific antigenic determinants of impaired tolerance [174]. Evidences also indicate the occurrence and expansion of antigen-specific tumor Treg clones in the TME. Selective suppression of such cells may provide a better therapeutic advantage compared to using monoclonal antibodies against a general pool of effector Tregs such as PD-1/PD-L1 or CTLA-4 [29].
Conclusion and future perspectives
Depletion or inhibition of Treg activity is considered a potential strategy for immunotherapy against various forms of cancer. Nevertheless, futuristic approaches need to be considered for tackling more serious and unknown threats. The following points are important to note.
-
1.
Combination of checkpoint therapy with chimeric antigen receptor-T cells (CAR-T), TCR-T cells, or vaccines.
-
2.
Maximization of combination therapy with different immune checkpoints may also minimize adverse effects.
-
3.
Exploring new target molecules and immune cells, optimizing dosing regimens, and combination therapies. Validation of monitoring system and identification of biomarkers predicting clinical responses and toxicities.
-
4.
Require specific molecular markers for Treg cells and to identify specific Treg targets and Treg-specific drug delivery strategies for Treg cell depletion and/or suppression.
-
5.
Metabolic and immunological profiling of Tregs in the TME areas based on gene alteration and precision immunotherapy against the disease exacerbates. Whole-genome sequencing and epigenetic analysis may assist in patient selection and individualized treatment with checkpoint inhibitors as backbone therapy. Sequential tumor biopsy analysis and peripheral blood sampling to detect novel, heterogeneous, and targetable resistance mechanisms.
-
6.
Novel biomarker selection for selecting patients and individualized treatments. Avoidance of weak immune response providers such as TIGIT, TIM-3, and LAG-3. Anti-PD-1 was combined with anti-TIGIT, anti-TIM-3, or anti-LAG-3 to avoid intolerable adverse events.
-
7.
Generation of the Treg egress mechanism in the TME to prevent homing of Tregs to lymph nodes and the immunosuppressive environment propagated by APCs. Application of a new generation of anti-CTLA-4 antibodies to deplete Tregs in the TME and prevent tumor cell adaptation, survival, proliferation and capacity to metastasis outside the cancer tissue.
-
8.
Understanding the mechanisms of Treg recruitment, differentiation, expansion, and immune suppression without causing severe adverse immune response including the onset of autoimmune reactions. A delicate balance between immunosuppression and immune surveillance is important in cancer.
-
9.
Biomarker analysis for patient-specific immunosuppression, expression of neoantigens, and antigen-specific T-cell responses relative to the expression of checkpoint receptors on effector cells. Serum lactate dehydrogenase is a critical parameter for the selection of novel checkpoint combinations against neoplasias.
-
10.
Fabrication of novel strategies such as chemokine-targeted polymeric nanoparticles or inorganic stealthy nanoparticles targeting checkpoint inhibitors such as PD-L1 and CTLA-4.
Data Availability
Not applicable.
Abbreviations
- APC:
-
Antigen-presenting cell
- CCL2:
-
Chemokine (C–C motif) ligand 2
- CCl7:
-
Chemokine (C–C motif) ligand 7
- CCR4:
-
C–C chemokine receptor type 4
- CCR4:
-
C–C chemokine receptor type 4
- CD25:
-
Interleukin-2 receptor alpha chain
- CD7:
-
Cluster of differentiation 7
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- CXCL12:
-
C-X-C motif chemokine ligand 12
- FR4:
-
Folate receptor 4
- GITR:
-
Glucocorticoid-induced tumor necrosis factor receptor-related protein
- ICOS:
-
Inducible T-cell COStimulator
- IL-10:
-
Interleukin 10
- IL-2:
-
Interleukin-2
- IL-35:
-
Interleukin 35
- LAG-3:
-
Lymphocyte-Activation Gene 3
- PD-1:
-
Programmed cell death protein-1
- PD-L1:
-
Programmed death ligand-1
- PGE2:
-
Prostaglandin E2
- TdLNs:
-
Tumor-draining lymph node
- TGF-β :
-
Transforming growth factor beta
- TIGIT:
-
T-cell immunoreceptor with immunoglobulin and ITIM domains
- TIM-3:
-
T-cell immunoglobulin mucin 3
- TME:
-
Tumor microenvironment
- Treg/Tregs:
-
Regulatory T-cell(s)
References
Tormoen GW, Crittenden MR, Gough MJ. Role of the immunosuppressive microenvironment in immunotherapy. Adv Radiat Oncol. 2018;3:520–6.
Burgos-Panadero R, Lucantoni F, Gamero-Sandemetrio E, de la Cruz-Merino L, Álvaro T, Noguera R. The tumour microenvironment as an integrated framework to understand cancer biology. Cancer Lett. 2019;461:112–22.
Rudensky AY, Regulatory T. Cells and Foxp3. Immunol Rev. 2011;241:260–8.
Wong HS, Park K, Gola A, Baptista AP, Miller CH, Deep D, et al. A local regulatory T cell feedback circuit maintains immune homeostasis by pruning self-activated T cells. Cell. 2021;184:3981-3997.e22.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61.
Chatila TA. Role of regulatory T cells in human diseases. J Allergy Clin Immunol. 2005;116:949–59.
Cools N, Ponsaerts P, Van Tendeloo VFI, Berneman ZN. Regulatory T Cells and human disease. Clin Dev Immunol. 2007;2007:1–10.
Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–1.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6.
Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–91.
Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, Volk H-D. Regulatory T cell-mediated anti-inflammatory effects promote successful tissue repair in both indirect and direct manners. Front Pharmacol. 2015. https://doi.org/10.3389/fphar.2015.00184.
Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med. 2013;5:200ra116.
Kuwahara T, Hazama S, Suzuki N, Yoshida S, Tomochika S, Nakagami Y, et al. Intratumoural-infiltrating CD4 + and FOXP3 + T cells as strong positive predictive markers for the prognosis of resectable colorectal cancer. Br J Cancer. 2019;121:659–65.
Shen Z, Zhou S, Wang Y, Li R, Zhong C, Liang C, et al. Higher intratumoral infiltrated Foxp3+ Treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J Cancer Res Clin Oncol. 2010;136:1585–95.
Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi M-B, Harpole DH, et al. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer. 2006;107:2866–72.
Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96.
Wang Y, Ma Y, Fang Y, Wu S, Liu L, Fu D, et al. Regulatory T cell: a protection for tumour cells. J Cell Mol Med. 2012;16:425–36.
Jiang X, Liu G, Li Y, Pan Y. Immune checkpoint: the novel target for antitumor therapy. Genes & Diseases. 2019 [cited 2020 Sep 13]; Available from: http://www.sciencedirect.com/science/article/pii/S2352304219301205.
Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Front Immunol. 2013;4:190.
Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci USA. 2009;106:1903–8.
Chen W, Jin W, Hardegen N, Lei K-J, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86.
Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–60.
Chen W, Jin W, Hardegen N, Lei K, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25− Naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–86.
Josefowicz SZ, Lu L-F, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64.
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–6.
Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–11.
Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92.
Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempel TR. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest. 2014;124:2425–40.
Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanović S, Robbins PF, et al. Tumor-infiltrating human CD4+ regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol. 2019;4:eaao4310.
Szymczak-Workman AL, Workman CJ, Vignali DAA. Cutting edge: regulatory T cells do not require stimulation through their TCR to suppress. J Immunol. 2009;182:5188–92.
Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–9.
Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. 2016;17:1322–33.
Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–65.
Klein M, Bopp T. Cyclic AMP represents a crucial component of Treg cell-mediated immune regulation. Front Immunol. 2016. https://doi.org/10.3389/fimmu.2016.00315.
Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. 2007;27:635–46.
Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5:2516–22.
Workman CJ, Vignali DAA. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J Immunol. 2005;174:688–95.
Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol. 2008;180:5916–26.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42.
Aksoylar H-I, Boussiotis VA. PD-1+ Treg cells: a foe in cancer immunotherapy? Nat Immunol. 2020;21:1311–2.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80.
Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018;15:366–81.
Schaaf MB, Garg AD, Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018;9:1–14.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70.
Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306.
Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22:679–84.
Bromley SK, Mempel TR, Luster AD. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat Immunol. 2008;9:970–80.
Whiteside TL. Clinical impact of regulatory T cells (Treg) in cancer and HIV. Cancer Microenviron. 2015;8:201–7.
Paluskievicz CM, Cao X, Abdi R, Zheng P, Liu Y, Bromberg JS. T Regulatory cells and priming the suppressive tumor microenvironment. Front Immunol. 2019;10:2453.
Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy. Immunol Res. 2011;51:170–82.
Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced regulatory T cells: their development, stability, and applications. Trends Immunol. 2016;37:803–11.
Li J, Shen C, Wang X, Lai Y, Zhou K, Li P, et al. Prognostic value of TGF-β in lung cancer: systematic review and meta-analysis. BMC Cancer. 2019;19:691.
Velasco-Velázquez M, Xolalpa W, Pestell RG. The potential to target CCL5/CCR5 in breast cancer. Expert Opin Ther Targets. 2014;18:1265–75.
Nishikawa H, Kato T, Tanida K, Hiasa A, Tawara I, Ikeda H, et al. CD4+ CD25+ T cells responding to serologically defined autoantigens suppress antitumor immune responses. Proc Natl Acad Sci USA. 2003;100:10902–6.
Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, et al. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 2001;409:97–101.
Nagase H, Takeoka T, Urakawa S, Morimoto-Okazawa A, Kawashima A, Iwahori K, et al. ICOS+ Foxp3+ TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with Helicobacter pylori. Int J Cancer. 2017;140:686–95.
Lee WS, Yang H, Chon HJ, Kim C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med. 2020;52:1475–85.
Tang T, Huang X, Zhang G, Hong Z, Bai X, Liang T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct Target Ther. 2021;6:72.
Oshimori N, Guo Y, Taniguchi S. An emerging role for cellular crosstalk in the cancer stem cell niche. J Pathol. 2021;254:384–94.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–50.
Marzagalli M, Ebelt ND, Manuel ER. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin Cancer Biol. 2019;59:236–50.
Wei F, Wang D, Wei J, Tang N, Tang L, Xiong F, et al. Metabolic crosstalk in the tumor microenvironment regulates antitumor immunosuppression and immunotherapy resisitance. Cell Mol Life Sci. 2021;78:173–93.
Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol. 2020;17:1–12.
Sarsembayeva A, Kienzl M, Gruden E, Ristic D, Maitz K, Valadez-Cosmes P, et al. Cannabinoid receptor 2 plays a pro-tumorigenic role in non-small cell lung cancer by limiting anti-tumor activity of CD8+ T and NK cells. Front Immunol. 2023. https://doi.org/10.3389/fimmu.2022.997115.
Ni L, Sun P, Zhang S, Qian B, Chen X, Xiong M, et al. Transcriptome and single-cell analysis reveal the contribution of immunosuppressive microenvironment for promoting glioblastoma progression. Front Immunol. 2023. https://doi.org/10.3389/fimmu.2022.1051701.
Aru B, Pehlivanoğlu C, Dal Z, Dereli-Çalışkan NN, Gürlü E, Yanıkkaya-Demirel G. A potential area of use for immune checkpoint inhibitors: targeting bone marrow microenvironment in acute myeloid leukemia. Front Immunol. 2023. https://doi.org/10.3389/fimmu.2023.1108200.
Lv J, Sun H, Gong L, Wei X, He Y, Yu Z, et al. Aberrant metabolic processes promote the immunosuppressive microenvironment in multiple myeloma. Front Immunol. 2022. https://doi.org/10.3389/fimmu.2022.1077768.
Guo H, Yang J, Wang H, Liu X, Liu Y, Zhou K. Reshaping the tumor microenvironment: the versatility of immunomodulatory drugs in B-cell neoplasms. Front Immunol. 2022. https://doi.org/10.3389/fimmu.2022.1017990.
Mocellin S, Wang E, Marincola FM. Cytokines and immune response in the tumor microenvironment. J Immunother. 2001;24:392–407.
Lebrun J-J. The dual role of TGFβ in human cancer: from tumor suppression to cancer metastasis. ISRN Mol Biol. 2012;2012: 381428.
Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017;25:1282-1293.e7.
Hsu P, Santner-Nanan B, Hu M, Skarratt K, Lee CH, Stormon M, et al. IL-10 potentiates differentiation of human induced regulatory T cells via STAT3 and Foxo1. J Immunol. 2015;195:3665–74.
Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7.
Hira SK, Rej A, Paladhi A, Singh R, Saha J, Mondal I, et al. Galunisertib drives Treg fragility and promotes dendritic cell-mediated immunity against experimental lymphoma. iScience. 2020;23:101623.
Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59:3128–33.
Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase Ia study of FoxP3+ CD4 Treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res. 2015;21:4327–36.
Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA. 2008;105:7797–802.
Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. 2018;19:665–73.
Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol. 2020;38:541–66.
Kitz A, Dominguez-Villar M. Molecular mechanisms underlying Th1-like Treg generation and function. Cell Mol Life Sci. 2017;74:4059–75.
Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci U S A. 2018;115:E6546–55.
Toor SM, Murshed K, Al-Dhaheri M, Khawar M, Abu Nada M, Elkord E. Immune checkpoints in circulating and tumor-infiltrating CD4+ T cell subsets in colorectal cancer patients. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.02936.
Kim HR, Park HJ, Son J, Lee JG, Chung KY, Cho NH, et al. Tumor microenvironment dictates regulatory T cell phenotype: upregulated immune checkpoints reinforce suppressive function. J Immunother Cancer. 2019;7:339.
Liu Y, Zheng P. Preserving the CTLA-4 checkpoint for safer and more effective cancer immunotherapy. Trends Pharmacol Sci. 2020;41:4–12.
Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers. Clin Cancer Res. 2019;25:1233–8.
Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc Effector function contributes to the activity of human Anti-CTLA-4 antibodies. Cancer Cell. 2018;33:649-663.e4.
Ingram JR, Blomberg OS, Rashidian M, Ali L, Garforth S, Fedorov E, et al. Anti-CTLA-4 therapy requires an Fc domain for efficacy. Proc Natl Acad Sci USA. 2018;115:3912–7.
Du X, Tang F, Liu M, Su J, Zhang Y, Wu W, et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018;28:416–32.
Sanseviero E, O’Brien EM, Karras JR, Shabaneh TB, Aksoy BA, Xu W, et al. Anti-CTLA-4 activates intratumoral NK cells and combined with IL15/IL15Rα complexes enhances tumor control. Cancer Immunol Res. 2019;7:1371–80.
Cari L, Nocentini G, Migliorati G, Riccardi C. Potential effect of tumor-specific Treg-targeted antibodies in the treatment of human cancers: a bioinformatics analysis. Oncoimmunology. 2018;7: e1387705.
Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, et al. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci USA. 2019;116:609–18.
Ren Z, Fu Y-X. Degradation of CTLA-4 balances toxicity and efficacy. Sci Bull. 2019;64:1388–9.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34.
Du X, Liu M, Su J, Zhang P, Tang F, Ye P, et al. Uncoupling therapeutic from immunotherapy-related adverse effects for safer and effective anti-CTLA-4 antibodies in CTLA4 humanized mice. Cell Res. 2018;28:433–47.
Albandar HJ, Fuqua J, Albandar JM, Safi S, Merrill SA, Ma PC. Immune-related adverse events (irAE) in cancer immune checkpoint inhibitors (ICI) and survival outcomes correlation: to rechallenge or not? Cancers. 2021;13:989.
Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chávez A, Keegan N, Khamashta MA, et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primers. 2020;6:38.
Zhang Y, Du X, Liu M, Tang F, Zhang P, Ai C, et al. Hijacking antibody-induced CTLA-4 lysosomal degradation for safer and more effective cancer immunotherapy. Cell Res. 2019;29:609–27.
Sharma N, Vacher J, Allison JP. TLR1/2 ligand enhances antitumor efficacy of CTLA-4 blockade by increasing intratumoral Treg depletion. Proc Natl Acad Sci USA. 2019;116:10453–62.
Dorta-Estremera S, Hegde VL, Slay RB, Sun R, Yanamandra AV, Nicholas C, et al. Targeting interferon signaling and CTLA-4 enhance the therapeutic efficacy of anti-PD-1 immunotherapy in preclinical model of HPV+ oral cancer. J Immunother Cancer. 2019;7:252.
Kvarnhammar AM, Veitonmäki N, Hägerbrand K, Dahlman A, Smith KE, Fritzell S, et al. The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J Immunother Cancer. 2019;7:103.
Goswami S, Apostolou I, Zhang J, Skepner J, Anandhan S, Zhang X, et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J Clin Invest. 2018;128:3813–8.
Kurtulus S, Sakuishi K, Ngiow S-F, Joller N, Tan DJ, Teng MWL, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015;125:4053–62.
Shen Y-C, Ghasemzadeh A, Kochel CM, Nirschl TR, Francica BJ, Lopez-Bujanda ZA, et al. Combining intratumoral Treg depletion with androgen deprivation therapy (ADT): preclinical activity in the Myc-CaP model. Prostate Cancer Prostatic Dis. 2018;21:113–25.
Langhans B, Nischalke HD, Krämer B, Dold L, Lutz P, Mohr R, et al. Role of regulatory T cells and checkpoint inhibition in hepatocellular carcinoma. Cancer Immunol Immunother. 2019;68:2055–66.
Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci USA. 2019;116:9999–10008.
Mahne AE, Mauze S, Joyce-Shaikh B, Xia J, Bowman EP, Beebe AM, et al. Dual roles for regulatory T-cell depletion and costimulatory signaling in agonistic GITR targeting for tumor immunotherapy. Cancer Res. 2017;77:1108–18.
Woods DM, Ramakrishnan R, Laino AS, Berglund A, Walton K, Betts BC, et al. Decreased suppression and increased phosphorylated STAT3 in regulatory T cells are associated with benefit from adjuvant PD-1 blockade in resected metastatic melanoma. Clin Cancer Res. 2018;24:6236–47.
Oweida AJ, Darragh L, Phan A, Binder D, Bhatia S, Mueller A, et al. STAT3 modulation of regulatory T cells in response to radiation therapy in head and neck cancer. J Natl Cancer Inst. 2019;111:1339–49.
DiDomenico J, Lamano JB, Oyon D, Li Y, Veliceasa D, Kaur G, et al. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology. 2018;7: e1448329.
Taylor NA, Vick SC, Iglesia MD, Brickey WJ, Midkiff BR, McKinnon KP, et al. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J Clin Invest. 2017;127:3472–83.
Liu Z, McMichael EL, Shayan G, Li J, Chen K, Srivastava R, et al. Novel effector phenotype of Tim-3+ regulatory T cells leads to enhanced suppressive function in head and neck cancer patients. Clin Cancer Res. 2018;24:4529–38.
Oweida A, Hararah MK, Phan A, Binder D, Bhatia S, Lennon S, et al. Resistance to radiotherapy and PD-L1 blockade is mediated by TIM-3 upregulation and regulatory T-cell infiltration. Clin Cancer Res. 2018;24:5368–80.
Liu J-F, Wu L, Yang L-L, Deng W-W, Mao L, Wu H, et al. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J Exp Clin Cancer Res. 2018;37:44.
Le K-S, Thibult M-L, Just-Landi S, Pastor S, Gondois-Rey F, Granjeaud S, et al. Follicular B lymphomas generate regulatory T cells via the ICOS/ICOSL pathway and are susceptible to treatment by anti-ICOS/ICOSL therapy. Cancer Res. 2016;76:4648–60.
Sim GC, Liu C, Wang E, Liu H, Creasy C, Dai Z, et al. IL2 variant circumvents ICOS+ regulatory T-cell expansion and promotes NK Cell activation. Cancer Immunol Res. 2016;4:983–94.
Pelicano H, Lu W, Zhou Y, Zhang W, Chen Z, Hu Y, et al. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res. 2009;69:2375–83.
Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci USA. 2016;113:8490–5.
Gianchecchi E, Fierabracci A. Inhibitory receptors and pathways of lymphocytes: the role of PD-1 in Treg development and their involvement in autoimmunity onset and cancer progression. Front Immunol. 2018;9:2374.
Miyara M, Chader D, Sage E, Sugiyama D, Nishikawa H, Bouvry D, et al. Sialyl Lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3high regulatory T cells in humans. Proc Natl Acad Sci USA. 2015;112:7225–30.
Foss F. Clinical experience with denileukin diftitox (ONTAK). Semin Oncol. 2006;33:S11-16.
Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012;4:134ra62.
Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366:2517–9.
Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1:32–42.
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–710.
Jensen SM, Maston LD, Gough MJ, Ruby CE, Redmond WL, Crittenden M, et al. Signaling through OX40 enhances antitumor immunity. Semin Oncol. 2010;37:524–32.
Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3:135–42.
Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol. 2010;184:6545–51.
Nishikawa H, Kato T, Hirayama M, Orito Y, Sato E, Harada N, et al. Regulatory T cell-resistant CD8+ T cells induced by glucocorticoid-induced tumor necrosis factor receptor signaling. Cancer Res. 2008;68:5948–54.
Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Rational design of anti-GITR-based combination immunotherapy. Nat Med. 2019;25:759–66.
Yamaguchi T, Hirota K, Nagahama K, Ohkawa K, Takahashi T, Nomura T, et al. Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity. 2007;27:145–59.
Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang L-P, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226–30.
Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci USA. 2013;110:17945–50.
Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. 2016;45:1122–34.
Barsheshet Y, Wildbaum G, Levy E, Vitenshtein A, Akinseye C, Griggs J, et al. CCR8+FOXp3+ Treg cells as master drivers of immune regulation. Proc Natl Acad Sci USA. 2017;114:6086–91.
Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A, Avramis E, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res. 2016;4:194–203.
Martinez M, Kim S, St. Jean N, O’Brien S, Lian L, Sun J, et al. Addition of anti-TIM3 or anti-TIGIT antibodies to anti-PD1 blockade augments human T cell adoptive cell transfer. OncoImmunology. 2021;10:1873607.
Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA. 2015;112:6140–5.
Wu L, Mao L, Liu J-F, Chen L, Yu G-T, Yang L-L, et al. Blockade of TIGIT/CD155 signaling reverses T-cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol Res. 2019;7:1700–13.
Ruffo E, Wu RC, Bruno TC, Workman CJ, Vignali DAA. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin Immunol. 2019;42: 101305.
Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73:3591–603.
Yoshida K, Okamoto M, Sasaki J, Kuroda C, Ishida H, Ueda K, et al. Anti-PD-1 antibody decreases tumour-infiltrating regulatory T cells. BMC Cancer. 2020;20:25.
Dodagatta-Marri E, Meyer DS, Reeves MQ, Paniagua R, To MD, Binnewies M, et al. α-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α-TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J Immunother Cancer. 2019;7:62.
Löffek S. Transforming of the tumor microenvironment: implications for TGF-β inhibition in the context of immune-checkpoint therapy. J Oncol. 2018;2018:9732939.
Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107:4275–80.
Wei SC, Anang N-AAS, Sharma R, Andrews MC, Reuben A, Levine JH, et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci USA. 2019;116:22699–709.
Chae YK, Arya A, Iams W, Cruz MR, Chandra S, Choi J, et al. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J Immunother Cancer. 2018;6:39.
Singh R, Gupta U, Srivastava P, Paladhi A, Sk UH, Hira SK, et al. γc cytokine-aided crosstalk between dendritic cells and natural killer cells together with doxorubicin induces a healer response in experimental lymphoma by downregulating FOXP3 and programmed cell death protein 1. Cytotherapy. 2022;24:1232–44.
Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. 2017;46:577–86.
Zou W, Wolchok JD, Chen L. PD-L1 (B7–H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4.
Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175–86.
Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276:97–111.
Shen P, Yue R, Tang J, Si H, Shen L, Guo C, et al. Preferential Tim-3 expression on Treg and CD8(+) T cells, supported by tumor-associated macrophages, is associated with worse prognosis in gastric cancer. Am J Transl Res. 2016;8:3419–28.
Gefen T, Castro I, Muharemagic D, Puplampu-Dove Y, Patel S, Gilboa E. A TIM-3 oligonucleotide aptamer enhances T cell functions and potentiates tumor immunity in mice. Mol Ther. 2017;25:2280–8.
Principe DR, Chiec L, Mohindra NA, Munshi HG. Regulatory T-cells as an emerging barrier to immune checkpoint inhibition in lung cancer. Front Oncol. 2021. https://doi.org/10.3389/fonc.2021.684098.
Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21:1346–58.
Wu S-P, Liao R-Q, Tu H-Y, Wang W-J, Dong Z-Y, Huang S-M, et al. Stromal PD-L1-positive regulatory T cells and PD-1-positive CD8-positive T cells define the response of different subsets of non-small cell lung cancer to PD-1/PD-L1 blockade immunotherapy. J Thorac Oncol. 2018;13:521–32.
Koh J, Hur JY, Lee KY, Kim MS, Heo JY, Ku BM, et al. Regulatory (FoxP3+) T cells and TGF-β predict the response to anti-PD-1 immunotherapy in patients with non-small cell lung cancer. Sci Rep. 2020;10:18994.
Singh R, Manna PP. Reactive oxygen species in cancer progression and its role in therapeutics. Explor Med. 2022;3:43–57.
Shields HJ, Traa A, Van Raamsdonk JM. Beneficial and detrimental effects of reactive oxygen species on lifespan: a comprehensive review of comparative and experimental studies. Front Cell Dev Biol. 2021;9: 628157.
Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. 2017;18:1332–41.
Mougiakakos D, Johansson CC, Jitschin R, Böttcher M, Kiessling R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood. 2011;117:857–61.
Lee K, Won HY, Bae MA, Hong J-H, Hwang ES. Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc Natl Acad Sci. 2011;108:9548–53.
Efimova O, Szankasi P, Kelley TW. Ncf1 (p47phox) is essential for direct regulatory T cell mediated suppression of CD4+ effector T cells. PLoS ONE. 2011;6: e16013.
Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature. 2019;576:143–8.
Kunisada Y, Eikawa S, Tomonobu N, Domae S, Uehara T, Hori S, et al. Attenuation of CD4+CD25+ regulatory T cells in the tumor microenvironment by metformin, a Type 2 diabetes drug. EBioMedicine. 2017;25:154–64.
Yu X, Lao Y, Teng X-L, Li S, Zhou Y, Wang F, et al. SENP3 maintains the stability and function of regulatory T cells via BACH2 deSUMOylation. Nat Commun. 2018;9:3157.
Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood. 2009;113:3542–5.
Uzhachenko R, Shanker A, Yarbrough WG, Ivanova AV. Mitochondria, calcium, and tumor suppressor Fus1: at the crossroad of cancer, inflammation, and autoimmunity. Oncotarget. 2015;6:20754–72.
Chamoto K, Chowdhury PS, Kumar A, Sonomura K, Matsuda F, Fagarasan S, et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci USA. 2017;114:E761–70.
Cichon MA, Radisky DC. ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-κB-dependent activation of Snail. Oncotarget. 2014;5:2827–38.
Srivastava P, Paladhi A, Singh R, Srivastava DN, Singh RA, Hira SK, et al. Targeting PD-1 in CD8+ T cells with a biomimetic bilirubin-5-fluoro-2-deoxyuridine-bovine serum albumin nanoconstruct for effective chemotherapy against experimental lymphoma. Mol Pharm. 2021;18:2053–65.
Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol. 2018;19:291–301.
Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature. 2017;546:421–5.
Zhang Q, Lu W, Liang C-L, Chen Y, Liu H, Qiu F, et al. Chimeric antigen receptor (CAR) Treg: a promising approach to inducing immunological tolerance. Front Immunol. 2018. https://doi.org/10.3389/fimmu.2018.02359.
Acknowledgements
RS thanks the Indian Council of Medical Council Research (ICMR), India (2021-12946/CMB-BMS), for a senior research fellowship.
Funding
This work was supported by IOE grant No. R/Dev/D/IOE/Incentive/2021–22/32275 to Partha Pratim Manna by Banaras Hindu University (BHU).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Singh, R., Srivastava, P. & Manna, P.P. Evaluation of regulatory T-cells in cancer immunotherapy: therapeutic relevance of immune checkpoint inhibition. Med Oncol 41, 59 (2024). https://doi.org/10.1007/s12032-023-02289-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-023-02289-y