
Abstract
Background
Disease prognosis after resection of lung cancer could be affected by pathological subtypes. In this study, we investigated the difference of gene variation and significantly altered pathways between adenocarcinoma in situ (AIS)/microinvasive adenocarcinoma (MIA) and invasive adenocarcinoma (IAC) subtypes to reveal the molecular mechanism of prognosis differences.
Methods
Sixty one tumor tissues were subjected to DNA extraction and customized 136 gene targeted next-generation sequencing. Comparisons between groups were performed with two-sided Fisher’s exact test for categorical variables and two-tailed unpaired t test for numerical variables.
Results
A total of 402 somatic mutations involved in 70 genes were detected in all these samples, and 74.29% of these genes were mutated in at least two samples. PMS2, ARID1A, EGFR, and POLE were the most frequently mutated genes. ALK_EML4 fusion was observed in one IAC patient and RET_ KIF5B fusion in one AIS patient. A significant higher proportion of patients with TP53 gene mutation was observed in the IAC group (P = 0.0057). The average onset age in IAC group is 62.48 years, which is greater than other subtypes (P = 0.0166). It revealed that mutations in genes involved in the mTOR signaling pathway (56.52% vs 26.32%, P = 0.0288) and Hippo signaling pathway (34.78% vs 10.53%, P = 0.0427) were significantly enriched in IAC subtypes, suggesting the key involvement of mTOR and Hippo signaling pathways in lung tumor development and malignant progression.
Conclusions
This study revealed the heterogeneity of gene mutations and significantly altered pathways between different lung cancer subtypes, suggesting the potential mechanism of different prognosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide [1]. And non-small-cell lung cancer (NSCLC) is the most common type, which accounts for approximately 85% of lung cancer patients [2]. Lung adenocarcinoma (LUAD) is a subtype of NSCLC, which accounts for 50% of all the diagnosed lung cancer patients [2, 3]. LUAD classification sponsored by the International Association for the Study of Lung Cancer (IASLC), American Thoracic Society (ATS), and European Respiratory Society (ERS) has been adopted for many years [4], which has been refined based on the invasion and predominant growth pattern in 2015 [3]. According to the invasion pattern, LUAD is composed of adenocarcinoma in situ (AIS), minimally invasive adenocarcinoma (MIA), invasive adenocarcinoma (IAC), etc. [3].
In general, surgery is unnecessary for AIS patients because of no interstitial invasion and regular follow-up is recommended [5]. It is a conventionally surgical approach to perform segmentectomy for AIS and MIA patients, which could reserve pulmonary function maximally. And IAC patients undergo lobectomy more often. Furthermore, lymph node dissection is necessary for IAC patients because of the higher probability of regional and distal metastasis. The clinical outcome of LUAD has been significantly improved based on the advances in surgery, radiotherapy, and systemic treatment [6, 7]. AIS and MIA patients have a similar surgical outcomes after complete resection [8], and the five-year survival rate after surgery is almost 100% [9,10,11]. However, the five-year survival rate after surgical resection for IAC patients decreases substantially to 75% [9,10,11,12]. Therefore, it is significantly valuable to distinguish the differences between AIS/MIA and IAC.
Mounting evidence has suggested that disease recurrence and prognosis after resection could be affected by pathological subtypes of lung cancer [13, 14]. Tremendous efforts have been focused on the molecular features of LUAD [15,16,17] in these years, which could help researchers understand its molecular heterogeneity. Meanwhile, large-scale sequencing studies have elucidated the complex genomic landscape of preinvasive LUAD subtypes [18,19,20], which benefited from the widespread implementation of the enhanced radiological techniques in lung cancer diagnosis.
However, the gene variation differences and the molecular mechanism of prognosis differences between AIS/MIA and IAC subtypes are still unclear. To achieve this, we focused on investigating the difference of gene alterations and significantly altered pathways between AIS/MIA and IAC subtypes in this study.
Materials and methods
Patient collection
A total of 61 tumor tissues (involved in 50 lung adenocarcinoma patients) from Renmin Hospital of Wuhan University between October 2021 and July 2022 were included in this study. Patients eligible for inclusion in this study were those with (a) 18–80 years old, (b) histologically proven IAC, or MIA, or AIS, or mixed AIS with MIA, (c) no other serious comorbidities of other organs. All pathology specimens were reviewed by experienced pathologists before analysis. Among these samples, individuals diagnosed with multiple lesions sharing the same pathological type were simultaneously included in this study. Informed consent was obtained from all participants. This study was approved by the Ethical Committee of Renmin Hospital of Wuhan University (approval number: WDRY2023-K027) and was performed in accordance with the ethical standards of the World Medical Association Declaration of Helsinki.
Targeted DNA sequencing
DNA samples were collected from formalin-fixed, paraffin-embedded (FFPE) tumor tissues with the QIAamp DNA FFPE Tissue Kit (56,404, Qiagen, Hilden, Germany). Then DNA libraries were constructed with KAPA Library Preparation Kit (Kapa Biosystems Inc., Wilmington, USA). The customized Agilent SureSelectXT DNA 136 panel was used to capture targeted region for 61 samples. Finally, high throughput sequencing was performed on the Illumina X10 platform (Illumina Inc., San Diego, CA, USA). Genes of 136 panel sequencing were shown in Supplementary file 1.
Putative somatic mutation calling and filtering
Low-quality sequencing reads were discarded, including reads containing adaptor sequences, > 5% unknown base ‘N,’ and > 15% bases with quality < = 19. The average sequencing depth was more than 2900 × . After filtration, we obtained 2.75 G high-quality clean bases on average, and the Q30 value was > 90% for samples. Then high-quality paired-end reads underwent mutation analysis and human genome build hg19 was used as the reference genome, which mapped to hg19 by Burrows-Wheeler Aligner (BWA version 0.7.15, default parameters, BWA-MEM algorithm). Software GATK MuTect2 (version 4.1, default parameters) was used to identify nonsilent somatic mutations. And mutations were annotated with software ANNOVAR (version 2016-02-01, default parameters).
Because of the unavailability for paired normal samples, the following criteria were utilized to filter the somatic variants: (a) retain nonsilent coding mutations that mutated in exonic or splicing region; (b) retain mutations that not included in public 1000 Genomes databases or the frequency of the alternative allele was < = 0.01; (c) retain mutations that the allele fraction in the tumor was > = 0.02; (d) retain mutations that called as damaging/deleterious for protein structure by at least one of two used prediction software algorithms, SIFT [21] and PolyPhen2 [22]; (e) remove mutations that recorded in dbSNP database; (f) retain mutations registered in the COSMIC database (version 94) [23] (while those mutations noted with ‘SNP’ in COSMIC needed to be discarded); and (g) retain mutations that contain > = 10 variant reads and detected in both forward and reverse DNA strands. The remaining mutations were identified as putative somatic mutations and were subsequently used for further analysis.
Gene fusion analysis
GeneFuse (version 0.6.0) [24] was used to verify critical gene fusions. A gene fusion was considered as true: (a) unique reads > 4, (b) verified as true by Integrative Genomics Viewer (IGV) software.
Statistical analysis and graphics drawing
Statistical analysis was performed by GraphPad Prism software (version 8.0). Comparisons between groups were performed with two-sided Fisher’s exact test for categorical variables and two-tailed unpaired t test for numerical variables. Data were presented as ‘Mean with Standard Deviation (SD)’ or as percentages of patients. In all analyses, P value < 0.0500 was considered statistically significant. Histogram and scatter diagram were plotted by GraphPad Prism software (version 8.0). Gene mutation spectrum was drawn with R package ‘maftools’ (version 2.12.0) or ‘ComplexHeatmap’ (version 2.12.1).
Results
Clinical characteristics and mutational profiles of lung adenocarcinoma patients
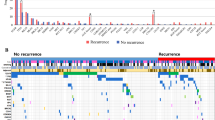
A total of 61 tumor samples from 50 lung adenocarcinoma patients were investigated in our study. All the clinical characteristics were shown in Supplementary file 2. All the patients were confirmed as unique pathological subtype, including IAC (23 samples/21 patients), MIA (6 samples/6 patients), AIS (29 samples/20 patients), or LMAC (3 samples/3 patients). All the clinical characteristics and somatic mutations are summarized in Fig. 1. Of these patients, the onset age ranges from 28 to 78 years (median, 59). The number of female patients was more than the number of male patients (31 vs 19). After filtering the mutations, a total of 402 somatic mutations (range 1–54, median 4) involved in 70 genes were detected in all these samples and 74.29% (52/70) of these genes were mutated in at least two samples. Of these mutations, 98.51% (396/402) mutations were observed as a SNV, whereas 1.49% (6/402) as an InDel type.

Clinical attributes and mutational profiles of 61 tumor tissues. Only genes with mutations in > = 4 samples are shown. Top column: mutation number represented in bottom landscape for each sample. Right column: number of mutated samples and gene mutation frequency for each gene. Bottom column: each column represents one sample, and each row shows one gene. IAC, invasive adenocarcinoma. MIA, minimally invasive adenocarcinoma. AIS, adenocarcinoma in situ. LMAC, lung mixed adenocarcinoma
As shown in Fig. 1, frequently mutated genes detected from this cohort included PMS2 (65.57%, 40/61), ARID1A (37.70%, 23/61), POLE (31.15%, 19/61), EGFR (27.87%, 17/61), MSH2 (24.59%, 15/61), ATM (19.67%, 12/61), TSC1 (18.03%, 11/61), FGFR1 (14.75%, 9/61), KRAS (14.75%, 9/61), AR (11.48%, 7/61), DNMT3A (11.48%, 7/61), CHEK2 (9.84%, 6/61), ERBB3 (9.84%, 6/61), MYC (9.84%, 6/61), ATR (8.20%, 5/61), BRCA2 (8.20%, 5/61), KIT (8.20%, 5/61), RB1 (8.20%, 5/61), TP53 (8.20%, 5/61), ERBB2 (6.56%, 4/61), MET (6.56%, 4/61), and SMO (6.56%, 4/61). Among them, PMS2, ARID1A, EGFR, and POLE were the most frequently mutated genes in this study. Additionally, two critical gene fusions were observed in one IAC patient (Pt17, ALK.exon20.chr2:29446349_EML4.intron19.chr2:42,546,586) and one AIS patient (Pt44, RET.intron11.chr10:43610997_KIF5B.intron15.chr10:32,316,372) (Supplementary file 3 and Supplementary Fig. 1).
Gene mutation and clinical feature comparison in hierarchical subgroups with different prognosis (IAC vs AIS/MIA/LMAC)
To explore the possible mechanism that attributes the difference of different lung adenocarcinoma group, gene mutations and clinical characteristics were compared among different subtypes. A significant higher proportion of patients with TP53 gene mutation was observed in the IAC group than that in the AIS/MIA/LMAC group (21.74% vs 0.00%, P = 0.0057) (Fig. 2A). There was no significant difference about other gene mutations between these two groups, including EGFR, KRAS, ERBB2, etc. (Supplementary Fig. 2). The average age of onset in IAC group is 62.48 years, which is greater than that 54.41 years in the AIS/MIA/LMAC group (P = 0.0166) (Fig. 2B).

Comparison of gene mutation (A) and age of onset (B) in different hierarchical subgroups with lung adenocarcinoma. Note: only genes mutated in > = 4 samples (frequency > = 5%) were considered. TP53wild, without TP53 mutation. TP53mut, TP53 mutation. IAC, invasive adenocarcinoma. MIA, minimally invasive adenocarcinoma. AIS, adenocarcinoma in situ. LMAC, lung mixed adenocarcinoma. Data in B were shown as ‘Mean with SD.’ Statistics in A: Fisher’s exact test. Statistics in B: unpaired t test
Significantly altered pathways between IAC and AIS/MIA/LMAC
Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to analyze the pathways that are distinct between the two groups. The results demonstrate that the major signaling pathways affected in IAC samples were mTOR signaling pathway (Fig. 3A, 56.52% vs 26.32%, P = 0.0288) and Hippo signaling pathway (Fig. 3B, 34.78% vs 10.53%, P = 0.0427). Genes involved in mTOR signaling pathway detected from our cohort included TSC1, KRAS, PIK3CA, TSC2, STK11, MAP2K1, BRAF, and MTOR. Mutations in TSC1 (n = 6), KRAS (n = 5), PIK3CA (n = 1), TSC2 (n = 2), STK11 (n = 2), MAP2K1 (n = 1), BRAF (n = 1), and MTOR (n = 1) were detected in 13 patients with IAC samples. Five samples in IAC group had concurrent mutations in at least two genes. Genes involved in the Hippo signaling pathway detected from our cohort included MYC, SMAD4, CTNNB1, APC, and YAP1. Taken together, these results indicate that IAC and AIS/MIA/LMAC subtypes are molecularly distinct based on the difference in the number and distribution of somatic mutation types detected in these tumors as well as the major pathways affected by these mutations.

KEGG analysis reveals distinct pathways in different hierarchical subgroups with lung adenocarcinoma. Two pathways with the significant enrichments in IAC samples as compared to AIS/MIA/LMAC samples. A: mTOR signaling pathway (56.52% vs 26.32%, P = 0.0288). B: Hippo signaling pathway (34.78% vs 10.53%, P = 0.0427). Mutated genes participating in any of the two pathways were listed. Each column represents a sample and each row represents a gene. Mutation types were denoted in different colors. Samples were grouped according to the histopathology. IAC invasive adenocarcinoma, MIA minimally invasive adenocarcinoma, AIS adenocarcinoma in situ, LMAC lung mixed adenocarcinoma. Data were shown as percentages of patients. Statistics: Fisher’s exact test
Discussion
This pilot study aimed to illustrate the difference of gene variants and clinical parameters between IAC and AIS/MIA/LMAC subtypes, an effort to help us perform precise medicine and treatment for different LUAD subtypes. In this study, a total of 402 somatic mutations were detected in all these 61 samples, and 74.29% genes were mutated in at least two samples. Of these mutations, PMS2 (mismatch repair system component) accounts for the highest percentage (65.57%), which is one of the critical genes in DNA mismatch repair (MMR) pathway with potential crucial roles in carcinogenesis [25, 26]. A high mutation rate of PMS2 demonstrates that the dysfunction in MMR pathway might impact the tumorigenesis of LUAD and may increase patients’ sensivivity to DNA damaging agents. ARID1A and EGFR genes were also detected with a high mutation rate. ARID1A gene, as a critical component of the switch/sucrose nonfermentable (SWI/SNF) complex, plays a role in cell cycle regulation, metabolic reprogramming, and epithelial–mesenchymal transition [27]. In recent years, potential ARID1A mutation-based therapeutic targets have been focused, including Poly (ADP-ribose) polymerase (PARP) inhibitors, enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) inhibitors, and immune checkpoint inhibitors [27]. EGFR gene encodes the epidermal growth factor receptor (EGFR) tyrosine kinase, and EGFR-mutated patients could benefit from EGFR tyrosine kinase inhibitors (TKIs). Besides, other new treatment strategies to overcome EGFR-TKIs resistance have also been researched in these years [28]. Additionally, our results showed that a significant higher mutation rate of TP53 was observed in IAC than that in AIS/MIA/LMAC, which was consistent with previous study [29]. It suggests the potential oncogenic activity of TP53 in IAC subtypes. Compared with AIS/MIA/LMAC, patients with IAC harbored an older onset age, which is in line with previous researches [29]. Besides, similar result that elder LUAD patients carried more TP53 mutations than young LUAD cohorts was observed in Yang’s study [30].
To further understand the difference of critical pathways involved in the disease progression for different LUAD subtypes, we performed pathway enrichment analysis. The results revealed that mutations in genes involved in the mTOR signaling pathway (56.52% vs 26.32%) and Hippo signaling pathway (34.78% vs 10.53%) were significantly enriched in IAC subtypes, suggesting the key involvement of mTOR and Hippo signaling pathways in lung tumor development and malignant progression. mTOR signaling pathway integrates a variety of biological cues, including intracellular and extracellular signals, to serve as a central regulator of cell metabolism, growth, proliferation and survival and to regulate organismal homeostasis [31]. mTOR signaling pathway can regulate many biological processes and is also associated with many pathological conditions, including cancer. Several reasons could be summarized for the importance of mTOR pathway in cancer pathogenesis [31]: (a) genes involved in PI3K signaling pathway (upstream of mTOR pathway) are often mutated in cancer, (b) TP53 mutation could promote mTOR activation [32], (c) mutations in genes of mTOR pathway always result in a superfluous phosphorylation, which could promote ribosome biogenesis, protein synthesis, and angiogenesis to support cell growth and proliferation or regulates cell cycle progression and survival [33, 34]. Hippo signaling pathway exerts critical roles in modulating cell proliferation, cell apoptosis [35], drug resistance [36] and has been demonstrated to contribute to the progression of various diseases including cancer [35]. Mutations or altered expression of gene components in Hippo pathway could promote the migration, invasion, and malignancy of cancer cells [37,38,39].
It will be very interesting to study how these pathways contribute to lung tumorigenesis, and it might be of great significance to development of small molecule or antibody drugs targeting core components in these pathways to provide new therapeutic strategies for future successful treatment of lung cancer. At present, there are many therapeutic agents that are designed to target the mTOR [40,41,42] and Hippo signaling pathway [43,44,45,46] that could serve as treatment options. Our results suggest the potential emerging therapeutic targets for IAC patients with mutations in genes involved in the mTOR and Hippo pathway.
Conclusion
In summary, we analyzed the differences in somatic mutations and clinical characteristics among different LUAD subtypes. Our results revealed the heterogeneity of gene mutations and significantly altered pathways between IAC and AIS/MIA/LMAC subtypes, suggesting the potential mechanism of different prognosis in different LUAD cohorts and individualized clinical management of patients is needed among different subtypes.
Data availability
The data that support the findings of this study are not publicly available due to their containing information that could compromise the privacy of research participants but are available from the corresponding author on reasonable request.
Abbreviations
- IAC:
-
Invasive adenocarcinoma
- MIA:
-
Microinvasive adenocarcinoma
- AIS:
-
Adenocarcinoma in situ
- LMAC:
-
Lung mixed adenocarcinoma
- NSCLC:
-
Non-small cell lung cancer
- LUAD:
-
Lung adenocarcinoma
- IASLC:
-
International Association for the Study of Lung Cancer
- ATS:
-
American Thoracic Society
- ERS:
-
European Respiratory Society
- FFPE:
-
Formalin-fixed, paraffin-embedded
- IGV:
-
Integrative Genomics Viewer
References
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. Ca Cancer J Clin. 2023;73(1):17–48.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J Clin. 2018;68(6):394–424.
Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JH, Beasley MB, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243–60.
Travis W, Brambilla E, Noguchi M, Nicholson A, Geisinger K, Yatabe Y, et al. The new IASLC/ATS/ERS international multidisciplinary lung adenocarcinoma classification. J Thoracic Oncol. 2011;6(2):244–85.
Kobayashi Y, Mitsudomi T. Management of ground-glass opacities: should all pulmonary lesions with ground-glass opacity be surgically resected? Translat Lung Cancer Res. 2013;2(5):354.
Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ Jr, Wu Y-L, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299–311.
Gettinger S, Horn L, Jackman D, Spigel D, Antonia S, Hellmann M, et al. Five-year follow-up of nivolumab in previously treated advanced non–small-cell lung cancer: results from the CA209-003 study. J Clin Oncol. 2018;36(17):1675–84.
Ishida H, Shimizu Y, Sakaguchi H, Nitanda H, Kaneko K, Yamazaki N, et al. Distinctive clinicopathological features of adenocarcinoma in situ and minimally invasive adenocarcinoma of the lung: a retrospective study. Lung Cancer. 2019;129:16–21.
Yotsukura M, Asamura H, Motoi N, Kashima J, Yoshida Y, Nakagawa K, et al. Long-term prognosis of patients with resected adenocarcinoma in situ and minimally invasive adenocarcinoma of the lung. J Thorac Oncol. 2021;16(8):1312–20.
Liu S, Wang R, Zhang Y, Li Y, Cheng C, Pan Y, et al. Precise diagnosis of intraoperative frozen section is an effective method to guide resection strategy for peripheral small-sized lung adenocarcinoma. J Clin Oncol. 2016;34(4):307–13.
Zhang J, Wu J, Tan Q, Zhu L, Gao W. Why do pathological stage IA lung adenocarcinomas vary from prognosis?: a clinicopathologic study of 176 patients with pathological stage IA lung adenocarcinoma based on the IASLC/ATS/ERS classification. J Thorac Oncol. 2013;8(9):1196–202.
Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6(2):244–85.
Caso R, Sanchez-Vega F, Tan KS, Mastrogiacomo B, Zhou J, Jones GD, et al. The underlying tumor genomics of predominant histologic subtypes in lung adenocarcinoma. J Thorac Oncol. 2020;15(12):1844–56.
Hung J-J, Yeh Y-C, Jeng W-J, Wu K-J, Huang B-S, Wu Y-C, et al. Predictive value of the International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification of lung adenocarcinoma in tumor recurrence and patient survival. J Clin Oncol. 2014;32(22):2357–64.
Shi J, Hua X, Zhu B, Ravichandran S, Wang M, Nguyen C, et al. Somatic genomics and clinical features of lung adenocarcinoma: a retrospective study. PLoS Med. 2016;13(12): e1002162.
Network CGAR. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543.
Wang Y, Liu B, Min Q, Yang X, Yan S, Ma Y, et al. Spatial transcriptomics delineates molecular features and cellular plasticity in lung adenocarcinoma progression. Cell Discovery. 2023;9(1):96.
Chen H, Carrot-Zhang J, Zhao Y, Hu H, Freeman SS, Yu S, et al. Genomic and immune profiling of pre-invasive lung adenocarcinoma. Nat Commun. 2019;10(1):1–6.
Wang S, Du M, Zhang J, Xu W, Yuan Q, Li M, et al. Tumor evolutionary trajectories during the acquisition of invasiveness in early stage lung adenocarcinoma. Nat Commun. 2020;11(1):1–8.
Sivakumar S, Lucas FAS, McDowell TL, Lang W, Xu L, Fujimoto J, et al. Genomic landscape of atypical adenomatous hyperplasia reveals divergent modes to lung adenocarcinoma. Can Res. 2017;77(22):6119–30.
Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–83.
Chen S, Liu M, Huang T, Liao W, Xu M, Gu J. GeneFuse: detection and visualization of target gene fusions from DNA sequencing data. Int J Biol Sci. 2018;14(8):843.
Luchini C, Bibeau F, Ligtenberg M, Singh N, Nottegar A, Bosse T, et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: a systematic review-based approach. Ann Oncol. 2019;30(8):1232–43.
de la Chapelle A, Hampel H. Clinical relevance of microsatellite instability in colorectal cancer. J Clin Oncol. 2010;28(20):3380.
Jin F, Yang Z, Shao J, Tao J, Reissfelder C, Loges S, et al. ARID1A mutations in lung cancer: biology, prognostic role, and therapeutic implications. Trends Mol Med. 2023;29(8):646–58.
Passaro A, Jänne PA, Mok T, Peters S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nature Cancer. 2021;2(4):377–91.
Xiang C, Ji C, Cai Y, Teng H, Wang Y, Zhao R, et al. (20202). Distinct mutational features across preinvasive and invasive subtypes identified through comprehensive profiling of surgically resected lung adenocarcinoma. Modern Pathol. 1–12.
Yang B, Li J, Li F, Zhou H, Shi W, Shi H, et al. Comprehensive analysis of age-related somatic mutation profiles in Chinese young lung adenocarcinoma patients. Cancer Med. 2019;8(4):1350–8.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–76.
Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci. 2005;102(23):8204–9.
Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55–61.
Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivitycancer-associated hyperactivating MTOR mutations. Cancer Discov. 2014;4(5):554–63.
Zheng Y, Pan D. The Hippo signaling pathway in development and disease. Dev Cell. 2019;50(3):264–82.
Zhao Y, Yang X. The Hippo pathway in chemotherapeutic drug resistance. Int J Cancer. 2015;137(12):2767–73.
Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29(6):783–803.
Lau AN, Curtis SJ, Fillmore CM, Rowbotham SP, Mohseni M, Wagner DE, et al. Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J. 2014;33(5):468–81.
Zhang W, Gao Y, Li F, Tong X, Ren Y, Han X, et al. YAP Promotes Malignant Progression of Lkb1-Deficient Lung Adenocarcinoma through Downstream Regulation of Survivin. Can Res. 2015;75(21):4450–7.
Tan AC. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thoracic cancer. 2020;11(3):511–8.
Iksen, Pothongsrisit S, Pongrakhananon V. Targeting the PI3K/AKT/mTOR signaling pathway in lung cancer: an update regarding potential drugs and natural products. Molecules. 2021;26(13):4100.
Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10(1):1–11.
Gobbi G, Donati B, Do Valle IF, Reggiani F, Torricelli F, Remondini D, et al. The Hippo pathway modulates resistance to BET proteins inhibitors in lung cancer cells. Oncogene. 2019;38(42):6801–17.
Dey A, Varelas X, Guan K-L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discovery. 2020;19(7):480–94.
Hsu P-C, Yang C-T, Jablons DM, You L. The crosstalk between Src and Hippo/YAP signaling pathways in non-small cell lung cancer (NSCLC). Cancers. 2020;12(6):1361.
Ghafouri-Fard S, Poornajaf Y, Hussen BM, Avval ST, Taheri M, Mokhtari M (2023). Deciphering the role of Hippo pathway in lung cancer. Pathol Res Pract. 154339.
Acknowledgements
The authors thank all the patients for participation. Also, we thank all the members in the department of pathology and clinical laboratory, Renmin Hospital of Wuhan University for their help.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities [Grant number 2042022kf1094].
Author information
Authors and Affiliations
Contributions
ZM: conceptualization, methodology, investigation, project administration, resources, funding acquisition, supervision, validation, writing-review and editing. SZ, PD and ZP: data curation, formal analysis, writing-original draft, writing and review and editing, visualization. QC and JZ: software, visualization. All the authors contributed to the article and approved the final version of the manuscript submitted for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
This study was approved by the Ethical Committee of Renmin Hospital of Wuhan University (approval number: WDRY2023-K027), and was performed in accordance with the ethical standards of the World Medical Association Declaration of Helsinki.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
12032_2023_2206_MOESM4_ESM.docx
Supplementary file4 The Integrative Genomes Viewer (IGV) snapshot of ALK_EML4 fusion (A) and RET_KIF5B fusion (B). (DOCX 125 KB)
12032_2023_2206_MOESM5_ESM.docx
Supplementary file5 Gene mutation comparison in hierarchical subgroups between IAC and AIS/MIA/LMAC. Only genes with mutations in >= 4 samples are shown. IAC, invasive adenocarcinoma. AIS, adenocarcinoma in situ. MIA, minimally invasive adenocarcinoma. LMAC, lung-mixed adenocarcinoma. Black column, samples with gene mutation. Grey column, samples without gene mutation. Statistics: Fisher’s exact test. (DOCX 354 KB)
12032_2023_2206_MOESM6_ESM.docx
Supplementary file6 Cancer-related pathways comparison between IAC and AIS/MIA/LMAC. IAC, invasive adenocarcinoma. AIS, adenocarcinoma in situ. MIA, minimally invasive adenocarcinoma. LMAC, lung mixed-adenocarcinoma. Blue, samples with gene mutation. Grey, samples without gene mutation. Statistics: Fisher’s exact test. (DOCX 297 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, S., Dong, P., Pan, Z. et al. Comparison of gene mutation profile in different lung adenocarcinoma subtypes by targeted next-generation sequencing. Med Oncol 40, 349 (2023). https://doi.org/10.1007/s12032-023-02206-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-023-02206-3