
Abstract
Drug resistance is one of the clinical challenges that limits the effectiveness of chemotherapy. Recent reports suggest that the unfolded protein response (UPR) and endoplasmic reticulum stress-adaptation signalling pathway, along with increased activation of its inositol-requiring enzyme 1α (IRE1α) arm, may be contributors to the pathogenesis of colorectal cancer (CRC). Here, we aimed to target the IRE1α/XBP1 pathway in order to sensitise CRC cells to the effects of chemotherapy. The CT26 colorectal cell line was treated with tunicamycin, and then was exposed to different concentrations of 5-fluorouracil (5-FU), either alone and/or in combination with the IRE1α inhibitor, 4µ8C. An MTT assay, flow cytometry and RT-PCR were performed to determine cell growth, apoptosis and IRE1α activity, respectively. In vivo BALB/c syngeneic colorectal mice received chemotherapeutic drugs. Treatment responses, tumour sizes and cytotoxicity were assessed via a range of pathological tests. 4µ8C was found to inhibit the growth of CRC, at a concentration of 10 µg/ml, without detectable cytotoxic effects and also significantly enhanced the cytotoxic potential of 5-FU, in CRC cells. In vivo experiments revealed that 4µ8C, at a concentration of 50 µM/kg prevented tumour growth without any cytotoxic or metastatic effects. Interestingly, the combination of 4µ8C with 5-FU remarkably enhanced drug responses, up to 40–60% and also lead to significantly greater inhibition of tumour growth, in comparison to monotherapy, in CRC mice. Targeting the IRE1α/XBP1 axis of the UPR could enhance the effectiveness of chemotherapy in both in vitro and in vivo models of CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is recognised as a significant health threat and is the second most fatal cancer globally [1]. Despite significant progress in prolonging overall survival via different treatments, the management of CRC remains a challenge [2, 3]. One of the current therapeutic regimes for CRC is 5-fluorouracil (5-FU)-based therapy; however, cases of drug resistance have been reported, which are among the main reasons for chemotherapeutic failure and tumor recurrence [4]. Therefore, finding new methods to reduce or eliminate drug resistance have always been key concerns for clinicians treating these patients [5]. One of the main cellular responses during chemotherapy and the development of drug resistance is ER stress (ERS) [6]. ERS is activated during changes in the pathology or physiology of all living cells such as nutrient deprivation, lack of oxygen, reactive oxygen species (ROS) and the accumulation of unfolded or misfolded proteins. The unfolded protein response (UPR) is a compensatory mechanism to counteract the effects of ERS [7]. The UPR is activated and orchestrated by three ER-localized proteins including inositol-requiring transmembrane kinase/endonuclease 1α (IRE1α), pancreatic ER kinase (PERK) and activating transcription factor 6 (ATF6).
IRE1α is a transmembrane protein consisting of two N- and C-terminal domains; the N-terminal domain known as ER luminal domain which interacts with unfolded proteins and the C-terminal domain, which is in the cytoplasmic region consisting of serine/threonine kinase and endoribonuclease (RNase) domains, which initiate the UPR signalling. In response to the accumulation of unfolded proteins, dimerisation and autophosphorylation of IRE1α is achieved by the kinase domain, with splicing and removal of a 26 nucleotide intron from XBP1 mRNA by the RNase domain of IRE1α [8]. The XBP1 spliced form (XBP1s) acts as transcription factor, and translocates to the nucleus to target the genes involved in ER-associated degradation (ERAD) of misfolded proteins, as well as protein entry to ER, folding capacity and biosynthesis of phospholipid [9]. The RNase domain of IRE1α is also required for degradation of the mRNAs encoding for ER proteins, through the regulated IRE1dependent decay (RIDD) pathway [10]. Overactivation of XBP1s is found in some diseases such as cancers and metabolic conditions, and has also been implicated in tumor progression [11, 12]. In this regard, inhibition of IRE1α expression, either by genetic or pharmacological means, may suppress the proliferation of cancerous cells, and thus this signal transducer may be considered as a suitable target for drug design [13, 14]. Pharmacological inhibitors of IRE1α have been designed using different strategies, based on the kinase or RNase domains. Compounds that target the RNase activity include salicylaldehyde analogs (e.g., MK0186893) and umbelliferones (e.g., 4µ8c) [15]. 4µ8c, but not MK0186893, specifically inhibits the RNase activity of IRE1α without impacting on its phosphorylation. Thus, 4µ8c is useful for studying specific activities of IRE1α, such as RIDD and XBP1 splicing [16]. Besides, 4µ8c as a type III inhibitor, has less toxicity, fewer effects on immune responses, and decreases cell proliferation in cancer cells [17]. The type I and II of IRE1α inhibitors include kinase-based compounds that inhibit or activate IRE1α signalling, mainly through kinase inhibition without directly targeting the RNase domain. However, due to the high toxicity and off-target activity (such as interactions with other kinases), these pharmacological inhibitors are less useful [17, 18].
In this study, we first aimed to reduce the growth of tumor cells by inhibiting the RNase domain of IRE1α with 4µ8C, and secondly to increase the degree of drug sensitivity by using 4µ8C and lower doses of 5-FU as chemotherapeutic drugs. Our findings underline the importance of targeting the IRE1/XBP1 pathway in colorectal cancer and also underline the therapeutic importance of this pathway and its contribution to drug resistance.
Materials and methods
Cell culture, cell viability and cell cytotoxicity
The CT26 colorectal cancer cell line was prepared from the cell bank of Pasteur Institute of Iran, Tehran. The cells were cultured in standard condition of RPMI1640 medium, (10%, v/v) of fetal bovine serum (FBS) and 1% of antibiotics (100 U/ml penicillin, and 100 µg/ml streptomycin). Then the cells were maintained at 37 °C incubator, 95% humidity and 5% CO2. To examine the CRC viability, a trypan blue exclusion test was applied followed by tunicamycin (TM) (1 µg/ml), and, also without TM treatment. TM causes ERS through inhibiting protein glycosylation. Briefly, the cells were seeded in an appropriate number, 24 h before treatment, then the different concentrations of 5-Fu (Eber pharma) (5,10, 20, 50, 100 µg/ml), 4µ8c (Cat no HY-19707 MCE, USA) (1,2, 5, 10, 20 µg/ml), and combination of them were used, then the cells were trypsinised and stained with 0.4% trypan blue. The alive and dead cells were counted under an inverted light microscope (Zeiss, Germany).
The cytotoxicity of the cells was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test. Briefly, the cells were treated with TM and different concentrations of 5-FU, either alone or in combination with 4µ8C, for a period of 24 h. The cells were then incubated for 4 h following by adding MTT solution (5 mg/ml) and dimethyl sulfoxide (DMSO) (100 µL). The optical density (OD) of the cells was read at 580 nm by ELISA reader (Thermo/Lab Systems Multiskan, USA). Then the cytotoxicity of the cells was calculated in comparison to control cells, as reported elsewhere [19].
Cell death analysis
Dead cells, such as apoptotic or necrotic cells, were assessed by annexin/PI staining and measured by flow cytometric analysis [20]. Briefly, both the treated and control cells were harvested and subsequently stained with 1X of binding buffer, 1 M Annexin V Fluorescein Isothiocyanate (FITC), and 1 mg/mL PI solutions. Then, the cells were kept in a dark place at room temperature for 10 min. The analysis should be done immediately less than one hour with a FACS Calibur flow cytometer (FACS Calibur, BD Bioscience, USA).
RT-PCR and gel electrophoresis
Total RNA from all the treated and control cells were extracted using RNX-Plus solution (CinaClon Co, Iran). The RNA concentration and optical density (OD) were measured by NanoDrop™ One C Spectrophotometer (Thermo Fisher Scientific Inc). A total of 1 µg of RNA was used for cDNA synthesis according to manufacturers’ instruction (Bio fact, South Korea). RT-PCR was used in a total reaction of 20 µl (cDNA template, forward and reverse primers, master mix and double distilled water) in the PCR set. The specific primers for both XBP1s and XBP1u were forward 5′ GAACACGCTTGGGAATGGACAC3′ and reverse 5′ AGAAAGGGAGGCTGGTAAGGAAC3′. The PCR program was run at 95 °C, 5 min, 38 cycles (95 °C, 25 s, 65 °C, 30 s and 72 °C 30 S), then 72 °C for 30 s. The product of PCR was run on 2% gel electrophoresis. The quantitative data were analyzed by image J 152 software and reported as quantitative information.
Animals
The 8 ± 12 week-old male BALB/C wild-type mice were obtained from the Pasteur Institute of Tehran, Iran. They were transferred to the animal care center and housed under standard conditions in a 12 h light and dark cycle and were given adequate mouse food and water. During their maintenance, and also during the experiment, the animals were excluded from the study whenever any problems, such as unusual weight loss or gain, were observed. The project was approved by the Ethical Committee at NIMAD institute, Iran, approval ID 1398.265.
Experimental CRC mice model and treatments
A total of 106 CT26 cells, in 100 µl RPMI media, were injected into the right flank of BALB/C wild-type (WT) mice. Following 1–2 weeks, when the tumor volume reached to 100–300 mm3, the mice were randomly classified into four groups, each containing four mice. The mice were received an intraperitoneal injection of 100 µl DMSO as control and therapeutic regimes containing 5-FU (20 mM/kg), 4µ8C (50 µM/kg) and a combination of them two days in a week for three weeks. Body weights and tumor volumes were measured every other day and calculated with the following formula-\(\frac{1}{2}*lengh*{\left(width\right)}^{2}\). Then the tumors and vital organs, such as kidneys and liver, were surgically removed 20 days after the last chemotherapy and kept in formalin for pathological analysis.
Pathologic complete response (pCR)
Pathologic complete response (pCR) was calculated by counting the number of cancerous cells in tissue samples after chemotherapy. Results of the scoring were reported as follows: RX means that the residual tumor cannot be distinguished; R0 means that no residual tumors was observed (100%); R1 indicated that single cells or rare small groups of cancer cells were observed (near complete response, 60–100%), R2 indicated residual cancer with evident tumor regression partial response, 40–60%), R3 represented extensive residual cancer being present with no evidence of tumour regression (poor or no response, 0–40%).
Metastases and toxicity assay
The tumours and vital organs including livers, kidneys were made as paraffin-fixed embedded tissues. Then, sections of 5–10 μm thickness were prepared from the paraffin block with a microtome. Sections were subsequently stained with H&E, according to routine laboratory protocol. The metastatic and mitotic cells were scored and counted in 1 mm2 of a microscopic frame, under light microscopy.
Statistical analyses
Results were obtained from three independent experiments, each performed in triplicate. The mean standard deviation (SD) and Student’s t-test were performed using SPSS software, Ver. 16 or GraphPad Prism 8.3.0. In all experiments p < 0.05 was considered to be statistically significant.
Results
Combination therapy of 5-FU and 4µ8C reduces cell proliferation and induces cell death in CT26 CRC cells more than monotherapy of 5-FU
To examine the IRE1 activation in colorectal cancer cells, IRE1 RNase activity was inhibited by 4µ8C, and cell viability determined. 4µ8C is an IRE1 Inhibitor III capable of inhibiting the IRE1α RNase activity. Proliferation of the CT26 cells was reduced and cell death increased following 5-FU chemotherapy. As depicted in Fig. 1a, in an unstressed situation, 5-FU could inhibit the cell proliferation significantly only at higher concentrations (100 µg/ml, p = 0.00014). However, following TM treatment (an ERS inducer), 5FU, 4µ8C and their use in combination reduced cell viability in a dose-dependent manner. For example, 5-FU at concentrations of 5, 10, 20, 50, and 100 µg/mL inhibited cell viability by 55.19% ± 3.16, 41.88% ± 1.36, 34.73% ± 2.09, 28.21% ± 2.07, and 21.88% ± 0.32, respectively. Similarly, 4µ8C at concentrations of 1, 2, 5, 10 and 20 µg/mL decreased the cell viability by 77.50% ± 5.30, 70.94% ± 0.8, 47.25% ± 20.31% ± 0.89, and 16.85% ± 0.69, respectively. Interestingly, the combination of 5-FU and 4µ8C (5/5, 5/10, 10/5, 10/10 µg/mL) led to more growth inhibition in CRC cells, by 72.34% ± 1.17 (p = 0.0019), 42.49% ± 5.8 (p = 0.00022), 76.24% ± 10.80 (p = 0.0382), and 15.26% ± 0.54 (p = 0.0000187), respectively (Fig. 1b). However, our results showed that 5-FU alone could induce ERS at higher concentrations, the combination of 5-FU with 4µ8C did not produce significant changes. Thus, we used TM to create an ERS model to obtain better visualisation of the results. The data have shown increased sensitivity of the cells to chemotherapy when cotreatment with 4µ8C was used.

Effect of different concentration of 5-FU and/or 4µ8C alone and in combination on the viability and cytotoxicity of CT26 CRC cells. The cells were treated by various concentrations of 5-FU (5, 10, 20, 50, 100 µg/ml), 4µ8C (1, 2, 5, 10, 20 µg/ml) and combination of them (5/5, 5/10, 10/5, 10/10 µg/mL) for 24 h, then viability and cytotoxicity were studied using trypan blue exclusion test and MTT assay, respectively (a) Un-stressed situation (b) Stressed situation by tunicamycin ™ induction (c) IC50 calculation for 5-FU (d) IC50 calculation for 4µ8C (e) IC50 calculation for 5-FU and 4µ8C together. The results are from triplicated of three independent experiments and represented as means ± SD. Statistically different results are indicated with *(p < 0.05), **(p < 0.01) and ***(p < 0.001) compared to untreated cells. They were analyzed by GraphPad Prism 8.3.0
An MTT assay was carried out to determine the IC50 of drugs, the concentration that inhibits the cell growth by 50%. As shown in Fig. 1c, d, e, the IC50 values for 5-FU and 4µ8C were 6.03, 3.7, respectively (Fig. 1c, d). The IC50 of the combination 5-FU and 4µ8C was 5 µg/ml for 5-FU and 5 µg/ml for 4µ8C, respectively (Fig. 1e).
Cell death analysis showed that the occurrence of apoptosis was not significant with the use of monotherapy. Although 5-FU at 5 µg/ml and 4µ8C at 10 µg/ml decreased proliferation of the CT26 cell line tested, neither drug induced cell death. However, a high percentage of apoptotic cells (74.6%) resulted from the combination therapeutic regime of 5-FU with 4µ8C (Fig. 2).

Effect of 5-FU and/or 4µ8C alone and in combination on cell death in CT26 CRC cells. Cells were treated with 5-FU (5 µ/ml), 4µ8C (10 µ/ml) and a combination of them for 24 h, then harvested and stained with PI/FITC for flowcytometric analyses by FACS Calibur flow cytometer (FACS Calibur, BD bioscience, USA).
Targeting IRE1 RNase domain decrease the splicing of XBP1 mRNA
4µ8C was used to target the RNase domain of IRE1 to study the splicing activity of IRE1. Following TM treatment, XBP1s mRNA expression was found to be increased whereas the mRNA XBP1u level was decreased. The level of xbp1s/xbp1u was decreased in the combination treatment of cells with 5-FU and 4µ8C in comparison to 5-FU alone, as observed in Fig. 3.

Tunicamycin ™ (1 µg/ml) is induced ER stress in CT26 CRC cell. Then the cells were treated by 5-FU (5 µg/ml) and IRE1 inhibitor, 4µ8C (10 µg/ml) alone and in combination together. RT PCR gel electrophoresis of XBP1u and XBP1s was performed. The band intensity of XBP1s and XBP1u was analyzed by image J software and reported as quantitative data
4µ8C could enhance the effectiveness of 5-FU, decrease the tumor growth, and increase the drug response in a mouse CRC model
We evaluated the effects of 5-FU alone, and in combination with 4µ8c, in the CRC mouse model in order to confirm our earlier findings. The doses of prescribed drugs were obtained from experimental results with no drug toxicity and metastasis. The combination therapy of 5-FU (20 mg/kg) and 4µ8C (50µM/kg) reduce the tumor size (Fig. 4a) and volume (Fig. 4b) compared to the control and monotherapy groups significantly, p < 0.05 from day 13 (Fig. 4). This decrease in tumor size of treated groups was positively correlated with the reduced mitotic cell counting. The mean mitotic rate was 14 ± 1 in the 5-FU group compared to 9 ± 1.2 in the 5-FU/4µ8C (p < 0.001). (Fig. 5a). The weights of all animals were checked during the experiment and there were no consequential discrepancies observed between the treatment and control groups. However, the variation of tumor volume to body weight (TV/BW) in different groups revealed that 5-FU/ 4µ8C reduced TV/BW more than 5-FU monotherapy, at 21-day (p < 0.001). (Fig. 5b). The response to treatment effect was 40–60% in combination therapy of 5-FU/ 4µ8C group in comparison to monotherapy with 5-FU, which was less than 30% (Fig. 6; Table 1.), suggesting that the combination of 5-FU/4µ8C enhanced the chemotherapeutic response in the in vivo model of CRC. The screening protocol of chemotherapy also confirmed that no signs of cytotoxicity or metastasis were detected in all treated groups (Fig. 7; Table 1).

The effect of 5-FU, and 4µ8C alone and in combination of them on tumor size and volume on BALB/C CRC model mice. Mice treated with 5-FU (20 mg/kg), 4µ8C (50 µM/kg) and combination of them. The tumour volume was measured every other day from 0–21 days of post 5-FU, 4µ8C and their combination administration (a) Tumor size and (b) tumor volume of different groups. In 5- FU group, during the chemotherapy, the tumor volume decreased in comparison to the control (p < 0.05 from day of 13); however, an increased rate was observed in compared to combination therapy. In 4µ8C group, although the tumor volume was diminished, an increasing trend was also observed in comparison to combination therapy from day 13. A significant reduction of tumour volume was observed in combination therapy group from day 7, in comparison to controls (p < 0.05 from day of 13) and also to monotherapy of 5-FU (p < 0.05 from day of 19). * for p < 0.05, ** for p < 0.01, *** for p < 0.001

The effects of 5-FU, and 4µ8C alone and in combination on mitotic and body weight of BALB/C CRC mice model (a) Mitotic cell counting in 1 mm2 of H&E staining slide in all treated and control group (b) The rate of tumor volume/body weight (TV/BW) of all treated and control animals. *p < 0.05, ** p < 0.01, *** p < 0.001

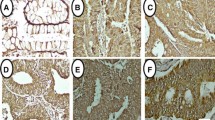
The effects of chemotherapy of 5-FU (20 mg/kg), and 4µ8C (50 µM/kg) alone and in combination on BALB/C CRC mice model

The cytotoxicity effect of chemotherapy of 5-FU, and 4µ8C alone and in combination on BALB/C CRC model mice. Kidneys and livers in all groups of animals were removed after 21st day of the last chemotherapy by surgery and monitored by H&E staining. There were no signs of toxicity and metastasis associated with treatments in all groups of single therapy of 5-FU and 4µ8C, and also with a combination of them
Discussion
5-FU is one of the most commonly-used drugs for the treatment of CRC. However, its prescription often leads to the development of drug resistance [21]. Recently, the association of chemoresistance with the molecular mechanism of ERS has been attracting in research attention [22]. Besides, it has also been shown that ERS and UPR activation can modulate the levels and activities of cell survival and autophagy-related regulators, thereby exerting crucial functions in controlling the drug resistance of cancer cells [22]. More importantly, 5-FU has also been linked to the stimulation of ERS in colon cancer [23]. The main goal of this study is focused on the enhancement of CRC susceptibility by targeting the IRE1α arm of UPR. Since the recognition of IRE1α as a suitable therapeutic target for many cancers, pharmacological inhibition of its activity has been seen to play an important role in therapeutic regimes [15]. Currently chemical manipulation of IRE1, such as targeting either the kinase domain or the RNase domain, is the main basis for drug development [15]. In this regard 4µ8C is an effective inhibitor of IRE1α RNase activity, with significant effects on the reduction of XBP1 splicing, the downstream effector of IRE1 and also on the reduced RIDD pathway [16]. Our results demonstrate that, in a controlled situation without using chemical inducers, 5-FU was able to diminish cell proliferation in CRC cells only at high doses (100 µg/ml). Furthermore, 5-FU could also induce ERS and XBP1 slicing in the CRC cell line, as reported before (21). Therefore, the ability of 5-FU to induce drug resistance should also be considered. In contrast, by using TM, 5-FU was found to decrease CRC proliferation in a dose-dependent manner (5–100 µg/ml). To enhance the 5-FU sensitivity, specific IRE1α inhibition, 4µ8C was used under both unstressed and stressed conditions. Our results show that although 4µ8C inhibited IRE1α RNase activity in CT26 cells under non-stressed situations, the inhibitory concentration was only obtained at high doses of 20 µg/ml. However, under TM treatment, this can enhance the susceptibility of CRC to 5-FU chemotherapy at 5 and 10 µg/ml, suggesting that this inhibition is likely to occur independently of IRE1α RNase activity, at higher concentrations, or may be due to off-target effects. This situation is similar to a report in hepatoma cells which revealed that higher concentrations of 4µ8c appear to be independent of IRE1α RNase activity [24]. Besides, the previous studies demonstrated the role of IRE1 in the regulation of cell growth and ER membrane expansion, in both unstressed and ER stressed-cells [25]. Our results here demonstrate that inhibition of the IRE1α-XBP1 branch promotes apoptosis in the CT26 CRC cell line, and therefore could be considered as potential target for chemotherapy. 4µ8C was found to successfully inhibit the IRE1 RNase activity and to enhance antitumor activity in CRC. The inhibition of RNase domain of IRE1 α is more stable than targeting kinase activity. There have also been some reports of pharmaceutical inhibitors that showed the importance of this domain in targeting IRE1 for cancer treatment- for example, using 4µ8C in hepatocellular carcinoma (HCC) or MKC8866 in triple-negative breast cancer [25, 26]. To further support our data on 4µ8C activity, we established the CRC mice model. The previous experiment has demonstrated that at increasing doses of 5-FU, the degree of apoptosis was raised, and, in addition, severe diarrhea, histopathological damage and an increased mortality rate were also found in the murine model [27]. In this study, we have chosen the optimal dosage by adhering to the suggested intestinal administration and by performing a pilot study. As a result, 20 mg/kg of 5-FU was used, either alone or in combination with an inhibitor, 4µ8C, and no toxicity was observed in any of the vital organs using this regimen. Different doses of 4µ8C were also selected according to the previous experience [28], in addition to the pilot study. Our results show that 50 µM/kg of 4µ8C is the most appropriate dose, which did not show toxicity but significantly reduced the tumour volume. Doses lower that 50 µM/kg failed to show any reduction in tumour size. Besides, the presence of 4µ8C did not promote cytoxicity in either of the treatment conditions, but higher concentrations of the inhibitor (60 µM) were associated with apparent off-target or compensatory responses that were not observed at 10 µM in an in vitro assay [24]. The tumour volume in treated mice confirmed the remarkable effects of combination therapy (5-FU and 4µ8C) on CRC mice, p ≤ 0.05, as observed from day 13. Indeed, the tumour volume of 5-FU/4µ8C combination therapy was significantly decreased in comparison to 5-FU monotherapy and in the control group. This treatment response also confirmed the efficacy of combination therapy, 40–60%. In addition, no metastases to either kidney or liver were reported in the therapeutic groups, pointing to the safety profiles of the doses used by intraperitoneal administration. These findings are in line with similar studies of IRE1 inhibition, used to enhance the chemotherapeutic effects in murine cancer models [26]. Although our results revealed that 4µ8C in combination with 5-FU did reduce tumour growth and improve the treatment response in the CRC mice model, the pharmacokinetics of 4µ8C limits the application of this particular IRE1inhibitor in animal models [16].
Conclusions
Our results reveal that 4µ8C can successfully inhibit the IRE1/XBP1 branch of UPR, and that co-treatment of 4µ8C with 5-FU may enhance the sensitivity of CRC in an experimental murine model in comparison to monotherapy.
References
Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019;14(2):89–103. https://doi.org/10.5114/pg.2018.81072.
Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;26(9863):303–12. https://doi.org/10.1016/S0140-6736(12)61900-X.
Falcone A, Ricci S, Brunetti I, Pfanner E, Allegrine G, Barbara C, et al. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: the gruppo oncologico nor. J Clin Oncol. 2007;1(13):1670–6. https://doi.org/10.1200/JCO.2006.09.0928.
Pouya FD, Gazouli M, Rasmi Y, Lampropoulou DI, Nemati M. MicroRNAs and drug resistance in colorectal cancer with special focus on 5-fluorouracil. Mol Biol Rep. 2022;49(6):5165–78. https://doi.org/10.1007/s11033-022-07227-1.
De Rosa M, Pace U, Rega D, Costabile V, Duraturo F, Izzo P, et al. Genetics, diagnosis and management of colorectal cancer (review). Oncol Rep. 2015;34(3):1087–96. https://doi.org/10.3892/or.2015.4108.
Cao S, Tang J, Huang Y, Li G, Li Z, Cai W, et al. The road of solid tumor survival: from drug-induced endoplasmic reticulum stress to drug resistance. Front Mol Biosci. 2021;8:620514. https://doi.org/10.3389/fmolb.2021.620514.
Nikesitch N, Lee JM, Ling S, Roberts TL. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin Transl Immunol. 2018;7(1):e1007. https://doi.org/10.1002/cti2.1007.
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is Induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91. https://doi.org/10.1016/s0092-8674(01)00611-0.
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. https://doi.org/10.1038/nrm3270.
Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69(2):169–81. https://doi.org/10.1016/j.molcel.2017.06.017.
Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–97. https://doi.org/10.1038/nrc3800.
Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol Mech Dis. 2015;10:173–94. https://doi.org/10.1146/annurev-pathol-012513-104649.
Xie Y, Liu C, Qin Y, Chen J, Fang J. Knockdown of IRE1ɑ suppresses metastatic potential of colon cancer cells through inhibiting FN1-Src/FAK-GTPases signaling. Int J Biochem Cell Biol. 2019;114:105572. https://doi.org/10.1016/j.biocel.2019.105572.
Jin C, Jin Z, Chen NZ, Lu M, Liu CB, Hu W, Le, et al. Activation of IRE1α-XBP1 pathway induces cell proliferation and invasion in colorectal carcinoma. Biochem Biophys Res Commun. 2016;470(1):75–81. https://doi.org/10.1016/j.bbrc.2015.12.119.
Raymundo DP, Doultsinos D, Guillory X, Carlesso A, Eriksson LA, Chevet E. Pharmacological targeting of IRE1 in Cancer. Trends in Cancer. 2020;6(12):1018–30. https://doi.org/10.1016/j.trecan.2020.07.006.
Cross BCS, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109:E869–78. https://doi.org/10.1073/pnas.1115623109.
Grandjean JMD, Wiseman RL. Small molecule strategies to harness the unfolded protein response: where do we go from here? J Biol Chem. 2020;295(46):15692–711. https://doi.org/10.1074/jbc.REV120.010218.
Siwecka N, Rozpędek-Kamińska W, Wawrzynkiewicz A, Pytel D, Diehl JA, Majsterek I. The structure, activation and signaling of IRE1 and its role in determining cell fate. Biomedicines. 2021;9(2):156. https://doi.org/10.3390/biomedicines9020156.
Adibzadeh R, Golhin MS, Sari S, Mohammadpour H, Kheirbakhsh R, Muhammadnejad A, et al. Combination therapy with TiO2 nanoparticles and cisplatin enhances chemotherapy response in murine melanoma models. Clin Transl Oncol. 2020;23(4):738–49. https://doi.org/10.1007/s12094-020-02463-y.
Mohammadalipour Z, Rahmati M, Khataee A, Moosavi MA. Differential effects of N-TiO2 nanoparticle and its photo‐activated form on autophagy and necroptosis in human melanoma A375 cells. J Cell Physiol. 2020;235(11):8246–59. https://doi.org/10.1002/jcp.29479.
Blondy S, David V, Verdier M, Mathonnet M, Perraud A, Christou N. 5-Fluorouracil resistance mechanisms in colorectal cancer: from classical pathways to promising processes. Cancer Sci. 2020;111(9):3142–54. https://doi.org/10.1111/cas.14532.
Bahar E, Kim JY, Yoon H. Chemotherapy resistance explained through endoplasmic reticulum stress-dependent signaling. Cancers (Basel). 2019;11(3):338. https://doi.org/10.3390/cancers11030338.
Kim JK, Kang KA, Piao MJ, Ryu YS, Han X, Fernando PMDJ, et al. Endoplasmic reticulum stress induces 5-fluorouracil resistance in human colon cancer cells. Environ Toxicol Pharmacol. 2016;44:128–33. https://doi.org/10.1016/j.etap.2016.05.005.
Stewart C, Estrada A, Kim P, Wang D, Wei Y, Gentile C, et al. Regulation of IRE1α by the small molecule inhibitor 4µ8c in hepatoma cells. Endoplasmic Reticulum Stress Dis. 2017;4(1):1–10. https://doi.org/10.1515/ersc-2017-0001.
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–29. https://doi.org/10.1038/nrm2199.
Pavlović N, Calitz C, Thanapirom K, Mazza G, Rombouts K, Gerwins P, et al. Inhibiting IRE1α-endonuclease activity decreases tumor burden in a mouse model for hepatocellular carcinoma. Elife. 2020;9:e55865. https://doi.org/10.7554/eLife.55865.
Zhang S, Liu Y, Xiang D, Yang J, Liu D, Ren X, et al. Assessment of dose-response relationship of 5-fluorouracil to murine intestinal injury. Biomed Pharmacother. 2018;1:106:910–6. https://doi.org/10.1016/j.biopha.2018.07.029.
Li X-X, Zhang H-S, Xu Y-M, Zhang R-J, Chen Y, Fan L, et al. Knockdown of IRE1α inhibits colonic tumorigenesis through decreasing β-catenin and IRE1α targeting suppresses colon cancer cells. Oncogene. 2017;36(48):6738–46. https://doi.org/10.1038/onc.2017.284.
Acknowledgements
M.A.M. appreciated the financial support of the National Institute of Genetic Engineering and Biotechnology.
Funding
This study has been funded and supported by National Institute for Medical Research Development (NIMAD), Grant No 978587.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Authors declares that they have no conflict of interest.
Ethical approval
The project was approved by the ethical committee at NIMAD, Iran, by 1398.265 approval ID.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Abbasi, S., Rivand, H., Eshaghi, F. et al. Inhibition of IRE1 RNase activity modulates tumor cell progression and enhances the response to chemotherapy in colorectal cancer. Med Oncol 40, 247 (2023). https://doi.org/10.1007/s12032-023-02105-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-023-02105-7