
Abstract
The first-line drug Imatinib (IM) has achieved a curative effect in most chronic myeloid leukemia (CML) patients, but drug resistance remains a problem. More alternative therapeutic strategies need to explore. In recent years, targeting dysregulated DNA repair mechanisms provided promising options for cancer treatment. Here, we discovered the versatile Mediator of DNA Damage Checkpoint 1 (MDC1) interacted with γ-H2AX and 53BP1 in the early stage of the DNA damage response of cells. MDC1 overexpressed in CML cell lines and patients’ bone marrow mononuclear cells. By knocking down MDC1, non-homologous end-joining pathways were mainly inhibited, leading to an intense accumulation of unrepaired intracellular DNA damage and an apparent cell apoptosis promotion. Notably, targeting MDC1 further enhanced drug sensitivity in IM-resistant CML cells. Our work revealed that MDC1 is a prospective target for CML treatment through regulating DNA damage repair mechanism, and also an alternative option for IM resistance dilemma. This study extends the understanding of regulating dysfunctional DNA repair mechanisms for cancer treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic myeloid leukemia (CML) is caused by t(9;22)(q34;q11) chromosomal translocation, forming the BCR-ABL fusion gene which produces BCR-ABL fusion protein with extremely constitutive tyrosine kinase activity and leads to abnormally activated downstream pathways [1,2,3]. Tyrosine kinase inhibitors (TKIs), particularly the first-generation TKI Imatinib (IM), show excellent effects. However, 25% of patients develop drug resistance and disease recurrence gradually [4, 5]. Alternative mechanisms to cope with the IM resistance or optimize CML treatment are needed to explore.
Genomic instability is one of the most common features in cancer, especially in hematological malignancy [6]. The dysregulation of DNA damage response (DDR) and repair mechanisms brings on chromosomal instability, and vice versa [7, 8]. Studies have found that BCR-ABL fusion protein promotes excessive DNA double-strand breaks (DSBs), which is the most notorious damage to cells [9]. Mammalian cells mainly prefer the non-homologous end-joining (NHEJ) to repair DSBs. Since it requires nucleotides processing at the junction, it is a relatively unfaithful repair pathway compared with homologous recombination (HR) [10]. DNA ends modification is prone to produce mutations associated with tumorigenesis, disease progression, and even drug-resistant phenotypes [11,12,13]. Hence, deciphering the proteins involved and targeting the dysfunctional DNA repair mechanisms provide new insights for CML treatment apart from TKIs.
Mediator of DNA Damage Checkpoint 1 (MDC1) comprises 2089 amino acid residues and contains a forkhead-associated (FHA) domain and two BRCA1 carboxy-terminal (BRCT) domains [14]. Those specific structures are shared by many key DDR-associated proteins. MDC1 is a key regulatory protein in DDR, which can activate S and G2/M phases cell cycle checkpoints to avoid the damaged cell entering the mitotic phase [15] and it can recruit downstream signaling proteins [16,17,18]. Normally, MDC1 is considered to play a protective role in cells. Whereas research showed that MDC1 was overexpressed in oral squamous cell carcinoma, and significantly correlated with lymph node metastasis. Low-level of MDC1 benefited when treated with surgery followed by radiation therapy [19, 20]. MDC1 was upregulation in ovarian cancer [21], and cervical cancer [22]. It indicates that the abnormally overexpressed MDC1 is highly correlated with cancers and could be a potential treatment target.
In this research, we explored MDC1’s role in DNA damage repair, and we tried to test whether it can be a target in CML treatment, especially in TKI-resistant CML cells. Our work demonstrated that MDC1 is a prospective target for future CML treatment.
Materials and methods
Cells and specimens
K562, K562/G01, KCL22, KU812, TK6, and THP1 (ATCC) cells were cultured in RPMI-1640. (Gibco, USA) K562/G01 is a high-degree IM-resistant cell line [23]. DMEM medium (Gibco, USA) was used to maintain 293T cells (SIBCB, China). SupB15 cells were grown in an IMDM medium (Cellmax, China). All cells were supplemented with 10% fetal bovine serum (FBS) at 37 °C in 5% (v/v) CO2 incubator. The bone marrow mononuclear cells (BMMCs) of normal individuals and CML patients were from the First and Second Affiliated Hospital of Chongqing Medical University. (The information of individuals was shown in Supplementary Table S1.) BMMCs were isolated by a human BMMCs isolation kit (Tbdscience, China).
Lentivirus infection
Genechem provided the small hairpin RNAs (shRNAs) (Shanghai, China). The sequence of shRNA oligonucleotide for the positive experiment group (shMDC1#1): 5′-GAGGCAGACUGUGGAUAAATT-3′; (shMDC1#2): GAGGCAGACUGUGGAUAAA; The negative control group (shControl) sequence: 5′-TTCTCCGAACGTGTCACGT-3′. The cells were infected with lentivirus-mediated shRNA targeting MDC1 for 72 h with 4 μL HitransGP (Genechem, China) and selected with 2 μg/mL puromycin (Sigma, USA) until all blank group cells died.
Apoptosis and cell cycle analysis
Use cold phosphate-buffered saline (PBS) wash the cells and suspend 2 × 105 in 100 μL of 1× binding buffer. Then, use Annexin PE/7-AAD Apoptosis Detection Kit (Vazyme, China) according to the manufacturer’s instructions. For cell cycle assay, 1 × 106 cells were washed and resuspended in cold PBS and immobilized in 70% ethanol overnight at 4 °C. Apoptosis and cell cycle were detected by Coulter FC500 flow cytometry (Beckman, USA).
Chemicals and antibodies
Calicheamicin-γ1 (Cali) was dissolved at 4 mM in ethanol and stored at − 80 °C. (MCE, China) IM (TargetMol, China) and KU55933—ATM kinase inhibitor (TargetMol, China). was kept at − 20 °C. Antibodies: anti-MDC1 (Beyotime; China), anti-MDC1 (Sigma, USA); anti-γ-H2AX, anti-p-c-ABL, and anti-PARP1, anti-Caspase3 (CST, MA); anti-53BP1, anti-RPA2 (Abcam, UK), anti-BRCA1, anti-ATM (Proteintech; China), and anti-β-Actin (zsbio, China). All antibodies are stored at − 20 °C.
Western blot
Whole cell protein extracts were separated by 8–12% SDS–PAGE, then transferred by polyvinylidene fluoride (PVDF) membranes (Millipore, MA). Super ECL plus Western blotting substrate was used for detection (Baoguang, China).
Colony-forming assay
100 cells/well were plated in 96-well plates with 100 μL complement RPMI-1640 to assess the proliferation ability. The results were checked after 7–10 days.
Immunofluorescence assay
Cells were fixed on slides with 4% paraformaldehyde solution. 1% Triton X-100 was used to permeabilize cells for 20 min. Goat serum was used to block (2 h, 4 °C). Then incubated with primary antibodies overnight. After incubation with a secondary antibody (Introvigen, MA) in darkness (1 h, 37 °C), the nucleus was stained with 4′,6 -diamidino-2-phenylindole (DAPI). The photos were acquired by a fluorescence microscope (Nikon, Japan).
Comet assay
100 μL preheated 1% agar was placed on the glass slide. Then 90 μL 0.5% low melting point agar was mixed with 10 μL cell suspension (1 × 104 cells). The mixture was added to the first layer of gel until solidification. The cells were lysed at 4 °C for 1 h, then DNA unwinding for 20 min and electrophoresis for 20 min. Stained with the SYBR GREEN solution and photo under a fluorescence microscope.
DSBs repair reporter assay
pDRGFP (Addgene, USA) was used for detecting HR, and pimEJ5GFP (Addgene, USA) was used for detecting NHEJ. After 48 h transfected pCBASce-I plasmid (Addgene, USA), GFP signal was detected 48 h post-transfection by flow cytometry. All plasmids were transfected by lipo8000 Reagent (Beyotime, China).
Co-immunoprecipitation
Cells were lysed with IP buffer (Beyotime, China) supplemented with 1% protease inhibitors for 30 min on ice. The supernatant after centrifuging is the whole-cell protein and incubated with MDC1 antibody-protein A/G magnetic beads (MCE, China) for 8 h at 4 °C. Antigen–antibody-magnetic bead complexes were washed three times by PBST. 30 μL 2×SDS–PAGE loading buffer was added to the complex and boil.
Statistical analysis
GraphPad Prism 8.0 software was performed for data analysis. The Student’s t-test was applied for calculating the main effect in two groups and One-Way ANOVA was used for that in three and more. p < 0.05 was considered statistically significant.
Results
MDC1 interacts with γ-H2AX and 53BP1 at the early stage of DDR
We used 44 nM DSBs inducer, Cali, to treat K562 and K562/G01 cells for one hour and performed Western blotting. Results showed that both MDC1 and γ-H2AX increased significantly after Cali treatment (Fig. 1A), which is consistent with the previous studies in that MDC1 participated in DDR [15]. Then Co-IP experiment was performed to verify the proteins that interact with MDC1 analyzed by STRING functional protein association networks (Supplementary Fig. S1B). Results confirmed the interaction between MDC1 with γ-H2AX and 53BP1 (Fig. 1B). Moreover, RPA2 was found to interact with MDC1 in our results which maintains telomere stability and plays a key role in the HR pathway.

MDC1 binds to the proteins that participate in DDR and regulates the HR and NHEJ pathways in cells. A 44 nM Cali was used to treat K562 and K562/G01 for 1 h, then we analyzed the change of γ-H2AX by Western blot. B Co-immunoprecipitation of lysates from K562/G01 was divided into three groups: UT (untreated), 0 h (exposed to Cali for 1 h), 8 h (exposed to Cali for 1 h, and released 8 h), then tested by Western blots. C K562/G01 was treated with KU55933 (10 μM) for 2, 8, 24 h and then analyzed by Western blot. D Western blot was used to examine MDC1 expression in 293T cells. E Knockdown efficiency of siRNA checked by Western blot. F Schematic of plasmid-based reporter assay. G, H The average proportion of GFP+ cells in Control and siMDC1 cell lines as a measurement for HR or NHEJ repair activity. *p < 0.05, **p < 0.01, and ***p < 0.001 versus controls
MDC1 is hyperactivated in an ATM kinases-dependent manner after DNA damage and rapidly relocalizes to damage foci [16]. So we treated the cells with KU55933 (an ATM kinase inhibitor) and found the inhibition of ATM leading to the subsequently reduced expression of MDC1 to some extent (Fig. 1C).
Those results demonstrated that MDC1 interacts with early stage DDR key proteins γ-H2AX and 53BP1 in an ATM kinases-dependent manner. Given the important role of MDC1 at the initial stage of DDR, interference with the expression of MDC1 may therefore affect the subsequent DNA repair mechanisms.
Knockdown of MDC1 compromises both HR and NHEJ repair pathways
To evaluate the compromised repair pathway after the knockdown of MDC1, we used DR-GFP and EJ5-GFP plasmid to establish the cell-based double vector system. Each plasmid has I-SceI sites that can generate I-SceI endonuclease-induced DSBs. If HR activity is functional, the DR-GFP transfected cells express GFP. So does the NHEJ pathway. The activity of HR or NHEJ repair events can be detected by GFP+ cell proportion [24, 25] (Fig. 1F). We detected that MDC1 shows a high expression in 293T cells (Fig. 1D), then we knockdown MDC1 by siRNA (Fig. 1E) and carry out the plasmid-repair experiment. The repair rate of the HR mechanism was reduced from 1.41 to 1.10%, and that of the NHEJ pathway was reduced from 13.11 to 9.81%. The efficiency of repair via HR and NHEJ decreased by 22.0% and 25.17%, respectively (Fig. 1G, H).
These results showed that the knockdown of MDC1 compromised the intracellular DNA repair activity. Particularly, the NHEJ repair was more disrupted based on the fact that it plays a major role in mammalian cells’ DNA repair.
Knockdown of MDC1 impacts cell proliferation and apoptosis
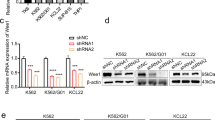
The data from Cancer Cell Line Encyclopedia (CCLE) indicate MDC1 over-expression in CML cells (Supplementary Fig. S1A). Our results confirmed that the expression of MDC1 in the BMMCs of CML patients was significantly higher than that in the control group at the mRNA level (Fig. 2A). There was also abnormal over-expression detected in CML cell lines (Fig. 2B, C). We used the lentivirus-mediated shRNAs to knock down the MDC1 in CML cells. The RT-qPCR results indicated that the efficiency of knockdown was significant and satisfying (Fig. 2D). Meanwhile, the knockdown efficiency is good at protein levels (Fig. 2E).

Expressions of MDC1 in CML patients’ BMMCs and CML cell lines. Relative MDC1 mRNA level was analyzed by RT-qPCR A in CML patients’ BMMCs, and B in leukemia cells. C The protein level of MDC1 in BCR-ABL-positive cell lines (K562, K562/G01, etc.) was analyzed by Western blot. D Knockdown efficiency of shRNA was validated by RT-qPCR analysis. E Western blot analysis. *p < 0.05, **p < 0.01, ***p < 0.001and ****p < 0.0001 versus controls
To test the effect of cell proliferation ability after MDC1 knockdown, we performed clone-formation assays. Cell proliferation was substantially suppressed by shMDC1 (Fig. 3A, B). Then flow cytometry (FCM) showed that cell apoptosis notably increased, especially in IM-resistant K562/G01 cells (Fig. 3C, D). The WB results were consistent with the FCM detection of apoptosis. The apoptosis-related protein PARP displayed an obvious cleavage band (Fig. 3E). Furthermore, we found that the expression of oncoprotein p-BCR-ABL was decreased after the knockdown of MDC1 (Fig. 3F).

MDC1 knockdown impairs cellular proliferation and promotes apoptosis in CML cells. A, B Colony-forming assay evaluated proliferate ability of cells treated with X-ray (4 Gy) or not. C, D The percentage of cell apoptosis was detected by FCM. E Western blot measured the expression of apoptosis-related proteins (PARP, cleaved-PARP). F Western blot analysis of the change of p-BCR-ABL. *p < 0.05, **p < 0.01, and ***p < 0.001 versus controls
These data suggested that disrupting MDC1 led to an anticancer manifestation, promoting apoptosis and inhibiting the proliferation of CML cells.
MDC1 knockdown CML cells accumulate more DSBs
Based on our findings, we speculated that MDC1-depleted cells are prone to accumulate more unrepaired DNA damage and correspondingly show more cell death. To investigate the underlying anticancer effect of knockdown MDC1, we used the immunofluorescence assay to verify the accumulation of γ-H2AX in MDC1 knockdown CML cells (Fig. 4A, B). We performed the comet assay to detect intracellular DNA damage. Results showed that after knockdown of MDC1, the comet tail distance significantly increased (Fig. 4C, D), as cells fail to deal with the excessive DNA damage, resulting in a high accumulation of DSBs. This result was consistent with the immunofluorescence assay. We further detected the repair capability in MDC1 knockdown cells after DNA damage. The MDC1 knockdown cells possessed an increased accumulation of γ-H2AX after ionizing radiation (IR) and failed to repair the damage even after 24 h (Fig. 4E), which explains the inhibition of proliferation and apoptosis enhancement in CML cells.

MDC1 deficiency causes genomic instability. A Immunofluorescence assay verifies the accumulation of γ-H2AX in MDC1 knockdown cells. B Graphical representation of (A). C, D The DNA damage was detected by a comet assay. CASP software was used to quantify the comet tail moment of 10 cells per group. That’s normalized to the control group. E K562 and K562/G01 were treated with X-ray (4 Gy) for 0, 4, 8, and 24 h. Western blot assay analyzed the protein change of γ-H2AX. ****p < 0.0001. versus controls
Silencing MDC1 promotes the sensitivity to IM and impedes the ability to deal with genotoxic damage in IM-resistant CML cells
A significant difference was found in the drug sensitivity test of K562/G01 cells that the half-maximal inhibitory concentration (IC50) value of the negative control group was about 4.7 µM, while the IC50 value of the shMDC1 group was 2.0 µM (Fig. 5A, B). There’s no significant change in IM-sensitive K562 cells (Supplementary Fig. S1D, E). The follow-up experiments primarily focus on K562/G01 cells, as targeting MDC1 displayed a more significantly anticancer effect in it. Then, the WB results showed apoptosis-related protein PARP and Caspase3 appeared obvious cleavage band after shMDC1 and combined with a low concentration of IM (2 μM) treatment (Fig. 5D). We also used the CCK-8 assay to test the ability of proliferation after treating the cells with very low concentrations of IM (1 μM). Results showed the proliferation ability of shMDC1 group cells was inhibited at 96 h (Fig. 5C). Therefore, silencing MDC1 improved the sensitivity of IM in K562/G01 cells.

Silencing MDC1 reduces the ability to deal with genotoxic damage in CML cells. A, B The cell viability and IC50 value were measured by CCK-8 assay after gradient concentration IM treatment in 48 h. C K562/G01 cells were transfected with shRNA alone or combined with IM (1 μM). Cell viability was assessed by CCK-8 assay. D Western blot was used to detect the change of apoptosis-related proteins (cleaved-PARP and cleaved-caspase3) after IM (2 μM) was treated. E, F FCM was performed to gauge the apoptosis rates of cells exposed to X-ray (4 Gy). *p < 0.05 **p < 0.01 ***p < 0.001 versus controls
Moreover, 4 Gy IR was used to induce DSBs in cells, and FCM was performed to detect the portion of cell apoptosis. Compared to control cells, the apoptosis rate in MDC1 knockdown combined with IR was further increased (Fig. 5E, F). The colony-forming assay showed similar results (Fig. 3A). The proliferation of CML cells was significantly suppressed after IR exposure. It is even more profound in K562/G01 cells, in which over 95% of cells died after IR treatment.
The results indicated that MDC1 knockdown can increase IM sensitivity and greatly reduce the ability to deal with genotoxic damage in K562/G01 cells.
The combination of MDC1 knockdown with IR leads to cell cycle arrest and chromosomes abnormality
After IR treatment, we found that the rate of blocked cells in the S phase was from 57.05 to 66.56% in MDC1 knocking down group, and a large portion of cells was blocked in the G2/M phase (Fig. 6A, B). This is probably due to the that knocking down MDC1 impaired the S, G2/M phase checkpoint function, promoting cells entering the S or G2/M phase [17, 18]. We speculated that DNA continued replicating but couldn’t further divide, forming the abnormal large polyploid cells (Supplementary Fig. S1C), and leading to cell death. Consistent with our speculation, the number of aneuploidies increased after the knockdown of MDC1 (Fig. 6A, C). This phenomenon aggravated after DNA damage caused by IR (Fig. 6C). We further examined the chromosomes of cells by Wright’s staining. It showed the abnormal chromosomes increased after knockdown MDC1 (such as polycentric, severely chromosome breakages), and these abnormalities were enhanced after IR. Unexpectedly, we found that the number of chromosomes in the MDC1 knockdown group highly increased (Fig. 6D), which is consistent with the aneuploidy test results.

Knockdown of MDC1 combined with IR impairs the chromosome integrity. A, B The cell cycle was detected and analyzed through FCM. C The number of aneuploids in cells. D Chromosome morphology analysis by Wright’s staining. E Schematic diagram of this research. Disrupting MDC1 made downstream proteins fail to be recruited, resulting in the accumulation of damage and then inducing cancer cell apoptosis.**p < 0.01 versus controls
Discussion
BCR-ABL fusion protein in CML has extremely strong tyrosine kinase activity and activates downstream signal transduction pathways [2, 26]. Recent studies suggested that the oncoprotein BCR‐ABL1 increases DSBs and enhances genetic instability. The latter lead to gene mutations and chromosomal translocation and is highly associated with hematological malignancy [10,11,12]. The NHEJ is predominantly used, which recognizes, excises, and ligates the damaged ends in a moderately inaccurate manner [27, 28]. This flexibility not only permits NHEJ to repair DNA junctions containing mutations [9, 29, 30], but also results in tumorigenesis, disease progression, and drug-resistant phenotypes [9]. Chromosome translocations are primarily caused by the NHEJ pathway, moreover, NHEJ pathway dysregulations are found in almost all types of leukemias [31, 32].
In our work, we decipher that MDC1 engaged in the initial stage of DNA damage repair by interacting with 53BP1 and γ-H2AX and its association with major DSBs repair mechanisms HR and NHEJ, especially with NHEJ. By disrupting the abnormal upregulation of MDC1 in CML, it exhibited an anticancer effect including triggering cell apoptosis and inhibition of cell proliferation. This finding is demonstrated by the apparent accumulation of intracellular DNA damage, the compromise of cell phase checkpoint function, and therefore the promotion of unrepaired cells entering the S or G2/M to increase the chromosome abnormality, eventually forming the abnormal large polyploid cells (Supplementary Fig. S1C). Notably, we found that these manifestations are more profound in the IM-resistant CML cell line. However, for the three groups in the Co-IP experiment, one (UT) was untreated, another (0 h) was treated with Cail, and a third (8 h) was treated with Cail and then released 8 h for repair (Fig. 1B). 0 h group pulled down the interacting protein successfully. But the proteins of the 8 h group show no obvious change after DNA repair. We suspected that possibly there was not sufficient time for DNA repair.
Our work illustrated that MDC1 is a prospective target for future CML adjuvant treatment, especially for its significant anticancer effect in the IM-resistant CML cell lines. We assumed that it may be relevant to the higher spontaneous DNA damage level in IM-resistant cells, which it’s more dependent on highly active repair mechanisms to keep cells thriving. Previous work has exhibited an increased number of γ-H2AX foci in K562/G01 than that in K562 and TK6 [33]. And it also showed an elevated γ-H2AX level in accelerated blast phase patients’ BMMCs as compared to the chronic period. CML patients’ samples have a greater level of expression than healthy controls [9]. Hence, it also explains that the higher the malignancy, the greater the effect shows. Although previous studies have confirmed that the level of DNA damage is related to the malignancy of CML, it is the first time that we have applied it in research and achieved positive results. This strategy appears feasible in other types of leukemia because of the over-expression of MDC1 in those cell lines, which is worth to be explored in the future.
Conclusion
In summary, we reported the MDC1 involvement with key proteins in the early stage of DNA damage. Suppression of abnormally upregulated MDC1 demonstrated an anticancer effect in CML treatment, especially in IM-resistant cells. Our finding not only identified a novel and promising target for the CML therapeutic options but also enriched the understanding of DNA repair mechanisms in clinical application.
References
de Klein A, van Kessel AG, Grosveld G, Bartram CR, Hagemeijer A, Bootsma D, Spurr NK, Heisterkamp N, Groffen J, Stephenson JR. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982;300:765–7.
Soverini S, Mancini M, Bavaro L, Cavo M, Martinelli G. Chronic myeloid leukemia: the paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol Cancer. 2018;17:49.
Bartram CR, de Klein A, Hagemeijer A, van Agthoven T, Geurts van Kessel A, Bootsma D, Grosveld G, Ferguson-Smith MA, Davies T, Stone M, et al. Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1983;306:277–80.
Clark RE. Tyrosine kinase inhibitor therapy discontinuation for patients with chronic myeloid leukaemia in clinical practice. Curr Hematol Malig Rep. 2019;14:507–14.
Braun TP, Eide CA, Druker BJ. Response and resistance to BCR-ABL1-targeted therapies. Cancer Cell. 2020;37:530–42.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Zhou H, Xu R. Leukemia stem cells: the root of chronic myeloid leukemia. Protein Cell. 2015;6:403–12.
Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45.
Popp HD, Kohl V, Naumann N, Flach J, Brendel S, Kleiner H, Weiss C, Seifarth W, Saussele S, Hofmann WK, Fabarius A. DNA damage and DNA damage response in chronic myeloid leukemia. Int J Mol Sci. 2020;21:1177.
Nowicki MO, Falinski R, Koptyra M, Slupianek A, Stoklosa T, Gloc E, Nieborowska-Skorska M, Blasiak J, Skorski T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104:3746–53.
Skorski T. BCR/ABL regulates response to DNA damage: the role in resistance to genotoxic treatment and in genomic instability. Oncogene. 2002;21:8591–604.
Skorski T. BCR/ABL, DNA damage and DNA repair: implications for new treatment concepts. Leuk Lymphoma. 2008;49:610–4.
Kim TD, Türkmen S, Schwarz M, Koca G, Nogai H, Bommer C, Dörken B, Daniel P, le Coutre P. Impact of additional chromosomal aberrations and BCR-ABL kinase domain mutations on the response to nilotinib in Philadelphia chromosome-positive chronic myeloid leukemia. Haematologica. 2010;95:582–8.
Ozaki T, Nagase T, Ichimiya S, Seki N, Ohiri M, Nomura N, Takada N, Sakiyama S, Weber BL, Nakagawara A. NFBD1/KIAA0170 is a novel nuclear transcriptional transactivator with BRCT domain. DNA Cell Biol. 2000;19:475–85.
Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–6.
Bartkova J, Horejsí Z, Sehested M, Nesland JM, Rajpert-De Meyts E, Skakkebaek NE, Stucki M, Jackson S, Lukas J, Bartek J. DNA damage response mediators MDC1 and 53BP1: constitutive activation and aberrant loss in breast and lung cancer, but not in testicular germ cell tumours. Oncogene. 2007;26:7414–22.
Wu L, Luo K, Lou Z, Chen J. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci USA. 2008;105:11200–5.
Nakanishi M, Ozaki T, Yamamoto H, Hanamoto T, Kikuchi H, Furuya K, Asaka M, Delia D, Nakagawara A. NFBD1/MDC1 associates with p53 and regulates its function at the crossroad between cell survival and death in response to DNA damage. J Biol Chem. 2007;282:22993–3004.
Dave JH, Vora HH, Ghosh NR, Trivedi TI. Mediator of DNA damage checkpoint protein 1 (MDC1) as a prognostic marker for patients with oral squamous cell carcinoma. J Oral Pathol Med. 2017;46:253–8.
Liu X, Qiu Z, Wang Z, Zuo W, Gong Z, Liu C, Zeng Q, Qian Y, Jiang L, Li Y, Bu Y, Hu G. NFBD1/MDC1 participates in the regulation of proliferation and apoptosis in human laryngeal squamous cell carcinoma. Clin Transl Oncol. 2018;20:534–41.
Liu X, Dong R, Jiang Z, Wei Y, Li Y, Wei L, Sun H, Li Y, Yang N, Yang Q, Liu Z, Kong B. MDC1 promotes ovarian cancer metastasis by inducing epithelial–mesenchymal transition. Tumour Biol. 2015;36:4261–9.
Yuan C, Bu Y, Wang C, Yi F, Yang Z, Huang X, Cheng L, Liu G, Wang Y, Song F. NFBD1/MDC1 is a protein of oncogenic potential in human cervical cancer. Mol Cell Biochem. 2012;359:333–46.
Zhu HL, Liu T, Meng WT, Jia YQ. Establishment of an Imatinib resistance cell line K562R and its resistant principia. Sichuan Da Xue Xue Bao Yi Xue. 2007;38:22–6.
Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4: e1000110.
Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–8.
Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–53.
Pannunzio NR, Watanabe G, Lieber MR. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J Biol Chem. 2018;293:10512–23.
Taleei R, Nikjoo H. The non-homologous end-joining (NHEJ) pathway for the repair of DNA double-strand breaks: I. A mathematical model. Radiat Res. 2013;179:530–9.
Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 2017;18:495–506.
O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60:547–60.
Ghezraoui H, Piganeau M, Renouf B, Renaud JB, Sallmyr A, Ruis B, Oh S, Tomkinson AE, Hendrickson EA, Giovannangeli C, Jasin M, Brunet E. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell. 2014;55:829–42.
Rassool FV. DNA double strand breaks (DSB) and non-homologous end joining (NHEJ) pathways in human leukemia. Cancer Lett. 2003;193:1–9.
Zhu Y, Dai H, Wang Y, Liang Y, Feng W, Yuan Y. Targeting FEN1 suppresses the proliferation of chronic myeloid leukemia cells through regulating alternative end-joining pathways. DNA Cell Biol. 2021;40:1101–11.
Acknowledgements
We appreciate Dr.Yonghong Wang and Dr.Yalin Zhu for their assistance with experiments. Dr. Chen of the Radiology Department in the First Affiliated Hospital of Chongqing Medical University for the radiology support.
Funding
This work is supported by the National Natural Science Foundation of China (Grant No. 81703095), Chongqing Natural Science Foundation (Grant No. cstc2021jcyj-msxmX0214), Youth Top Science and Technology Talent Fund Project of The First Affiliated Hospital of Chongqing Medical University (Grant No. BJRC2020–04), and Innovation Support Program for Overseas Students of Chongqing (Grant No. cx2018142).
Author information
Authors and Affiliations
Contributions
YY and YL conceived and designed the experiments. YL and YQ completed the experiments and wrote the draft. YL, YQ, WF, and YY analyzed the data and results. YY, WF, and GJ supervised the project and revised the manuscript. The final version of the manuscript was read and approved by all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval and consent to participate
The research conformed to the standard stipulated by the Declaration of Helsinki and was performed with the approval of the Ethics Committee of Chongqing Medical University (ID:2022059).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
12032_2022_1821_MOESM2_ESM.tif
Supplementary file2 (TIF 5166 KB)—Figure S1. (A) Relativity mRNA expression of MDC1 in various cancer cells, data from CCLE datasets. (B) Network analysis of known protein interaction partners of MDC1, from STRING website. (C) Left. Abnormal large K562/G01 cells (200×); Right. enlarged cell image. (D, E) Cell viability was detected by the CCK-8 assay. Data are expressed as the mean ± SD.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liang, Y., Qin, Y., Jiang, G. et al. Targeting MDC1 promotes apoptosis and sensitizes Imatinib resistance in CML cells by mainly disrupting non-homologous end-joining repair. Med Oncol 39, 226 (2022). https://doi.org/10.1007/s12032-022-01821-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-022-01821-w