
Abstract
Glioblastomas are aggressive and usually incurable high-grade gliomas without adequate treatment. In this study, we aimed to investigate the potential of desloratadine to induce apoptosis/autophagy as genetically regulated processes that can seal cancer cell fates. All experiments were performed on U251 human glioblastoma cell line and primary human glioblastoma cell culture. Cytotoxic effect of desloratadine was investigated using MTT and CV assays, while oxidative stress, apoptosis, and autophagy were detected by flow cytometry and immunoblot. Desloratadine treatment decreased cell viability of U251 human glioblastoma cell line and primary human glioblastoma cell culture (IC50 value 50 µM) by an increase of intracellular reactive oxygen species and caspase activity. Also, desloratadine decreased the expression of main autophagy repressor mTOR and its upstream activator Akt and increased the expression of AMPK. Desloratadine exerted dual cytotoxic effect inducing both apoptosis- and mTOR/AMPK-dependent cytotoxic autophagy in glioblastoma cells and primary glioblastoma cell culture.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas are tumors of central nervous system that arise from glial cells. They make up about 30% of all brain and central nervous system tumors and 80% of all malignant brain tumors. According to the World health organization (WHO) gliomas can be classified by cell type, grade, origin, and location [1]. Inaccessible localisation as well as chemo- and radio-resistance of glioma represents a huge problem in therapeutic treatment.
Glioblastoma represents a most aggressive glioma type and has very poor prognosis with survival time period of approximately 1 year [2]. In addition, glioma cells are prone to acquire drug resistance systems. Therefore, there is a need to identify agents which could overcome tumor chemotherapeutic resistance.
Biogenic amine histamine is an organic nitrogenous compound that acts through specific histaminic receptors (H1–H4). Histamine is involved in local immune response as well as in regulation of physiological function in the gastrointestinal tract and neurotransmission. Some previous studies have reported inverse association between tumorigenesis and allergic conditions [3] but also possible protumorigenic histamines’ effect. In this line, some data have shown antitumor effects related to H1-receptor antagonists among different tumor types [4,5,6,7] that made us to examine potential antitumor effect of desloratadine. Desloratadine is one of H1-receptor antagonists that is frequently used to suppress itch in pruritic diseases, such as urticaria [8]. On the other hand it was shown that it can induce apoptosis in cutaneous T-cell lymphoma in vitro [9] and express anticancer effects when investigated in vivo [10].
When anticancer effect of potentially new chemotherapeutic agent is examined, it is preferred that it does not induce inflammation as a general response to cell death. Therefore, it would be preferable that potentially anticancer agent induces apoptosis and/or autophagy as the cell death mechanisms that are not accompanied by inflammation [11].
Apoptosis (programmed cell death type I) is an active process by which cells activate intracellular death program that is mediated by caspase activation and leads to self-destruction [12]. It is widely considered to be one of the most important mechanisms by which anticancer agents accomplish its cytotoxic effect. Therefore, any deficiency in the apoptotic cascade could lead to a drug-resistant phenotype.
Beside apoptosis, as a most investigated mechanism of cell death induced by H1-receptor antagonist [3, 4], some data revealed that H1 antagonists can also induce autophagy [13, 14]. Macroautophagy (referred to here after as autophagy) is a catabolic process used for degradation and recycling of the cell’s own unnecessary or dysfunctional components [15]. Autophagy is controlled by AMP-activated protein kinase (AMPK), which is a key energy sensor that regulates cellular metabolism in order to maintain energy homeostasis in cells. During nutrient deprivation, hypoxia, oxidative stress, DNA damage, and other stress conditions, autophagy provides energy for maintaining essential cellular metabolism and can interfere with apoptotic/necrotic cell death pathways [16]. Keeping that in mind, it was shown that there is a cross-talk between apoptosis and autophagy so that, in certain situations, autophagy can also stimulate apoptosis [17] or can function as an alternative cell death pathway (programmed cell death type II) [18]. On the other hand, mammalian target of rapamycin (mTOR), cell growth regulator, is main autophagy repressor which is activated by phosphoinositide 3-kinase (PI3K)/Akt pathway [15]. One of most commonly used indicator of autophagy vesicles formation is increased expression of LC3II [19]. There are data showing that H1 antagonists like diphenhydramine and AST–HIS induce autophagy in astrocytes and MCF-7 cells, respectively [13, 14], while deptropin blocks basal autophagy in human hepatoma cells [20]
Taking into account the great antiapoptotic potential of brain tumor cells [21, 22] and the fact that almost all available anticancer treatments induce cytotoxic effects by means of apoptosis [22], it would be of great importance to investigate also a possible proautophagic effects of potential novel therapeutic treatment.
Since, to the best of our knowledge, there are not any data about potential cytotoxic effect of H1 antagonists on glioblastoma cells, we aimed to investigate the potential cytotoxic effect of tricyclic antihistamine, desloratadine, on glioblastoma cells in culture, as well as the mechanism underlying its potential cytotoxic effect.
Material and methods
Cell culture and chemicals
All chemicals were from Sigma-Aldrich unless otherwise stated.
U251 (human glioblastoma cell line) was obtained from European Collection of Cell Cultures ECACC. This cell line is not listed as a commonly misidentified cell line by International Cell Line Authentication Committee. Cells were maintained at 37 °C degrees, in a humidified atmosphere with 5% CO2. Cells were cultured in 20-mM Hepes-buffered RPMI-1640 cell culture medium supplemented with 5% fetal calf serum and 2-mM glutamine. All cells were passage 10–15 and for experiments were prepared using the conventional trypsinization procedure with trypsin/EDTA. After the trypsinization cells were seeded in 96-well flat bottom plates (15 × 104 cells/well) for viability assays and in 6 well flat bottom plates (3 × 105 cells/well) for flow cytometry analysis. For immunoblot analysis cells were seeded in Petri dishes (10 cm, 2.5 × 106 cells). After seeding, cells were rested for 24 h and then treated with desloratadine which was dissolved in cell culture medium (concentrations were from 7.8 to 1000 µM) and/or autophagy inhibitors bafilomycin A1, hydroxychloroquine, and NH4Cl, as described in Results and Figure legends. All experiments were performed after 24-h cells’ treatment except Western Blot analysis which was performed after 4-h, 6-h, 18-h and 24-h treatment with desloratadine.
Primary glioblastoma cell line
The primary glioblastoma cell culture was established from the tumor tissue collected from patients with WHO grade IV glioblastoma (Clinic of Neurosurgery, Department of Neurooncology, Clinical Center of Serbia, Belgrade). Prior informed consent was obtained in written form from all patients, and all procedures were approved by the institutions’ Ethical Committee in accordance with the Declaration of Helsinki. Isolation of human primary glioblastoma cells was performed according to the previously described procedure [23].
Cell culture medium (20-mM Hepes-buffered RPMI-1640 supplemented with 10% fetal calf serum and 2 mM glutamine) was changed after 24 h and then every 48 h until the cells reached 80%. Cells were detached by conventional trypsinization procedure and passaged 2–3 times in this manner, before the experimental procedure. Cells were rested for 24 h in cell culture medium and then treated with desloratadine for further analysis.
Determination of cell viability
Cell viability was assessed using Crystal violet (CV) assay, to stain viable, adherent cells and MTT assay to measure the activity of mitochondrial dehydrogenases [24]. The absorbances were measured in an automated micro-plate reader at 570 nm (Sunrise, TECAN, UK) and the results are presented as relative to the control value (untreated cells).
Cell shape analysis
Cell shape analysis was performed on micrographs obtained with inverted fluorescent microscope (Leica Microsystems DMIL, Wetzlar, Germany) using Leica Microsystems DFC320 camera and Leica Application Suite software (version 2.8.1) on 200 × magnification. Ten images per cell culture were used for analysis. Cell shape of U251 or glioblastoma cells was analyzed using shape descriptors (circularity, aspect ratio, roundness, and solidity) in ImageJ 1.49.
Circularity is a degree to which the cell shape is similar to a circle and is defined as follows:
with a value of 1 indicating a perfect circle and value of 0 indicating an increasingly noncircular shape [25, 26].
The aspect ratio is the ratio of the width to the height of an ellipse fitted to the region of interest and is calculated using formula [25]:
Roundness is the ratio of the cell to the calculated circular area formed by the major axis:
As the shape of the cell has an increasingly elongated shape, the value of roundness approaches 0 [27].
The measurement of the overall concavity, solidity, of a cell was defined as follows:
As the image area and convex hull area approach each other, resulting in a solidity value of 1, the more solid cell shape is [25].
Reactive oxygen species (ROS) measurement
Intracellular production of ROS was determined by measuring the intensity of green fluorescence, emitted by redox-sensitive dye dihydrorodamine 123 (DHR; Invitrogen), using flow cytometry analysis (FACSCalibur flow cytometer, BD, Heidelberg, Germany). The results are presented as dot plots and represent mean ± SD values of three measurements. Corresponding dot plots (FSC/SSC) are presented in supplementary material. In order to underline the effect of desloratadine treatment, fold increase of fluorescence intensity change relative to control cells (arbitrarily set as 1) was calculated and presented in the text (not presented in the figures).
Caspase activity
Caspase activation was measured using flow cytometry after labeling the cells with a cell-permeable FITC-conjugated PAN caspase inhibitor (ApoStat R&D systems) according to manufacturer’s instructions. The results are presented as dot plots and represent mean ± SD values of three measurements. Corresponding dot plots (FSC/SSC) are presented in supplementary material. In order to underline the effect of desloratadine treatment, fold increase of fluorescence intensity change relative to control cells (arbitrarily set as 1) was calculated and presented in the text (not presented in the figures).
Measurement of intracellular acidification
The acidic vesicles (i.e., lysosomes, autophagolysosomes) were visualized by supravital stain acridine orange (AO) under the inverted fluorescent microscope (Leica Microsystems DMIL, Wetzlar, Germany) using Leica Microsystems DFC320 camera and Leica Application Suite software (version 2.8.1). Alternatively, acridine orange-stained cells were also analyzed on a FACSCalibur flow cytometer using Cell Quest Pro software. Accumulation of acidic vesicles was quantified as red/green fluorescence ratio (mean FL3/FL1) and dot plot was presented as mean ± SD values of three measurements. Corresponding dot plots (FSC/SSC) are presented in supplementary material.
Apoptosis determination
Apoptotic cell death was analyzed by double staining with annexin (V–FITC) and propidium iodide (PI) that emitted green (FL1) and red (FL2) fluorescence, respectively. Staining was performed according to the manufacturer’s instructions (Abcam, UK). A fluorescence of annexin/PI- and PI-stained cells was analyzed using a FACS Calibur flow cytometer (BD, Heidelberg, Germany). The percentages of viable (annexin − /PI −), early apoptotic (annexin + /PI −), and late apoptotic/necrotic (annexin + /PI +) cells were determined using Cell Quest Pro software (BD, Heidelberg, Germany). Representative dot plots from three measurements are presented.
Immunoblot analysis
After incubation and treatment cells were lysed and prepared for immunoblot analysis [23]. Membranes were incubated with primary antibody for LC3II (Cat no 2775S), phospho-AMPK (Thr172) (Cat no 2531S), AMPK, phospho-AKT (Ser 473) (Cat no 9271S), AKT (Cat no 9272S), phospho-mTOR (Ser 2448) (Cat no 2971S), mTOR (Cat no 2983), actin (Cat no 4970S), and peroxidase-conjugated goat anti-rabbit IgG (Cat no 7074) (all from Cell Signal Technology), as the secondary antibody. Specific protein bands were visualized using Amersham ELC reagent (GE Healthcare). The protein levels were quantified using densitometry (Image Lab program) and expressed relative to actin (LC3II) or corresponding total protein signal (phospho-AMPK, phospho-AKT, phospho-mTOR). The results are presented as fold change in signal intensity compared to that of untreated (control) cells at the same time point, which was arbitrarily set to 1.
Statistical analysis
The statistical significance of the differences was analyzed by Student’s t test or one-way analysis of variance (ANOVA) followed by Student–Newman Keuls test. A p value of less than 0.05 was considered significant.
Results
Desloratadine exerts antitumor activity against U251 cells
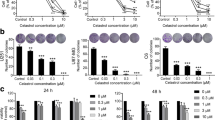
When used in different concentrations (7.8–1000 µM), desloratadine decreased U251 cell viability in dose-dependent manner, with IC50 value of 50 µM after 24 h incubation. There was no statistical significant difference between results obtained with both viability tests (CV and MTT) (Fig. 1a).

Desloratadine reduces viability of U251 glioma cells in dose-dependent manner. U251 cells were incubated for 24 h with different concentrations of desloratadine (7.8–1000 μM) and the cell viability was estimated by MTT and CV assays (a). Cell morphology was examined by phase contrast microscopy (10 × magnification) after 24-h treatment with different concentrations of desloratadine (b). Data are presented as mean ± SD values of three independent experiments (*p < 0.05 compared to control cells; **p < 0.01 compared to control cells)
Cytotoxic effect of desloratadine against U251 cells was further verified using phase contrast microscopy (Fig. 1b). Morphological analysis of cells treated with desloratadine showed decrease in the number of live cells (polygonal, dark, adherent cells) and an increase in the number of dead cells (round, white non-adherent cells). Untreated control cells had typical cobblestone-like appearance.
Cell shape analysis of U251 cells showed that cell circularity is significantly increased in all treated cells in dose-dependent manner (control 0.44 ± 0.07; DSL 25 µM 0.48 ± 0.06; DSL 50 µM 0.61 ± 0.06; DSL 100 µM 0.90 ± 0.03, p < 0.01). Aspect ratio was significantly decreased only in DSL 100 µM-treated cells (1.26 ± 0.12, p < 0.01), whereas there was no statistical difference between other groups (control 2.38 ± 0.34; DSL 25 µM 2.44 ± 0.44; DSL 50 µM 2.49 ± 0.73). Roundness was significantly increased only in DSL 100 µM-treated cells (0.82 ± 0.05, p < 0.01), whereas there was no statistical difference between other groups (control 0.57 ± 0.05; DSL 25 µM 0.55 ± 0.06; DSL 50 µM 0.55 ± 0.08). Solidity is significantly increased in all treated cells in dose-dependent manner (control 0.72 ± 0.06; DSL 25 µM 0.75 ± 0.06; DSL 50 µM 0.88 ± 0.04; DSL 100 µM 0.99 ± 0.01, p < 0.01) except between control cell and DLS 25 µM-treated cells.
Desloratadine induce oxidative stress and caspase-dependent apoptosis of U251 cells
Desloratadine significantly increased production of ROS in dose-dependent manner that was verified with DHR staining (Fig. 2a), suggesting that oxidative stress is a contributing factor of its observed cytotoxic effect. When fluorescence intensity of control cells was arbitrarily set as 1, calculated increase against control cells was 3.3- and 4.1-fold, in desloratadine treatment (25 µM and 50 μM, respectively).

Desloratadine induce oxidative stress and caspase-dependent apoptosis in U251 cells. Cells were treated with desloratadine for 24 h and ROS production; (a) caspase activation (b) and apoptotic/necrotic cell death (c) were examined by flow cytometry using DHR, Apostat, and annexin/PI staining, respectively. Data are presented as mean ± SD values of three measurements (*p < 0.05, **p < 0.01, compared to control cells)
Measurement of caspase activation, using fluorescent-labeled pan-caspase inhibitor ApoStat after 24-h treatment, showed significant caspase activation. When cells were treated with 25 µM and 50 µM of desloratadine, the caspase activity increased 3.6 and 5.9 times relatively to control cells, respectively (Fig. 2b). These results could indicate that desloratadine induces apoptosis which is caspase dependent.
Further, apoptotic cell death was investigated and verified using Ann/PI staining. We showed that desloratadine induced an increase in the percentage of Ann-positive cells in dose-dependent manner. Namely, total percent of Ann-positive cells in early (lower right) and late apoptosis (upper right quadrant) treated with 12.5 µM, 25 µM, and 50 µM of desloratadine was 8.65%, 16.79%, and 47.08%, respectively (Fig. 2c). These data indicate that desloratadine induces apoptotic cell death in U251 cells.
Desloratadine induce AMPK/mTOR-dependent autophagy of U251 cells
The presence of autophagy was investigated first qualitatively by fluorescent microscopy. U251 cells treated with desloratadine (50 µM) had increased number of intracytoplasmic acidic vesicles (lysosomes/autophagolysosomes) that correspond to orange–red fluorescent spots in green cytoplasm, compared to control (untreated cells) (Fig. 3a).

Desloratadine induce AMPK/mTOR-dependent autophagy in U251 cells. The presence of intracytoplasmic acidic vesicles after 24-h treatment with desloratadine (50 μM) was verified by fluorescent microscopy (20 × magnification) after AO staining (a), while quantification of acidic vesicles content through FL3/FL1 ratio was analyzed by flow cytometry (b). The levels of phosphorylated/total Akt, mTOR, and AMPK relative to total corresponding protein expression (c) and LC3II relative to actin (d) were determined by immunoblotting for the indicated time periods and signal intensity are presented below the protein bands. Representative blots from three independent experiments are presented. To analyze the nature of autophagy induced with desloratadine, cell viability of U251 cells simultaneously treated with desloratadine (50 µM) and one of autophagy inhibitors: bafilomycin (5 nM), chloroquine (10 µM), and ammonium chloride (5 mM) was measured using MTT assay (e). Data are presented as mean ± SD values of three measurements (**p < 0.01 compared to control cells)
To quantify acidic vesicles content, we measured the increase of red/green (FL3/FL1) fluorescence ratio, using flow cytometry. After 24-h treatment with desloratadine (50 µM) flow cytometry analysis revealed significant increase in FL3/FL1 fluorescence ratio (FL3/FL1 = 1.7) comparing to control cells (FL3/FL1 = 1) (Fig. 3b). Given data suggest the increase in acidic intracytoplasmic vesicles content.
As AO staining is not specific for autophagolysosomes only, since it can label also other acidic vesicles, we further performed immunoblot analysis to confirm the presence of autophagy-related proteins and to define intracellular signaling pathways involved in autophagy induction. Therefore, we measured the expression of LC3II protein, which is a constituent of autophagosome membrane. It is formed through the conversion of soluble cytoplasmic form of LC3I to membrane form, LC3II [19], during the maturation of autophagosomes [28]. Also, proteins of mTOR signaling pathway were measured. After 4-h, 6-h, and 18-h treatment with desloratadine (50 µM) expression of pAKT, pmTOR, and pAMPK were analyzed, while expression of LC3II was analyzed following 18-h and 24-h treatment with desloratadine (25 µM and 50 µM). Immunoblot analysis revealed that, under the treatment with desloratadine (50 µM), the expression of main autophagy repressor mTOR and its upstream activator Akt were decreased, while main mTOR inhibitor, AMPK, was increased after 18-h incubation (Fig. 3c). Increase in one of autophagy markers, LC3II was observed after 18-h and 24-h incubation (Fig. 3d). These results indicate that desloratadine induces AMPK/mTOR-dependent autophagy.
To define whether the autophagy is the type of cell death or cell protection mechanism, U251 cells were simultaneously treated with desloratadine (50 μM) and one of well-known autophagy inhibitors: bafilomycin (5 nM), chloroquine (10 mM), or ammonium chloride (5 mM). After 24-h incubation, the presence of inhibitors reduced cytotoxicity of desloratadine suggesting that autophagy in U251 cells contributed to its cytotoxic effects (Fig. 3e).
Desloratadine exerts cytotoxic effect and induces oxidative stress and autophagy in primary glioblastoma cells
Additionally, antitumor effect of desloratadine was also confirmed on primary human glioblastoma cell culture. Viability assays revealed decrease in cell viability in dose-dependent manner, with IC50 value of 50 µM (Fig. 4a), while phase contrast microscopy revealed same morphological changes as in U251 cell line that indicated an increase in the number of dead cells (round, white non-adherent cells) (Fig. 4b).

Desloratadine exerts cytotoxic effect and induces oxidative stress and autophagy in primary human glioblastoma cells. Primary human glioblastoma cells were incubated for 24 h with different concentrations of desloratadine (7.8–1000 μM) and the cell viability was estimated by MTT and CV assays (a). Cell morphology was examined by phase contrast microscopy (10 × magnification) after 24-h treatment with desloratadine (50 μM) (b). Acidic vesicle content was verified by FL3/FL1 ratio using flow cytometry (c), while the presence of intracytoplasmic acidic vesicles was verified by fluorescent microscopy after AO staining (20 × magnification) (d). The levels of autophagy-related protein LC3II relative to actin (e) in cells treated with desloratadine (50 μM) was determined by immunoblotting for the indicated time periods and signal intensity are presented below the protein bands. Representative blots from three independent experiments are presented. ROS production (f) was measured by DHR staining using flow cytometry. Data (a, c, f) are presented as mean ± SD values of three measurements (*p < 0.05 compared to control cells; **p < 0.01 compared to control cells)
Cell shape analysis of primary glioblastoma cells showed that cell circularity is significantly increased in treated cells (control 0.28 ± 0.04; DSL 50 µM 0.35 ± 0.05, p < 0.01). There were no statistical difference in aspect ratio (control 3.42 ± 0.34; DSL 50 µM 3.73 ± 0.55) and roundness (control 0.42 ± 0.03; DSL 50 µM 0.39 ± 0.06) in-between control and DSL-treated cells. Solidity is significantly increased in DSL-treated cells (control 0.61 ± 0.07; DSL 50 µM 0.71 ± 0.04, p < 0.01).
Acidic vesicles content was quantified after AO staining as the increase of FL3/FL1 (red/green fluorescence ratio), using flow cytometry analysis. After 24-h treatment with desloratadine (25 µM) flow cytometry analysis revealed significant increase in FL3/FL1 fluorescence ratio (FL3/FL1 = 1.4) compared to control cells (FL3/FL1 = 1), while 50 µM of desloratadine significantly decreased it (FL3/FL1 = 0.6) comparing to control cells. (Fig. 4c). The presence of acidic vesicles was further confirmed by fluorescent microscopy of AO-stained glioblastoma cells which revealed an increased number of intracytoplasmic acidic vesicles (lysosomes/ autophagolysosomes) after 24-h treatment with desloratadine (50 µM) (Fig. 4d).
Additionally, the presence of autophagy vesicles was confirmed by measuring the expression of LC3II protein. After 4-h, 6-h, and 18-h treatment with desloratadine (50 µM) increase in LC3II level was observed (Fig. 4e).
Concentrations of 25 µM and 50 µM of desloratadine significantly increased production of ROS (3.2-fold increase compared to control in both concentrations). (Fig. 4f).
Unfortunately, using Ann/PI and pan-caspase inhibitor, we did not detect either apoptosis either caspase activity under the treatment of human glioblastoma primary cell culture with desloratadine (data not shown).
Discussion
In the present study we demonstrate that H1 antagonist desloratadine decreases viability of glioblastoma cells in culture, through various mechanisms, including increase in reactive oxygen species production, induction of apoptosis, and autophagy. Aside from a report that other H1 antagonists can induce apoptosis and autophagy [3, 6, 13, 14], this is the first study clearly demonstrating desloratadine-induced activation of Akt/mTOR-dependent cytotoxic autophagy in tumor cells.
Keeping in mind that glioblastoma is an aggressive tumor that has the highest rate of mortality among all malignant brain tumors [29] there is a huge requirement to identify novel target molecules to which more effective therapeutic approaches can be developed. It was already shown that H1 antagonists can exert cytotoxic effects through the induction of both apoptosis and autophagy [3, 6, 13, 14, 30]. Even the histamine deficiency can induce apoptosis [31] suggesting the role of histamine receptor activity in cell survival. In the current study, we investigated the potential cytotoxic effects of H1-receptor antagonist, desloratadine, on glioblastoma cell line and primary human glioblastoma cell culture.
Glioblastoma cells express great chemoresistance [32] that can be supported by a presence of membranous ABC (ATP-binding Cassette) superfamily of efflux pumps located on blood–brain barrier [33]. Also, genetic instability caused with numerous mutations, deletions, and genetic amplifications contributes its chemoresistance [34]. Having in mind all this complex glioblastoma characteristics that lead to their great biological diversity, there is a need for combined therapeutic approaches, targeting more than one signaling pathway. The results of our study contribute to this approach, since desloratadine affected both, apoptosis and autophagy, as two different programmed cell death mechanisms. In addition to already known desloratadine cytotoxic effect against cutaneous T-cell lymphoma cell lines, EJ and SW780 cells [9, 30] our results clearly shown dose-dependent cytotoxic activity of desloratadine against U251 cell line. Nevertheless, the obtained IC50 value for desloratadine is in accordance with its cytotoxic activity on non-small cell lung cancer (NSCLC) cell lines [10]. Its cytotoxic effects are, even partially, achieved through the induction of ROS overproduction which is a well-known trigger that could induce cell death [35]. Moreover, we found that desloratadine induced the externalization of phosphatidylserine in cell membrane, together with an increase in caspase activation, both hallmarks of apoptosis. Given data are in accordance with studies taken by Plekhova et al., and Ma et al., where Ann + macrophages as well as bladder cancer EJ and SW780 cells were detected after desloratadine treatment, respectively [30, 36]. Also, it was shown that desloratadine can induce apoptosis in cutaneous T-cell lymphoma cell lines through inhibition of STAT3 and c-Myc activities. Additionally, caspase-dependent proapoptotic activity was previously reported for other H1 antagonists, such as diphenhydramine and meclizine [5, 7].
It is known that autophagy in tumor cells can either promote apoptosis or serve as mechanism of programmed cell death type II. Several reports provided evidence that spontaneous autophagy is reduced in glioblastoma [35, 37] suggesting that this could be responsible for its considerable malignant potential. In contrast, there are data that show autophagy as a prosurvival mechanism that protects cancer cells from apoptotic or necrotic cell death induced by different anticancer drugs [17]. In light of all the foregoing considerations we were interested to examine this process using glioblastoma cell line.
We have shown that desloratadine induces autophagy in U251 glioblastoma cells through increased expression of acidic cytosolic vesicles, both qualitatively and quantitatively. Given data are additionally confirmed with increased expression of autophagosome-associated LC3II protein. Ma et al. also impaired expression of autophagy-related proteins such as Beclin 1, p62, and LC3I/II in EJ and SW780 cells after treatment with desloratadine [30]. After diphenhydramine treatment acidic vacuoles were detected in astrocytes, which is confirmed with increased LC3I to LC3II conversion [13] Autophagy followed with increased expression of LC3II protein was also reported for some other H1 antagonists [6, 14]. Furthermore, we looked toward the intracellular signaling pathway underlying autophagic process of U251 glioblastoma cells induced by desloratadine. AMPK is one of the positive regulators of autophagy that stimulates autophagy in response to energy depletion (increased AMP/ATP ration) [38] and that way connecting cellular energy homeostasis and autophagy. Beside AMPK, we looked toward activity of AMPK downstream target, mTOR, as well as prosurvival signaling molecule Akt involved in stimulation of protein synthesis and cell proliferation. Akt signaling pathway is usually regarded as independent signaling route from AMPK signaling pathway and as alternative pathway that regulates autophagy [39]. In our experimental model we observed the upregulation of AMPK and downregulation of mTOR and Akt indicating that desloratadine-induced autophagy of U251 cells is mTOR/AMPK dependent. Astemizol is the other histamine antagonist that induced autophagy in breast cancer cells [14]. Beside autophagy and similarly to desloratadine, astemizol also induced apoptosis through caspase activation and increased production of ROS. On the other hand, there are data indicating that antihistamine deptropin blocked basal autophagy which is experimentally confirmed through increased LC3II expression, while Akt, AMPK, vacuolar protein sorting 34 (VPS34), and Atg7 expression were not changed after treatment in human hepatoma cells [20]. Having in mind that autophagy could be either prosurvival or cell death mechanism, using simultaneous treatment with different autophagy inhibitors and desloratadine, we have shown that the autophagy in glioblastoma cells induced by desloratadine is cytotoxic. Together with previously reported finding that glioblastomas cells have reduced spontaneous autophagy; this induction of cytotoxic autophagy with desloratadine could be of great importance in modulation of current chemo-radiation treatment. The effect of autophagy modulation on glioblastoma radio sensitivity was intensively investigated previously [40] and, even still not completely clear, it is evident that autophagy modulation offers a promising, novel approach to glioblastoma treatment.
In addition to desloratadine effects observed in U251 glioblastoma cell line, we showed also similar patterns of desloratadine action against human glioblastoma primary cell culture. Its cytotoxic effect was performed through induced ROS production that possibly initiated autophagy verified through an increase in number of acidic vesicles as well as an increase in LC3II autophagosome-associated protein.
The observed results clearly demonstrate that desloratadine induces apoptosis accompanying with cytotoxic mTOR/AMPK-dependent autophagy. This dual antitumor potential of desloratadine is particularly important for tumor cells that express great antiapoptotic potential, such as cells of glioblastoma.
Data availability
Data from this manuscript are available and you can contact corresponding author whenever you are interested.
References
Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23:1231–51. https://doi.org/10.1093/neuonc/noab106.
DeAngelis LM. Brain tumors. N Engl J Med. 2001;344:114–23. https://doi.org/10.1056/NEJM200101113440207.
Pouchieu C, Raherison C, Piel C, et al. Allergic conditions and risk of glioma and meningioma in the CERENAT case-control study. J Neurooncol. 2018;138:271–81. https://doi.org/10.1007/s11060-018-2816-6.
Hadzijusufovic E, Peter B, Gleixner KV, et al. H1-receptor antagonists terfenadine and loratadine inhibit spontaneous growth of neoplastic mast cells. Exp Hematol. 2010;38:896–907. https://doi.org/10.1016/j.exphem.2010.05.008.
Jangi S-M, Díaz-Pérez JL, Ochoa-Lizarralde B, et al. H1 histamine receptor antagonists induce genotoxic and caspase-2-dependent apoptosis in human melanoma cells. Carcinogenesis. 2006;27:1787–96. https://doi.org/10.1093/carcin/bgl021.
Nicolau-Galmés F, Asumendi A, Alonso-Tejerina E, et al. Terfenadine induces apoptosis and autophagy in melanoma cells through ROS-dependent and -independent mechanisms. Apoptosis : Int J Program Cell Death. 2011;16:1253–67. https://doi.org/10.1007/s10495-011-0640-y.
Lin J-C, Ho Y-S, Lee J-J, et al. Induction of apoptosis and cell-cycle arrest in human colon cancer cells by meclizine. Food and Chem Toxicol: An Int J Publ British Ind Biol Res Assoc. 2007;45:935–44. https://doi.org/10.1016/j.fct.2006.11.016.
Cevikbas F, Steinhoff M, Fau - Ikoma A and Ikoma A,. Role of spinal neurotransmitter receptors in itch: new insights into therapies and drug development. CNS Neurosci Ther. 2011;17:742–9. https://doi.org/10.1111/j.1755-5949.2010.00201.x.
Döbbeling U, Waeckerle-Men Y, Zabel F, et al. The antihistamines clemastine and desloratadine inhibit STAT3 and c-Myc activities and induce apoptosis in cutaneous T-cell lymphoma cell lines. Exp Dermatol. 2013;22:119–24. https://doi.org/10.1111/exd.12086.
Ellegaard A-M, Dehlendorff C, Vind AC, et al. Repurposing cationic amphiphilic antihistamines for cancer treatment. EBioMedicine. 2016;9:130–9. https://doi.org/10.1016/j.ebiom.2016.06.013.
Hengartner MO. Apoptosis: corralling the corpses. Cell. 2001;104:325–8. https://doi.org/10.1016/s0092-8674(01)00219-7.
Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15.
Hu W-W, Yang Y, Wang Z, et al. H1-antihistamines induce vacuolation in astrocytes through macroautophagy. Toxicol Appl Pharmacol. 2012;260:115–23. https://doi.org/10.1016/j.taap.2012.01.020.
Jakhar R, Paul S, Bhardwaj M, et al. Astemizole-Histamine induces Beclin-1-independent autophagy by targeting p53-dependent crosstalk between autophagy and apoptosis. Cancer Lett. 2016;372:89–100. https://doi.org/10.1016/j.canlet.2015.12.024.
Ravanan P, Srikumar IF, Talwar P. Autophagy: the spotlight for cellular stress responses. Life Sci. 2017;188:53–67. https://doi.org/10.1016/j.lfs.2017.08.029.
Periyasamy-Thandavan S, Jiang M, Schoenlein P, et al. Autophagy: molecular machinery, regulation, and implications for renal pathophysiology. Am J Physiol Renal Physiol. 2009;297:F244–56. https://doi.org/10.1152/ajprenal.00033.2009.
Fimia GM, Piacentini M. Toward the understanding of autophagy regulation and its interplay with cell death pathways. Cell Death Differ. 2009;16:933–4. https://doi.org/10.1038/cdd.2009.47.
Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. https://doi.org/10.1038/nrm2239.
Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–18. https://doi.org/10.1016/j.biocel.2004.05.009.
Liang Y-C, Chang C-C, Sheu M-T, et al. The antihistamine deptropine induces hepatoma cell death through blocking autophagosome-lysosome fusion. Cancers. 2020;12:1610. https://doi.org/10.3390/cancers12061610.
Sharma A, Boise LH, Shanmugam M. Cancer metabolism and the evasion of apoptotic cell death. Cancers. 2019;11:1144. https://doi.org/10.3390/cancers11081144.
Pfeffer CM, Singh ATK. Apoptosis: a target for anticancer therapy. Int J Mol Sci. 2018;19:448. https://doi.org/10.3390/ijms19020448.
Pantovic A, Bosnjak M, Arsikin K, et al. In vitro antiglioma action of indomethacin is mediated via AMP-activated protein kinase/mTOR complex 1 signalling pathway. Int J Biochem Cell Biol. 2017;83:84–96. https://doi.org/10.1016/j.biocel.2016.12.007.
Kaludjerović GN, Miljković D, Momcilović M, et al. Novel platinum(IV) complexes induce rapid tumor cell death in vitro. Int J Cancer. 2005;116:479–86. https://doi.org/10.1002/ijc.21080.
Lobo J, See EY, Biggs M, et al. An insight into morphometric descriptors of cell shape that pertain to regenerative medicine. J Tissue Eng Regen Med. 2016;10:539–53. https://doi.org/10.1002/term.1994.
Pantic I, Nesic D, Basailovic M, et al. Chromatin fractal organization, textural patterns, and circularity of nuclear envelope in adrenal zona fasciculata cells. Microsc Microanal. 2016;22:1120–7. https://doi.org/10.1017/s1431927616011910.
Koo HJ, Sung YS, Shim WH, et al. Quantitative computed tomography features for predicting tumor recurrence in patients with surgically resected adenocarcinoma of the lung. PLoS ONE. 2017;12:e0167955. https://doi.org/10.1371/journal.pone.0167955.
Reggiori F, Klionsky DJ. Autophagy in the eukaryotic cell. Eukaryot Cell. 2002;1:11–21. https://doi.org/10.1128/ec.01.1.11-21.2002.
Krex D, Klink B, Hartmann C, et al. Long-term survival with glioblastoma multiforme. Brain: J Neurol. 2007;130:2596–606. https://doi.org/10.1093/brain/awm204.
Ma J, Qi J, Li S, et al. Desloratadine, a novel antigrowth reagent for bladder cancer. Technol Cancer Res Treat. 2020;19:1533033820926591–1533033820926591. https://doi.org/10.1177/1533033820926591.
Deng L, Hong T, Lin J, et al. Histamine deficiency exacerbates myocardial injury in acute myocardial infarction through impaired macrophage infiltration and increased cardiomyocyte apoptosis. Sci Rep. 2015;5:13131–13131. https://doi.org/10.1038/srep13131.
Quezada C, Garrido W, Oyarzún C, et al. 5’-ectonucleotidase mediates multiple-drug resistance in glioblastoma multiforme cells. J Cell Physiol. 2013;228:602–8. https://doi.org/10.1002/jcp.24168.
Efferth T. The human ATP-binding cassette transporter genes: from the bench to the bedside. Curr Mol Med. 2001;1:45–65. https://doi.org/10.2174/1566524013364194.
Ferguson LR, Chen H, Collins AR, et al. Genomic instability in human cancer: molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin Cancer Biol. 2015;35:S5–24. https://doi.org/10.1016/j.semcancer.2015.03.005.
Kanzawa T, Germano IM, Komata T, et al. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448–57. https://doi.org/10.1038/sj.cdd.4401359.
Plekhova NG, Eliseeva EV, Dubnyak IN. Antihistamines modulate functional activity of macrophages. Bull Exp Biol Med. 2021;170:759–62. https://doi.org/10.1007/s10517-021-05150-2.
Paglin S, Hollister T, Delohery T, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Can Res. 2001;61:439–44.
Kahn BB, Alquier T, Carling D, et al. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. https://doi.org/10.1016/j.cmet.2004.12.003.
Memmott RM, Dennis PA. Akt-dependent and-independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009;21:656–64. https://doi.org/10.1016/j.cellsig.2009.01.004.
Sharma K, Goehe R, Beckta JM, et al. Autophagy and radiosensitization in cancer. EXCLI J. 2014;13:178–91.
Acknowledgements
We would like to express our gratitude to Mrs. Nada Bojović for help in biochemical analyses. Financial support for this study was granted by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Project 200110).
Funding
Ministarstvo Prosvete, Nauke i Tehnološkog Razvoja, 200110, Sasenka Vidicevic-Novakovic.
Author information
Authors and Affiliations
Contributions
AI designed the study. SV-N, JT, KK, NT, and JZ collected data, prepared figures, and analyzed and interpreted results. TM did morphometry and statistics. DG, RI, and SR collected glioblastoma tissue samples. AI, ZS, JT, and SV-N wrote the main manuscript text.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vidicevic-Novakovic, S., Stanojevic, Z., Tomonjic, N. et al. Proapoptotic and proautophagy effect of H1-receptor antagonist desloratadine in human glioblastoma cell lines. Med Oncol 40, 241 (2023). https://doi.org/10.1007/s12032-023-02117-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-023-02117-3