
Abstract
Clear cell renal cell carcinoma (ccRCC) is the most common subtype of renal cell carcinoma. P4HA3 is a key enzyme in collagen biosynthesis and has emerged as important molecules in regulation of proliferation, invasion, and metastasis in various tumor types. The role of P4HA3 in the development of ccRCC has remained to be elucidated. Genes expression, prognostic, and enrichment analyses were carried out with bioinformatics analysis. The efficiency of P4HA3 knockdown was confirmed by real-time quantitative PCR and Western blotting. The cellular functions were analyzed by CCK-8, EdU, wound healing, and transwell assays. The levels of related proteins expression were analyzed by Western blotting. P4HA3 was highly expressed in ccRCC compared with normal tissue samples from the TCGA database. Kaplan–Meier curves results showed that the expression level of P4HA3 was significantly negatively correlated with overall survival of patients. P4HA3 expression knockdown inhibited the proliferation, migration, and invasion of ccRCC cells, as demonstrated by in vitro experiments. In addition, GSEA results revealed that P4HA3 may be related to EMT and involved in the PI3K-AKT-GSK3β pathway in ccRCC; this was tentatively confirmed through Western blotting. P4HA3 may induce ccRCC progression via the PI3K-AKT-GSK3β signaling pathway and could represent a potential therapeutic target.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Renal cell carcinoma (RCC) is a common cancer and the leading cause of cancer-related death globally [1]. The most common pathological subtype of RCC is clear cell RCC(ccRCC), which accounts for about 80% of cases and is characterized by large amounts of lipid droplets in the cytoplasm [2]. RCC can be successfully treated with surgery at an early stage; however, approximately one-third of patients develop local recurrence or distant metastases within several years [3]. In addition, RCC progresses slowly and lacks specific clinical manifestations; therefore, about 20–30% of patients are diagnosed with advanced cancer (distant metastasis) at the initial examination [4]. Molecular targeted agents, such as tyrosine kinase inhibitors are used as first-line therapy for metastatic RCC; however, owing to the highly dynamic, adaptive, and heterogeneous tumor microenvironment (TME) of RCC, drug resistance develops readily [5], and most patients will eventually die because of therapy resistance and metastasis [6]. Therefore, it is of great necessity to elucidate the essential mechanisms driving ccRCC and to identify novel molecular biomarkers with high prognostic value and effective therapeutic targets.
The development and progression of tumors are not only related to the characteristics of the tumor itself but are also closely correlated with the TME [7]. The TME is composed of various cellular and non-cellular components. One of the most important non-cellular components, the extracellular matrix (ECM), provides structural and biochemical support to surrounding cells. The rigidity of the ECM is thought to play a critical part in regulating cell growth, differentiation, and maintenance of tissue homeostasis, and malignant tumor initiation, invasion, and metastasis are frequently associated with changes in the ECM [8]. Collagen is the main component of the ECM. As the scaffold for cell growth, collagen can induce the proliferation, differentiation, and migration of epithelial cells. It also plays important parts in maintaining intercellular adhesion, restoring tissue integrity, and providing structural support and protection for organs. Dysregulation of collagen metabolism leads to diverse disorders, including invasive cancer [9]. Thus, a better understanding of the metabolic mechanisms of collagen and how collagen affects the progression of malignancies will be beneficial for prognostic management of cancer patients and the development of new therapeutics.
P4HA3, a member of prolyl 4-hydroxylases (P4Hs) family, is a key enzyme in collagen biosynthesis and catalyzes post-translational modification and modulate protein folding and stability. The P4Hs family is composed of five different isoforms: P4HA1, P4HA2, P4HA3, P4HB and P4HTM [10]. In recent years, several studies have reported that the P4Hs family is up-regulated in multiple tumors and related to poor outcomes of patients [11]. For example, P4HA1 can indirectly promote cancer progression by increasing the stability of HIF-1α, enhancing tumor cell proliferation and angiogenesis; [12] P4HA2 promotes tumor invasion and metastasis by depleting vitamin C [13, 14], contacts with HBV [15], and increasing collagen deposition [16]; P4HB is associated with activation of the MAPK signaling cascade [17] and downregulation of tumor chaperone BiP [18, 19]. P4HA3 was the last member of the P4H family to be discovered, and several studies in recent years have found that P4HA3 participates in the progression of various neoplastic diseases, including head and neck squamous cell carcinoma (HNSCC) [20], gastric cancer [21], lung carcinoma [22], pituitary adenoma [23], and colon cancer [24]. Taken together, these results indicate that P4Hs family genes are widely involved in the progression of various tumors, suggesting potential roles as markers and therapeutic targets in tumor patients.
However, P4HA3 has rarely been studied in detail in the context of ccRCC. In this study, therefore, we performed a multi-level analysis of P4HA3 in ccRCC patients, including bioinformatics analysis and in vitro experiments, aiming to identify potential therapeutic value and reveal the biological role of P4HA3 in ccRCC.
Materials and methods
P4HA3 expression analysis in ccRCC
RNA-sequencing (RNA-seq) expression profiles and clinical data were downloaded from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/) for 539 kidney renal clear cell carcinoma (KIRC) tissues and 72 adjacent non-tumor tissues. In addition, the GSE53757 dataset was downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), as a complement to the KIRC data from TCGA. P4HA3 expression in KIRC was analyzed using the R software (version.3.6.3) based on downloaded data in HTSeq-FPKM (fragment per kilobase per million) format from the TCGA database. To analyze the expression profiles, RNA-seq data in FPKM format were converted to TPM (transcripts per million reads) format and logged. Mann–Whitney U test was used to compare unpaired samples, and paired sample t-test was used for paired samples. Next, the HPA (Human Protein Atlas) online tool was used to explore the protein expression of P4HA3 in KIRC tissues and normal kidney tissues.
Survival analysis and clinical characteristics of P4HA3 in ccRCC
RNA-seq and survival data downloaded from TCGA were used for survival analysis. The cut-off value for high and low P4HA3 expression was the median value of P4HA3 expression. Several clinical characteristics including pathologic stage, T stage, and histologic grade were also analyzed.
Differential expression and enrichment analyses
The KIRC data were separated into two groups based on the median value of P4HA3 expression. The “limma” algorithm was used to identify DEGs (differentially expressed genes) [25]. |Fold change (FC)|> 1.5 and p < 0.05 were used as the thresholds for differential expression of mRNAs. To understand the underlying oncological features of P4HA3, the DEGs were analyzed by functional enrichment, including GO (gene ontology), KEGG (Kyoto Encyclopedia of Genes and Genomes) analyses, and gene set enrichment analysis (GSEA). The R packages “clusterProfiler” [26] and “ggplot2” were included.
Cell culture
Five cell lines, OSRC2, 786-O, ACHN, 769-P, and Caki-1(Procell, China), were cultured in RPMI-1640 medium (Gibco, USA) with 10% FBS (fetal bovine serum) (Gibco, USA) in a cell incubator (37 °C, 5% CO2). These cell lines were not contaminated by mycoplasma or bacteria and were authenticated by short tandem repeat analysis.
Short interfering RNA (siRNA) transfection
Three siRNAs were designated by RIBOBIO (Guangzhou, China) with the following target sequences: #1 (GGATGAGGCAAGTCTAGAA), #2 (GGACACTGTTGACCCAAAA), #3 (GGAGTACCGCATCAGCAAA). OSRC2 and 769-P cells were seeded in six-well plates for 24 h. When the cell density reached 70%, cells were transfected with a mixture containing 125 μl Opti-MEM (Gibco, USA), 100 pmol siRNA, and 4 μl Lipo-8000(Beyotime, China) per well. The medium was replaced after 6 h. The efficiency of P4HA3 knockdown was confirmed by real-time quantitative PCR (qRT-PCR, after 48 h) and Western blotting (after 96 h).
qRT-PCR
Total RNA was isolated from OSRC2 and 769-P cell samples using SteadyPure Quick RNA Extraction Kit (Agbio, China). A NanoDrop2000 spectrophotometer (Thermo Scientific, USA) was used to measure the concentration of isolated RNA. RNA reverse transcription (total RNA 1000 ng) was performed with a PrimeScript™ RT reagent kit (Takara, Japan). The cDNA products were amplified by a two-step PCR method using a CFX Connect system (Bio-Rad, USA) with SYBR Green™ Premix Ex Taq™ II(Takara, Japan). The primers for β-actin and P4HA3 were synthesized by Tsingke Biotechnology Co., Ltd. (Beijing, China). The relative mRNA values were calculated by the 2 − ΔΔCt method, where ΔΔCt = ΔCt sample − ΔCt reference. β-actin was chosen as the internal reference gene. The primer sequences were as follows:
-
β-actin, forward ACAACTTTGGTATCGTGGAAGG;
-
reverse GCCATCACGCCACAGTTTC;
-
P4HA3, forward GGAACAAGCCCTAAGGCAACT;
-
reverse TGCTGATATACCGCATAGCTCAA.
Western blot analysis
Total protein extract was prepared with RIPA lysis buffer with PMSF (protease inhibitor) and phosphatase inhibitor at a ratio of 100:1:1. The total protein concentration was measured by BSA (bovine serum albumin) standard curves using a BCA protein assay kit (Solarbio, China). The appropriate primary antibodies were incubated at 4℃ overnight. Then, after washing with TBST three times, the PVDF membranes were incubated with secondary antibodies for 1 h at room temperature. The antibody details and dilution ratios are provided in Supplementary Table 1. Last, the membranes were washed with TBST three times, and protein bands were visualized using a CLINX ChemiScope S6 (Clinx, China) with ECL (enhanced chemiluminescent) (FG grade) (Sorfabio, SS1701, China).
Cell proliferation assays
Cell culture conditions were the same as described above. For Cell counting kit-8 (CCK-8) assay, OSRC2 and 769-P cells were seeded on 96-well plates at a density of 4000 cells per well. After 0, 24, 48, 72, and 96 h of incubation, 10 μl CCK-8 reagent (APExBIO, USA) was added into each well for 2 h at 37 °C. The absorbance was measured at 450 nm by a Varioskan LUX Multimode Microplate Reader (Thermo Fisher Scientific, MA, USA).
For EdU assay, Cell-Light EdU Apollo In Vitro Kit (RIBOBIO, Guangzhou, China) was used following the manufacturer’s protocol. Then, OSRC2 and 769-P cells were observed and photographed in EdU (Cy5) and nucleus (DAPI) fluorescence with inverted fluorescence microscope (Nikon ECLIPSE Ti, Japan).
Wound healing assay
We used six-well plates for scratch experiments. Cell culture conditions were the same as described above. The monolayer was scratched using a 200 μl (for OSRC2 cells) or 10 μl (for 769-P cells) pipette tip when the cell density reached about 90%. After cells had been washed twice with phosphate-buffered saline (PBS), the cell culture medium was replaced with basic RPMI-1640 medium. Then cell scratches were photographed with an inverted microscope at 0 and 24 h. Image J and GraphPad Prism 8 (GraphPad Software, Inc.) were used to calculate wound areas and for statistical data analysis, respectively.
Migration and invasion assays
The cell migration and invasion assays were performed in 24-well plates with 8 μm transwell chambers (Jet Biofil, China). For the migration assay, 500 μl basal medium with 10% FBS was added to each well. Then 5 × 104 tumor cells were seeded in each chamber with 200 μl of basic RPMI-1640 medium. About invasion assay, before addition of cells, the insides of the transwell chambers were evenly spread with Matrigel (Corning, USA) (50 μl per chamber), and the plates were placed in an incubator at 37 °C for 2 h. Otherwise, the procedure was the same as for the migration assay. After culture in the incubator for 12 h, the cells remaining on the inside of the chambers were removed with cotton buds. Then, the cells on the outside of the chambers were fixed in 4% multi-formaldehyde for 20 min and stained with 0.5% crystal violet for 30 min at room temperature. Three fields of view were randomly selected to calculate the numbers of migrating and invading cells.
Statistical analysis
All experiments were repeated three times and all results from multiple independent experiments are given as mean ± SD (standard deviation), unless otherwise specified. Differences between two groups were analyzed by Student’s t-test. GraphPad Prism 8 was used for data analysis. p < 0.05 was considered to indicate statistical significance in all experiments. ns, p ≥ 0.05, *, p < 0.05, **, p < 0.01, ***, p < 0.001.
Results
P4HA3 is highly expressed in ccRCC and associated with poor prognosis of patients
In the KIRC cohort, the expression of P4HA3 in tumor tissues (n = 539) was higher than that in normal kidney tissues (n = 72), and there was a statistically significant difference in both unpaired and paired comparisons (Fig. 1A, B). As supplementary data, we downloaded the GSE53757 dataset from the GEO database, including 72 ccRCC samples and paired normal samples. Through unpaired and paired expression analysis, it was further demonstrated that P4HA3 was up-regulated in ccRCC (Fig. 1C, D). In addition, the protein expression of P4HA3 in ccRCC tissue and normal renal tubular epithelial tissue was verified using the HPA database. It is worth noting that P4HA3 protein was mainly distributed in the cytoplasm, and its protein expression in tumor tissue was notably higher than that in non-tumor tissue (Fig. 1F, G). These results signified that mRNA and protein expression of P4HA3 are significantly increased in ccRCC tissues compared with normal kidney tissues. Furthermore, in order to explore the relationship between the mRNA expression of P4HA3 and the survival time of patients with ccRCC, we constructed Kaplan–Meier survival curve based on TCGA data. The expression level of P4HA3 was significantly negatively correlated with overall survival of patients (Fig. 1E).

P4HA3 level is increased in ccRCC and associated with poor prognosis. A, B The expression of P4HA3 among multiple tumors and corresponding normal tissues in TCGA database. C, D The expression of P4HA3 between tumor and adjacent normal tissues in unpaired (C) and paired (D) analyses based on GSE53757 dataset. E Correlation among overall survival and P4HA3 expression of ccRCC based on TCGA database. F, G IHC results of P4HA3 in ccRCC (G) and normal kidney tissues (F) from HPA database. *p < 0.05, **p < 0.01, ***p < 0.001
GO and KEGG analysis and GSEA of DEGs
To further investigate the oncogenic mechanism of P4HA3 in ccRCC, a differential analysis between high and low P4HA3 expression groups was performed based on TCGA data. When filter conditions were set to |FC |≥ 1.5 and p < 0.05, we identified 498 up-regulated and 254 down-regulated DEGs. The GO enrichment analysis showed that the up-regulated DEGs were mainly enriched in ECM structure and metabolism, whereas the down-regulated DEGs were mostly enriched in apical plasma membrane and membrane transport (Supplementary Fig. 1C, D). Furthermore, KEGG enrichment analysis showed that the up-regulated DEGs were mainly enriched in PI3K-AKT signaling pathway, focal adhesion, and protein digestion and absorption (Fig. 2A), while the down-regulated DEGs were mainly enriched in the PPAR signaling pathway, carbon metabolism, and bile secretion (Supplementary Fig. 1E). Moreover, we performed hallmark of GSEA. The positive correlations with P4HA3 expression were mainly related to tumor progression, including EMT, the TNF-α signaling pathway, and hypoxia (Fig. 2B). The negative correlations were primarily related to cell metabolism (Supplementary Fig. 1F).

KEGG analysis and GSEA of up-regulated DEGs. A The KEGG enrichment analysis of the up-regulated DEGs. B The hallmark of GSEA of the up-regulated DEGs
P4HA3 knockdown inhibits proliferation, migration, and invasion of ccRCC cells
As P4HA3 was abundantly expressed in OSRC2 and 769-P cell lines (Fig. 3A, B), these two cell lines were chosen for further exploration the effects of P4HA3 knockdown on cell biological function. To this end, we designed three siRNAs that were transiently transfected into OSRC2 and 769-P cells to silence the expression of P4HA3. As shown in Fig. 3C–E, the knockdown efficiency of the third siRNA was the highest, so we chose this siRNA for subsequent tumor biology experiments. The results of the CCK-8 and EdU assay showed that high P4HA3 expression had positive effects on ccRCC cell proliferation (Fig. 4A–D). Then, the results of the wound healing and transwell assay (including migration and invasion assays) showed that inhibition of P4HA3 expression reduced the migration and invasion abilities of OSRC2 and 769-P cells (Fig. 4E–H). These results suggest that P4HA3 may promote ccRCC migration and invasion in vitro.

P4HA3 expression of five ccRCC cell lines and verification of knockdown efficiency. A, B The mRNA and protein levels of P4HA3 in ccRCC cell lines, named OSRC2,769-P, Caki-1, ACHN and 786-O measured by RT-PCR (A) and Western blotting (B). C, D The knockdown efficiencies of P4HA3 in OSRC2 (C) and 769-P (D) cells were detected by RT-PCR. E The knockdown efficiency of P4HA3 in OSRC2 cells was detected by Western blotting

P4HA3 knockdown inhibits proliferation, migration, and invasion of ccRCC cells. A, B The CCK-8 assay of OSRC2 (A) and 769-P (B) cells. C, D The EdU assay of OSRC2 (C) and 769-P (D) cells. E, F Wound healing assay of OSRC2 (E) and 769-P (F) cells. G, H Transwell assay (including detection of migration and invasion abilities) of OSRC2 (G) and 769-P (H) cells. Yellow bar = 50 μm. *p < 0.05, **p < 0.01, ***p < 0.001
P4HA3 may promote the migration and invasion of ccRCC via EMT
The results of the GSEA showed that the highest normalized enrichment score (NES) was that of EMT; therefore, we focused on the mechanism of EMT. We selected the top 50 EMT-related genes from the EMTome database (http://www.emtome.org/) [27] and analyzed their correlations with the expression of P4HA3. As shown in Supplementary Fig. 2, 45 EMT-related genes were significantly related to the expression of P4HA3; therefore, we speculated that P4HA3 might enhance the migration and invasion capability of ccRCC via EMT progression. Western blotting was performed to verify the protein expression of common EMT marker proteins (Fig. 5A). The results showed that expression levels of mesenchymal proteins zinc finger E-box binding 1(Zeb-1), N-cadherin, Slug, and Snail were decreased to varying degrees in the P4HA3 knockdown group, whereas levels of epithelial protein E-cadherin were relatively increased, compared with the control group. Taking the above data together, we speculate that P4HA3 may be involved in EMT, which in turn promotes migration and invasion of ccRCC.

P4HA3 links to EMT and PI3K-AKT-GSK3β pathway in ccRCC. A Western blot analysis of Zeb-1, N-cadherin, E-cadherin, vimentin, slug and snail expression in OSRC2 cells. B Western blot analysis of PI3K, p-PI3K, AKT, p-AKT, GSK3β and p-GSK3β expression in OSRC2 cells. The relative expression of the target proteins was shown on the right. *p < 0.05, **p < 0.01, ***p < 0.001
P4HA3 may promote EMT through the PI3K/AKT/GSK-3β cascade
Since the KEGG enrichment analysis showed that the up-regulated DEGs were mainly enriched in PI3K-AKT signaling pathway (Fig. 2A), we performed Western blotting to verify whether the expression of PI3K and AKT proteins (both in total and phosphorylation) changed. The results showed that the knockdown of P4HA3 decreased the phosphorylation of PI3K and also AKT. In addition, several studies have found that the transcriptional activity of SNAIL1 is mainly mediated by the phosphorylation of GSK-3β 28,29,30,31], which is a downstream protein of the PI3K-AKT signaling pathway. Therefore, we also verified the expression of GSK-3β proteins (both in total and phosphorylation). Western blotting results showed that the expression of phosphorylation of GSK-3β decreased with knockdown of P4HA3 (Fig. 5B). Therefore, next, we treated OSRC2 cells in P4HA3 knockdown group with GSK-3β inhibitor 1 (MedChemExpress, China), and Western blotting results showed that the originally inhibited EMT-related proteins, including N-cadherin, E-cadherin, and snail, reverted to varying degrees compared to knockdown group (Fig. 6A), indicating that GSK-3β could inhibit EMT. All together, these data suggested P4HA3 might promote EMT through the PI3K/AKT/GSK-3β cascade.

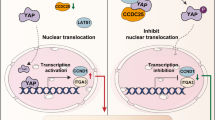
P4HA3 regulates EMT via PI3K-AKT-GSK3β signaling pathway in ccRCC. A Western blot analysis of N-cadherin, E-cadherin, and snail expression in OSRC2 cells after treated with GSK-3β inhibitor 1 (5 μM) for 36 h. The relative expression of the target proteins was shown on the right. B At the bottom of the figure, most of the DEGs involved in PI3K-AKT-GSK3β signaling pathway belong to the parts of ECM, integrin, growth factor (GF) and receptor tyrosine kinase (RTK).This schematic diagram was created with Biorender.com. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
Hydroxylation of proline residues is vital for the stability of triple-helical collagen structure, and regulation of collagen hydroxylation has a significant effect on the properties of the ECM and thus the behavior of cells [32]. As key enzymes in collagen biosynthesis, members of the P4H family have emerged in recent years as important molecules in the regulation of proliferation, invasion, and metastasis in several tumor types. Although P4HA3 was the last member of the P4H family to be discovered, many studies have found that it is extensively involved in the advancement of several non-neoplastic diseases, such as atherosclerosis, atrial fibrosis and pulmonary fibrosis [33, 34], as well as various neoplastic diseases, including lung cancer, HNSCC, gastric cancer, colon cancer and pituitary adenoma. For example, Nakasuka et al. demonstrated that TGF-β could regulate amino acid metabolism by promoting the expression of P4HA3, which may ultimately enhance the malignant behavior of lung cancer cells in both vitro and vivo [22]. In vitro studies have also shown that P4HA3 can promote tumor progression via EMT in HNSCC [20]. In gastric cancer, P4HA3 has been proved to enhance motility and invasiveness of cancer cells [21], leading to unfavorable OS of patients. In colon cancer, Zhou et al. found that miR-1266-3p could inhibit colon cancer growth and EMT by targeting P4HA3 [35]. At the same time, P4HA3 could promote the growth and metastasis of colon cancer by affecting the TGF-β/Smad signaling pathway [24]. In pituitary adenomas, data obtained by Long et al. suggest that COL6A6 could block the PI3K-AKT signaling pathway by inhibiting P4HA3, thereby preventing pituitary adenoma growth and invasion [23]. However, the role of P4HA3 has not been well studied in ccRCC. In this study, based on bioinformatics analysis, supplemented by in vitro validation, we demonstrate that P4HA3 can be considered as an emerging cancer therapeutic target.
In this study, we observed higher expression of P4HA3 in ccRCC tissues compared with normal samples based on both TCGA and GEO data (Fig. 1A–D). Using Kaplan–Meier prognostic curve (Fig. 1E), we confirmed that the mRNA expression of P4HA3 was negatively correlated with survival time of ccRCC patients. We then performed in vitro experiments and found that knockdown of P4HA3 resulted in a certain inhibition of the proliferation, migration, and invasion capabilities of tumor cells (Fig. 4A–H). To further study the detailed role of P4HA3, we carried out KEGG analyses and GSEA on the DEGs of P4HA3. KEGG pathway enrichment analysis demonstrated that these DEGs were mainly involved in ECM, cell adhesion, and the PI3K-AKT signaling pathway (Fig. 2A). GSEA demonstrated significant enrichment for EMT among the DEGs (Fig. 2B). In addition, there is evidence that collagen biosynthesis and deposition are closely related to the EMT process in cancer progression [36]; as P4HA3 is one of the key enzymes in collagen biosynthesis, we speculated that P4HA3 might be associated with EMT in ccRCC. To this end, we examined some common EMT-linked proteins to determine whether they were altered in P4HA3 knockdown cells through Western blotting analyses. The results showed that expression of the epithelial marker E-cadherin was increased, whereas that of mesenchymal markers (N-cadherin, Snail, Slug, and Zeb-1) was decreased, although vimentin expression did not seem to change significantly (Fig. 5A). These results provide preliminarily verification of the involvement of P4HA3 in EMT. Previously, Wang et al. demonstrated in vitro that P4HA3 could promote HNSCC cell proliferation, migration, and invasion by inducing EMT [20]. Nakasuka et al. demonstrated that TGF-β promotes the malignant behavior of NSCLC cells by promoting the expression of P4HA3 [22]. Zhou et al. found that P4HA3 could promote EMT by affecting the TGF-β/Smad signaling pathway [24]. Therefore, we speculate that P4HA3 may promote the invasion and metastasis of ccRCC through EMT.
When epithelial cells undergo EMT, they cells lose their junctions and apical-basal polarity, and undergo cell shape remodeling and gene expression reprogramming, which increases the motility of tumor cells and enables the development of aggressive phenotypes [37]. Therefore, EMT is closely related to tumor invasion and metastasis. The progression of EMT is regulated by several signaling pathways, including TGF-β, PI3K–AKT, MAPK, JNK, Wnt/β-catenin, Notch, Hedgehog, and JAK-STAT. These signaling pathways interact and together regulate epithelial plasticity responses that may progress to EMT, a prerequisite for cancer cell invasion and dissemination [38]. In pituitary adenomas, data from Long et al. suggest that P4HA3 promotes tumor EMT through the PI3K–AKT pathway [23]. Our KEGG analysis showed that the up-regulated DEGs were mainly enriched in the PI3K–AKT signaling pathway (Fig. 2A), suggesting that P4HA3 may promote EMT by participating in the PI3K–AKT signaling pathway, and thus promote the metastasis of ccRCC.
In addition, gene expression changes that contribute to suppressing epithelial phenotypes and activating mesenchymal phenotypes are regulated by multiple transcription factors, including SNAIL, TWIST, and ZEB. Their expression is activated in the early stage of EMT and thus they have a central role in tumor EMT progression [39]. SNAIL1 (the coding protein is snail) and SNAIL2 (the coding protein is slug) repress the transcription of epithelial genes by fastening to E‑box DNA sequences, while activating the transcription of mesenchymal genes, which in turn play an important role in tumorigenesis to activate the EMT program [40]. The transcriptional activity of SNAIL1 is mainly mediated by the phosphorylation of glycogen synthase kinase-3β (GSK-3β) [28], and various signaling pathways can indirectly increase the activity of SNAIL1 by affecting GSK-3β-mediated phosphorylation. The WNT and PI3K-AKT pathways inhibit SNAIL1 phosphorylation through promoting GSK-3β phosphorylation [31], while Notch and nuclear factor-κ B (NF‑κB) signaling disrupt GSK-3β-SNAIL1 interactions [29, 30]. They all increase the stability of SNAIL1, which in turn makes SNAIL1 continue to play a transcriptional regulatory part in the nucleus. Like SNAIL1, MAPKs can increase the activity of TWIST1 through phosphorylation at Ser68 [41]. Moreover, TWIST1 and SNAIL1 can synergistically induce the expression of ZEB1 [42]. The results of Western blotting experiments in this study indicated that three transcription factor proteins SNAIL, SLUG, and ZEB1 may be involved in the process by which the PI3K-AKT signaling pathway promotes EMT. Moreover, the expression of phosphorylation of PI3K, AKT, and GSK3β decreased with knockdown of P4HA3 (Fig. 5B). After treated with GSK-3β inhibitor, the expression of N-cadherin, E-cadherin, and Snail in P4HA3 knockdown group was somewhat restored (Fig. 6A). All together, we drew a possible schematic diagram of the regulation of EMT by P4HA3 via the PI3K-AKT-GSK3β signaling pathway in ccRCC (Fig. 6B); however, this requires subsequent experimental verification.
This study had some limitations. First, the expression and prognostic significance of P4HA3 were analyzed using online databases; more local clinical data are needed to validate the results. Second, only in vitro experiments were performed; the results were not verified in vivo. Finally, the specific mechanism by which P4HA3 promotes tumor migration and invasion through EMT has not yet been explored.
Conclusions
Taken together, our results suggest that: (1) P4HA3 is highly expressed in ccRCC and has negative correlation with prognosis of ccRCC patients; (2) decreased P4HA3 expression inhibits tumor proliferation, invasion, and metastasis in KIRC; and (3) P4HA3 may induce EMT by participating in the PI3K-AKT-GSK3β signaling pathway. In conclusion, our study suggests that P4HA3 may induce tumor progression through the PI3K-AKT-GSK3β signaling pathway and that this may be a key step in the development of ccRCC. Therefore, P4HA3 has potential as an emerging target for the treatment of ccRCC.
Data availability
All data generated or analyzed during this study are included in this article. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics. CA Cancer J Clin. 2022;72(1):7–33. https://doi.org/10.3322/caac.21708.
Leibovich BC, Lohse CM, Crispen PL, Boorjian SA, Thompson RH, Blute ML, Cheville JC. Histological subtype is an independent predictor of outcome for patients with renal cell carcinoma. J Urol. 2010;183(4):1309–15. https://doi.org/10.1016/j.juro.2009.12.035.
Jonasch E, Gao J, Rathmell WK. Renal cell carcinoma. BMJ. 2014;349:g4797. https://doi.org/10.1136/bmj.g4797.
Alt AL, Boorjian SA, Lohse CM, Costello BA, Leibovich BC, Blute ML. Survival after complete surgical resection of multiple metastases from renal cell carcinoma. Cancer. 2011;117(13):2873–82. https://doi.org/10.1002/cncr.25836.
Heidegger I, Pircher A, Pichler R. Targeting the tumor microenvironment in renal cell cancer biology and therapy. Front Oncol. 2019;9:490. https://doi.org/10.3389/fonc.2019.00490.
Linehan WM, Ricketts CJ. Decade in review-kidney cancer: discoveries, therapies and opportunities. Nat Rev Urol. 2014;11(11):614–6. https://doi.org/10.1038/nrurol.2014.262.
Vuong L, Kotecha RR, Voss MH, Hakimi AA. Tumor microenvironment dynamics in clear-cell renal cell carcinoma. Cancer Discov. 2019;9(10):1349–57. https://doi.org/10.1158/2159-8290.CD-19-0499.
Mohan V, Das A, Sagi I. Emerging roles of ECM remodeling processes in cancer. Semin Cancer Biol. 2020;62:192–200. https://doi.org/10.1016/j.semcancer.2019.09.004.
Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15(12):786–801. https://doi.org/10.1038/nrm3904.
Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. 2010;45(2):106–24. https://doi.org/10.3109/10409231003627991.
Shi R, Gao S, Zhang J, Xu J, Graham LM, Yang X, Li C. Collagen prolyl 4-hydroxylases modify tumor progression. Acta Biochim Biophys Sin (Shanghai). 2021;53(7):805–14. https://doi.org/10.1093/abbs/gmab065.
Zhou Y, Jin G, Mi R, Zhang J, Zhang J, Xu H, Cheng S, Zhang Y, Song W, Liu F. Knockdown of P4HA1 inhibits neovascularization via targeting glioma stem cell-endothelial cell transdifferentiation and disrupting vascular basement membrane. Oncotarget. 2017;8(22):35877–89. https://doi.org/10.18632/oncotarget.16270.
Gilkes DM, Chaturvedi P, Bajpai S, Wong CC, Wei H, Pitcairn S, Hubbi ME, Wirtz D, Semenza GL. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res. 2013;73(11):3285–96. https://doi.org/10.1158/0008-5472.Can-12-3963.
Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, O’Connor K, Lane AN, Xu R. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1 alpha stabilization and TNBC chemoresistance. Nat Commun. 2018. https://doi.org/10.1038/s41467-018-06893-9.
Feng GX, Li J, Yang Z, Zhang SQ, Liu YX, Zhang WY, Ye LH, Zhang XD. Hepatitis B virus X protein promotes the development of liver fibrosis and hepatoma through downregulation of miR-30e targeting P4HA2 mRNA. Oncogene. 2017;36(50):6895–905. https://doi.org/10.1038/onc.2017.291.
Pankova D, Jiang Y, Chatzifrangkeskou M, Vendrell I, Buzzelli J, Ryan A, Brown C, O’Neill E. RASSF1A controls tissue stiffness and cancer stem-like cells in lung adenocarcinoma. Embo J. 2019. https://doi.org/10.15252/embj.2018100532.
Sun S, Kiang KMY, Ho ASW, Lee D, Poon M-W, Xu F-F, Pu JKS, Kan ANC, Lee NPY, Liu X-B, et al. Endoplasmic reticulum chaperone prolyl 4-hydroxylase, beta polypeptide (P4HB) promotes malignant phenotypes in glioma via MAPK signaling. Oncotarget. 2017;8(42):71911–23. https://doi.org/10.18632/oncotarget.18026.
Banach A, Jiang Y-P, Roth E, Kuscu C, Cao J, Lin RZ. CEMIP upregulates BiP to promote breast cancer cell survival in hypoxia. Oncotarget. 2019;10(42):4307–20. https://doi.org/10.18632/oncotarget.27036.
Li JZ, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6(1):45–54. https://doi.org/10.2174/156652406775574523.
Wang T, Wang Y-X, Dong Y-Q, Yu Y-L, Ma K. Prolyl 4-hydroxylase subunit alpha 3 presents a cancer promotive function in head and neck squamous cell carcinoma via regulating epithelial-mesenchymal transition. Arch Oral Biol. 2020. https://doi.org/10.1016/j.archoralbio.2020.104711.
Song H, Liu L, Song Z, Ren Y, Li C, Huo J. P4HA3 is epigenetically activated by slug in gastric cancer and its deregulation is associated with enhanced metastasis and poor survival. Technol Cancer Res Treat. 2018. https://doi.org/10.1177/1533033818796485.
Nakasuka F, Tabata S, Sakamoto T, Hirayama A, Ebi H, Yamada T, Umetsu K, Ohishi M, Ueno A, Goto H, et al. TGF-beta-dependent reprogramming of amino acid metabolism induces epithelial-mesenchymal transition in non-small cell lung cancers. Commun Biol. 2021;4(1):782. https://doi.org/10.1038/s42003-021-02323-7.
Long RQ, Liu ZH, Li JH, Yu HL. COL6A6 interacted with P4HA3 to suppress the growth and metastasis of pituitary adenoma via blocking PI3K-Akt pathway. Aging-Us. 2019;11(20):8845–59. https://doi.org/10.18632/aging.102300.
Zhou H, Zou J, Shao C, Zhou A, Yu J, Chen S, Xu C. Prolyl 4-hydroxylase subunit alpha 3 facilitates human colon cancer growth and metastasis through the TGF-beta/Smad signaling pathway. Pathol Res Pract. 2022;230:153749. https://doi.org/10.1016/j.prp.2021.153749.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. https://doi.org/10.1093/nar/gkv007.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7. https://doi.org/10.1089/omi.2011.0118.
Vasaikar SV, Deshmukh AP, den Hollander P, Addanki S, Kuburich NA, Kudaravalli S, Joseph R, Chang JT, Soundararajan R, Mani SA. EMTome: a resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br J Cancer. 2021;124(1):259–69. https://doi.org/10.1038/s41416-020-01178-9.
Zhou BHP, Deng J, Xia WY, Xu JH, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3 beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6(10):931. https://doi.org/10.1038/ncb1173.
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA. 2008;105(17):6392–7. https://doi.org/10.1073/pnas.0802047105.
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappa B is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15(5):416–28. https://doi.org/10.1016/j.ccr.2009.03.016.
Yook JI, Li X-Y, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, et al. A Wnt-Axin2-GSK3 beta cascade regulates snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8(12):1398-U1357. https://doi.org/10.1038/ncb1508.
Rappu P, Salo AM, Myllyharju J, Heino J. Role of prolyl hydroxylation in the molecular interactions of collagens. Essays Biochem. 2019;63(3):325–35. https://doi.org/10.1042/EBC20180053.
Van den Diepstraten C, Papay K, Bolender Z, Brown A, Pickering JG. Cloning of a novel prolyl 4-hydroxylase subunit expressed in the fibrous cap of human atherosclerotic plaque. Circulation. 2003;108(5):508–11. https://doi.org/10.1161/01.Cir.0000080883.53863.5c.
Xiao Z, Reddy DPK, Xue C, Liu X, Chen X, Li J, Ling X, Zheng S. Profiling of miR-205/P4HA3 following angiotensin II-induced atrial fibrosis: implications for atrial fibrillation. Front Cardiovasc Med. 2021;8:609300. https://doi.org/10.3389/fcvm.2021.609300.
Zhou H, Huang S, Shao C, Zou J, Zhou A, Yu J, Xu C. miR-1266–3p suppresses epithelial-mesenchymal transition in colon cancer by targeting P4HA3. Anal Cell Pathol (Amst). 2022;2022:1542117. https://doi.org/10.1155/2022/1542117.
Shintani Y, Hollingsworth MA, Wheelock MJ, Johnson KR. Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH(2)-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res. 2006;66(24):11745–53. https://doi.org/10.1158/0008-5472.CAN-06-2322.
Thiery JP, Acloque H, Huang RYJ, Angela Nieto M. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90. https://doi.org/10.1016/j.cell.2009.11.007.
Katsuno Y, Lamouille S, Derynck R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25(1):76–84. https://doi.org/10.1097/CCO.0b013e32835b6371.
Peinado H, Olmeda D, Cano A. Snail, ZEB and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–28. https://doi.org/10.1038/nrc2131.
Barrallo-Gimeno A, Nieto MA. The snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132(14):3151–61. https://doi.org/10.1242/dev.01907.
Hong J, Zhou J, Fu J, He T, Qin J, Wang L, Liao L, Xu J. Phosphorylation of serine 68 of twist1 by MAPKs stabilizes twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011;71(11):3980–90. https://doi.org/10.1158/0008-5472.Can-10-2914.
Dave N, Guaita-Esteruelas S, Gutarra S, Frias A, Beltran M, Peiro S, Garcia de Herreros A. Functional cooperation between snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. 2011. https://doi.org/10.1074/jbc.M110.168625.
Li M, Wang Q, Zheng Q, Wu L, Zhao B, Wu Y. Prognostic and diagnostic roles of prolyl 4-hydroxylase subunit α members in breast cancer. Biomark Med. 2021;15(13):1085–95. https://doi.org/10.2217/bmm-2020-0323.
Li Q, Shen Z, Wu Z, Shen Y, Deng H, Zhou C, Liu H. High P4HA1 expression is an independent prognostic factor for poor overall survival and recurrent-free survival in head and neck squamous cell carcinoma. J Clin Lab Analy. 2020. https://doi.org/10.1002/jcla.23107.
Zhang J, Guo S, Wu Y, Zheng Z-c, Wang Y, Zhao Y. P4HB, a novel hypoxia target gene related to gastric cancer invasion and metastasis. Biomed Res Int. 2019. https://doi.org/10.1155/2019/9749751.
Funding
This work was supported by National Natural Science Foundation of China (Grant No. 81801507), Research Program of Natural Science Foundation in Chongqing (cstc2021jcyj-msxmX0484), and Kuanren Talent Program of the Second Affiliated Hospital of Chongqing Medical University (KY2019Y004).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by RZ and YZ. The first draft of the manuscript was written by ZZ and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interests
The authors have no relevant financial or non-financial interests to disclose.
Consent for publication
The informed consent was obtained from study participants.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Z., Zhang, Y. & Zhang, R. P4HA3 promotes clear cell renal cell carcinoma progression via the PI3K/AKT/GSK3β pathway. Med Oncol 40, 70 (2023). https://doi.org/10.1007/s12032-022-01926-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-022-01926-2