
Abstract
Background
Colorectal cancer (CRC) is among the leading cause of cancer-related morbidity and mortality worldwide. Aerobic glycolysis, as a metabolic hallmark of cancer, plays an important role in CRC progression. Enolase 3 (ENO3) is a glycolytic enzyme that catalyzes 2-phosphoglycerate into phosphoenolpyruvate, while its role in CRC is still unknown.
Methods
Bioinformatics analysis was performed to examine the expression changes and roles of ENO3 in CRC patients from public databases. Then, ENO3 expression was validated in CRC tissues using Quantitative real-time PCR (qRT-PCR), immunohistochemical (IHC) analysis, and western blot. Overexpression and silencing models were constructed using plasmid and lentivirus transfection. Cell viability, proliferation, and migration in vitro were applied to evaluate the protumoral effects of ENO3 on CRC. RNA sequencing and GO enrichment analysis of differentially expressed genes (DEGs) were performed to explore the underlying molecular mechanisms of ENO3 in CRC progression. The ATP and lactate production level were detected to assess cell glycolysis.
Results
ENO3 was significantly up-regulated in CRC. High ENO3 expression was positively correlated with poor prognosis and higher clinical stages of CRC patients. ROC curve demonstrated the diagnostic value of ENO3 for CRC with the AUC of 0.802. Gain- and loss-of function experiments demonstrated that ENO3 significantly enhanced the proliferation and migration ability of CRC cells in vitro. After ENO3 knockdown, RNA sequencing screened out a list of DEGs which were enriched in the regulation of the glycolytic process. The detection of lactate production and ATP level verified the role of ENO3 in the glycolytic process.
Conclusion
Our findings illustrate that ENO3 could promote the progression of CRC by the enhancement of cell glycolysis, indicating the potential value of ENO3 as a novel biomarker and therapeutic target for CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC), the most common gastrointestinal malignant tumor, and the second leading cause of cancer-related death worldwide, is a heavy health burden nowadays [1]. The popularity of colonoscopy screening, radical surgical resection, and novel therapeutic strategies have prolonged the overall survival (OS) and progress-free survival (PFS) of CRC patients. However, the overall mortality rate of CRC remains high [2]. Hence, a deeper understanding of the underlying mechanisms of CRC initiation and progression is urgently required to enhance the prevention, diagnosis, and personalized therapy for patients.
Even with enough oxygen, tumor cells prefer to use glycolysis rather than oxidative phosphorylation for energy generation with increased lactate release which is referred to as Warburg effect, the metabolic characteristics of nearly all tumors [3, 4]. Aerobic glycolysis promotes tumor growth and migration with sufficient energy and generation of massive multiple substrates [5, 6]. Therefore, targeting glycolysis is an effective strategy in the therapeutic work-up of tumor.
Enolase (ENO), also known as phosphopyruvate hydratase, is a key glycolytic enzyme that catalyzes Mg2 + -dependent conversion of 2-phosphoglycerate into phosphoenolpyruvate. In mammals, enolase mainly includes three isoforms, possessing different biological functions [7]. α-enolase (ENO1), the most extensively studied one, is considered to be an oncogenic factor in multiple cancers, including CRC [8,9,10]. γ-enolase (ENO2), which is mainly stained in the cytoplasm of neuroendocrine cells, was reported to be overexpressed in tumors derived from neurogenic and neuroendocrine origin, like small cell lung cancer [11]. Recently, ENO2 was also identified to be an oncogenic factor in CRC [7]. As for β-enolase (ENO3), it was recently reported to be over-expressed with poor prognosis in CRC from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases [12, 13], while its function and mechanism in CRC progression remains unknown.
In this study, we validated that ENO3 was upregulated and associated with tumor progression in CRC. Besides, the biological role of ENO3 in CRC and the possible underlying mechanisms were explored. Taken together, our data revealed the oncogenic roles of ENO3 in CRC progression, indicating the potential value of ENO3 as a novel biomarker and therapeutic target for CRC.
Materials and methods
Bioinformatics analysis
Expression data of mRNA and clinical information of CRC patients were obtained from the public TCGA and GEO databases. Besides, the level of HTSeq-FPKM was converted into Transcripts Per Million (TPM) for the further analysis. Boxplots were used to evaluate ENO3 expression in normal and CRC tissues from TCGA and GEO datasets. Receiver operating characteristic curves (ROC) and the area under the curve (AUC) were performed to assess the diagnostic value of ENO3 in CRC patients. Kaplan–Meier method was used to explore the association between ENO3 expression level and OS or PFS of CRC patients.
Western blot
Cell or tissue total proteins were extracted with RIPA lysis buffer (Beyotime, China). Total proteins were loaded on 10% SDS-PAGE gel and electrophoretically transferred to PVDF membranes. After blocked with 5% BSA solutions, membranes were incubated using then ENO3 antibody (1:1000, Proteintech, USA) at 4℃ overnight. β-actin (1:2000, Abways, China) was used as an endogenous control. Afterward, horseradish peroxidase (HRP)-conjugated secondary antibodies were incubated for 2 h at room temperature. Specific immune complexes were exposed with ECL Western Blotting Substrate (NCM Biotech, China), and detected using ChemiDoc™ Touch Imaging System (Bio-Rad, USA) and GE Amersham Imager 600 (USA).
RNA isolation and qRT-PCR
Tissues and cells total RNAs were extracted with TRIzol Reagent (Invitrogen, China) and cDNA was reversed by using a Reverse Transcription Kit (Applied Biosystems). qRT-PCR was performed with SYBR Green Kit (Vazyme, China) using Roche LightCyclerTM 480 according to the manufacturer’s protocols. The data was analyzed by 2-ΔΔCT method. β-actin was used as the endogenous control. Sequence of all primers used was provided in Supplementary Table 1.
Cell culture and transfection
Human CRC cell lines (LoVo, HCT116, RKO, HT29) and HEK293T were purchased from American Type Culture Collection (ATCC, USA), and routinely maintained at 37 °C in a 5% CO2 incubator with recommended culture medium.
The ENO3 over-expression plasmid and shRNA were designed and synthesized by Tsingke Biotechnology Co., Ltd. Detail sequence of shRNAs was presented in Supplementary Table 2. Lenti-viruses were generated by co-transfecting the pLKO.1- copGFP-puro vectors, del8.9 (Beyotime, China), and vesicular stomatitis virus-G (Beyotime, China) into HEK293T cells. The viral supernatants were harvested and enriched via centrifugation. PolyJet (Thermo Scientific, USA) was used for plasmid transfection. The transfection efficiency of viral infection was examined by western blot and qRT-PCR.
Cell proliferation assay
For cell viability assays, 3000 cells per well were seeded in 96-well plates. At 6 h of culture, as well as 24-, 48-, 72-, and 96 h after that, cells were measured using CCK-8 assay reagents (Meilun Biotechnology, China) and the cell growth curves were then obtained.
For plate colony formation assay, 800 cells per well were plated into 6-well plates and maintained for 2 weeks at 37℃ in 5% CO2. At the end point, colonies were stained with 0.05% crystal violet (Sigma, USA) for 30 half an hour after fixation by 4% PFA for 20 min. The result was determined by counting the stained colonies number and detecting the 570-nm absorbance of the stained colonies eluent.
Cell migration and wound healing assay
Cell migration assay was conducted using transwell chambers (Corning, USA). Briefly, 20 × 104 cells in 100 μl of serum-free medium were plated into the upper chamber with 700 μl of 10% FBS medium added to the lower chamber. After 24 h, cells migrated to other side of the membrane were fixed by 4% PFA and stained with 0.05% crystal violet, and finally photographed under the optical microscope.
For wound healing assay, same amount of CRC cells was seeded into 6-well plates. Then a pipette tip to draw gaps on the cell monolayers. After this, we used serum-free DMEM to allow cells to migrate. The cells that migrated into these gaps were observed at 0 h, 12 h, and 24 h using an inverted microscope (Leica, DMI1, China).
Immunohistochemical (IHC) analysis
Tissue samples were fixed in 4% PFA and embedded in paraffin. Antigen retrieval was enforced in citrate buffer (pH 6.5) for 20 min. Tissue slices were incubated overnight at 4℃ with anti-ENO3 antibody (1:1000, Proteintech, USA), followed by secondary antibody incubation using Immunohistochemical secondary antibody kit (ZSGB-Bio, China). Antigen–antibody complexes were then determined with a DAB kit (ZSGB-Bio, China). Slices were lightly counterstained with hematoxylin to visualize nuclei. The data analysis was performed as previously described [14].
RNA sequencing
Collected cell samples were added Trizol reagent and then handed over to a biotechnology company (LC-Bio Technology, China) for the subsequent transcriptome sequencing. The sequencing results were obtained as the FPKM (fragment per kilobase of exons per million reads). The DEGs were selected with p < 0.05 and fold change > 2 by R software (package DESeq2), and then the differentially expressed genes was further used for GO enrichment analysis.
Detection of Lactate production and ATP
Lactate of culture medium was detected by the Lactate Assay kit (Solarbio, China). ATP of cell were assessed by a luciferase-based ATP assay kit (Beyotime, China). Lactate and ATP level were normalized to protein concentration, which was determined using BCA Kit (FDbio Science, China).
Statistical analysis
The statistical significance of Kaplan–Meier survival plot was determined by log-rank test. To compare the means between two groups, student t-test or non-parametric Mann–Whitney test was used according to data normality and homoscedasticity. Two-way ANOVA was used to evaluate the statistical differences of multiple groups. Graphpad Prism 8.0 and R software (version 3.6.3) was used for statistical analysis. p < 0.05 was considered to be statistically significant.
Results
ENO3 was upregulated and associated with tumor progression in CRC
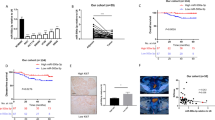
Based on the analysis of GEO datasets, we found that the mRNA expression of ENO3 in 566 CRC samples were higher than those in 19 normal samples (p < 0.01; Fig. 1A). In another 40 pairs of CRC and normal samples from TCGA datasets, ENO3 was also significantly upregulated in CRC tissues (p < 0.001; Fig. 1B). After correlated its expression with pathological stages, ENO3 was increased in the advanced N stage (p < 0.05; Fig. 1C). Furthermore, the AUC of ENO3 was 0.802, indicating a significantly altered expression level of ENO3 between CRC and normal tissues (Fig. 1D). We further analyzed the survival time of CRC patients and found that CRC patients with a higher ENO3 expression showed a poorer OS (p < 0.001; Fig. 1E) and PFS (p = 0.003; Fig. 1F). Baseline clinical characteristics of patients from TCGA database was shown in Supplementary Table 3 and censor data for OS was shown in Supplementary Table 4. In addition to CRC, ENO3 was also significantly higher expressed in breast infiltrating carcinoma (BRCA), esophageal carcinoma (ESCA), pleomorphic glioma (GBM), lung squamous cell carcinoma (LUSC) and adenocarcinoma (LUAD), prostate cancer (PRAD), pancreatic cancer (PAAD), endometrial cancer (UCEC), and gastric cancer (STAD) (Supplementary Fig. S1).

ENO3 is upregulated and correlated with poor survival outcomes in CRC from TCGA and GEO datasets. A ENO3 expression in normal and CRC tissues from GEO datasets. B ENO3 expression in normal and CRC tissues from TCGA datasets. C ENO3 expression level in different N stages in TCGA datasets. D ROC curve showing the role of ENO3 in discriminating CRC from normal tissues. E Kaplan–Meier plots of OS in CRC patients stratified by ENO3 levels in TCGA datasets. F Kaplan–Meier plots of PFS in CRC patients stratified by ENO3 levels in TCGA datasets. *p < 0.05, **p < 0.01
Then, IHC analysis of 84 pairs of clinical CRC tissues also confirmed that the ENO3 expression was significantly upregulated in CRC (p < 0.01; Fig. 2A, B). QRT-PCR (14 pairs of CRC samples, p < 0.05; Supplementary Fig. S2A) and western blot (5 pairs, Supplementary Fig. S2B) showed consistent results with the IHC analysis. Besides, ENO3 expression was also significantly increased in the advanced T stage (p < 0.05; Fig. 2C), N stage (p < 0.01; Fig. 2D), M stage (p < 0.01; Fig. 2E), and AJCC stage (p < 0.05; Fig. 2F). Baseline clinical characteristics of patients used in immunohistochemical analysis was shown in Supplementary Table 5.

ENO3 was upregulated and associated with tumor progression in CRC A Representative images of IHC staining of ENO3 in CRC tissues and adjacent normal tissues. B IHC analysis of 84 pairs of clinical CRC tissues. C–F IHC analysis showed that ENO3 expression was significantly increased in advanced AJCC/TNM stage. Scale bar = 200 μm. *p < 0.05, **p < 0.01
Knockdown of ENO3 suppressed the proliferation and migration of CRC cells
First, the protein expression of ENO3 in four wild colon cancer cell lines were measured by immunoblot (Supplementary Fig. S3). Based on this, ENO3 knockdown was performed in HCT116 and RKO for loss-of-function assay, while ENO3 overexpression in LoVo for gain-of-function assay.
ENO3 shRNA lentivirus was first transfected to stably knockdown ENO3 in HCT116 and RKO cells (Fig. 3A, B). The result of CCK-8 assay showed that ENO3 knockdown significantly suppressed the growth of HCT116 and RKO cells (Fig. 3C, D). In addition, the colony formation abilities of HCT116 and RKO cells were also greatly reduced by ENO3 knockdown (Fig. 3E, F). As shown by the cell migration assay, ENO3 knockdown decreased migrated cells via transwell assays (Fig. 4A, B). In wound healing assay, HCT116 and RKO cells with ENO3 knockdown also showed a markedly inhibited migration rate (Fig. 4C, D).

Knockdown of ENO3 suppressed the proliferation of CRC cells. A The mRNA level of ENO3 in HCT116 and RKO cells after lentiviral transduction of ENO3 shRNA or nonspecific shRNA (shCtrl) by qRT-PCR. B Expression of ENO3 in HCT116 and RKO cells after lentiviral transduction of ENO3 shRNA or shCtrl by western blot. C–D The cell growth curves of ENO3 silenced CRC cells were obtained from CCK-8 assays in HCT116 and RKO cells. E–F Colony formation assays of ENO3 silenced HCT116 and RKO cells. The top panel shows representative colony formation images, while the bottom panel shows bar graphs of the number of colonies per well (mean ± SD) and absorbance (570 nm) of the cell eluent per well (mean ± SD). *p < 0.05, **p < 0.01

Knockdown of ENO3 suppressed the migration of CRC cells. A Representative crystal violet staining results of transwell migration in HCT116 and RKO cells with ENO3 knockdown and control cells. B Statistical bar graphs of the number of migration cells per well. C, D Wound healing assays of HCT116 and RKO cells after being transfected with ENO3 shRNA or nonspecific shRNA (shCtrl). The top panel shows representative wound healing images for 0 h, 12 h, and 24 h, while the bottom panel shows bar graphs of the relative migration rate of per well for 12 h and 24 h. *p < 0.05, **p < 0.01 and ns, not significant
Overexpression of ENO3 promoted CRC cell proliferation and migration
To further validate the function of ENO3 in the proliferation and migration of CRC cells, LoVo cells were transfected with ENO3 expression plasmid (Fig. 5A). We found that ENO3 overexpression could enhance the cell proliferation ability in LoVo cells, as assessed by CCK-8 assay (Fig. 5B) and colony formation assay (Fig. 5C, D). In addition, increased migrated CRC cells were observed in our transwell and wound healing assay (Fig. 5E, F), indicating that ENO3 could enhance the cell migration ability of CRC.

Overexpression of ENO3 promoted CRC cell proliferation and migration. A The ENO3 mRNA and protein expression were detected by qRT-PCR and Western blotting, respectively after transfecting with ENO3 over-expression plasmid. β-actin was used as the internal control. B The cell growth curves of ENO3 overexpression (oe-ENO3) or control CRC cells (vector) in LoVo cells were obtained from CCK-8 assays. C Colony formation assays of ENO3 overexpression and control cells in LoVo cells. D Statistical bar graphs of the number of colonies per well (mean ± SD) and absorbance (570 nm) of the cell eluent per well (mean ± SD). E Representative crystal violet staining results of Transwell migration in ENO3 overexpressed LoVo cells and control cells. F Wound healing assays of LoVo after being transfected with over-expression plasmid or vector plasmid. *p < 0.05, **p < 0.01
Potential downstream targets of ENO3 in CRC
To explore the underlying mechanisms of ENO3 in CRC progression, RNA sequencing was performed. A total of 55 upregulated and 59 downregulated genes were selected after ENO3 was knockdown in HCT116 cells (p < 0.05, fold change ≥ 2) (Fig. 6A). Furthermore, GO enrichment analysis indicated that differential genes were significantly enriched in glycolytic process (Fig. 6B).

RNA sequencing to explore downstream genes and potential mechanisms regulated by ENO3 in CRC. A Volcano plot of differential genes of HCT116 cells after being transfected with shRNA lentivirus of ENO3. B GO enrichment of differentially expressed genes in HCT116 cells. C qRT-PCR was performed to detect the expression of indicated genes in HCT116 and RKO cells. D, E Extracellular lactate production, ATP level was measured in HCT116 and RKO cells after being transfected with ENO3 shRNA or nonspecific shRNA. F Extracellular lactate production, ATP level was measured in LoVo cells transfected with ENO3 expression plasmid or vector. Lactate and ATP level was normalized to protein concentration. *p < 0.05, **p < 0.01 and ns, not significant
Then, non-coding genes, genes with FPKM of < 1 in all samples or p ≥ 0.01 were removed from further analysis. In final, nine downregulated (SOSC7, NAFT5, MAFF, SLC25A43, DCTN4, SLC6A6, YTHDF3, SKA1, IER2) and four upregulated (ASB1, POLR3G, KITLG, SLC10A3) genes were selected for further qRT-PCR validation. As presented in Fig. 6C, the mRNA level of NAFT5, MAFF, and SLC25A43 were downregulated > 1.5 fold change after the knockdown of ENO3 in both HCT116 and RKO cells and validated as the potential downstream genes.
ENO3 enhanced cell glycolysis in CRC cells
Then, lactate levels in the culture media and ATP levels in CRC cells were detected to verify the role of ENO3 in glycolytic process. It showed that knockdown of ENO3 in HCT116 and RKO cells can decrease the levels of lactate production and ATP (Fig. 6D, E). Consistently, ENO3 overexpression significantly promoted the generation of lactate and ATP (Fig. 6F). These results indicated that ENO3 enhance cell glycolysis in CRC cells.
Discussion
Human ENO family includes ENO1-3, possessing different physiological functions in humans. Several bioinformatics analysis-based researches revealed that high ENO3 expression of was associated with poor outcome in CRC [13, 15]. In this study, we revealed that the ENO3 expression was significantly upregulated in CRC, both in online databases and our own clinical samples. Besides, we found that ENO3 had a relatively high AUC of 0.802 for CRC patients from TCGA database. Nevertheless, additional prospective researches are needed to validate the diagnostic value of ENO3 in CRC.
Recently, Hu et al. reported that CD47 could enhance glycolysis and ERK activity in CRC cells by stabilizing ENO1, and knockdown of ENO1 diminished CD46-induced cell proliferation and migration, showing a great potential of targeting glycolysis in the management of CRC [16]. Another study showed that ENO2 was activated in BRAF V600E-mutated CRC and could promote CRC progression through MAPK and PI3K/Akt pathways [7]. When compared to other members, the investigation on the role of ENO3 in cancer is relatively rare. To investigate the potential value of ENO3 as a therapeutic target in CRC, we evaluated the function of ENO3 in CRC cell proliferation, migration in vitro and found that ENO3 might be a valuable biomarker of CRC aggressiveness. To our knowledge, our study is the first to investigate the function of ENO3 in CRC.
Both Oxidative phosphorylation and extra-mitochondrial glycolysis are the two main way for normal cells to generate energy, and cancer cells primarily utilize glycolysis to generate energy [17,18,19]. The rapid proliferation of cancer cells contributes to local hypoxia tumor microenvironment, and cancer cells adapt to hypoxia microenvironment through enhanced glycolysis efficiency and lactate secretion. In addition, the accumulation of lactate could further promote the proliferation, migration, and immune escape of cancer cells [20, 21]. Recently, Shen et al. revealed that N6-methyladenosine (m6A) modification participated in the glucose metabolism of CRC, and targeting METTL3 offered a novel therapeutic target in CRC patients with high glycolysis efficiency [22]. Our study revealed that the enforced ENO3 expression in CRC cells led to increased lactate and ATP production, while knockdown of ENO3 showed the opposite effect, indicating that ENO3 play its role in CRC progression by enhancing extra-mitochondrial glycolysis.
As a glycolytic enzyme, ENO3 is predominantly found in muscle tissues, having a vigorous energy metabolism [23]. It has been demonstrated that ENO3 deficiency may lead to metabolic myopathies [24]. Lu et al. revealed that ENO3 could promote the progression of non-alcoholic steatohepatitis (NASH) progression through negatively regulating ferroptosis via elevating the expression of GPX4 and the accumulation of lipid, and was identified as a novel therapeutic target for NASH [25]. Besides, ENO3 has also been involved in various cancers. For example, ENO3 is up-regulated in CRC while down-regulated in hepatocellular cancer [13, 26]. Park et al. demonstrated that ENO3 had a protumoral effect on the progression of lung adenocarcinoma (LUAD) with STK11 mutations and was a potential therapeutic target [27]. Cui et al. uncovered that low expression of ENO3 was associated with poor prognosis of HCC patients and ENO3 acted as a tumor inhibitor in HCC development [28].
For the potential upstream of ENO3, Hif-1α is a key regulator of glycolysis, which can activate many key enzymes in glucose metabolism and catalyze irreversible rate-limiting steps [29]. So, as a glycolytic enzyme, whether ENO3 is mediated by Hif-1α deserves further exploration. Zheng et al. revealed that insulin-like growth factor-1 could stimulate ENO2 activity, thereby promoting liver metastasis in pancreatic ductal adenocarcinoma [30]. Peng et al. found that miR-7-5p knockdown could upregulate ENO2, which contributed to the radioresistance of nasopharyngeal carcinoma cells [31]. Both ENO3 and ENO2 belong to the ENO family and share the homology domains, whether the role of ENO3 in CRC is regulated by a similar upstream mechanism needs future confirmation.
To explore the potential downstream genes of ENO3 in CRC, RNA sequencing was performed in our study. According to GO annotation, ENO3 might influence the progression of CRC by enhancing the glycolytic process of CRC cells. Furthermore, NAFT5, MAFF, and SLC25A43 genes were successfully validated as the potential downstream genes. Zhang et al. had reported that silencing of NFAT5 suppressed proliferation of tumor cells, and phosphoglycerate kinase 1 (PGK-1), the key enzyme generating ATP in glycolysis, was identified as a downstream target of NFAT5 [32]. Another study reported that Circ_0001944 (Circular RNA) could also promote the progression and glycolysis of Non-Small Cell Lung Cancer (NSCLC) cells by upregulating NFAT5 in both vivo and vitro [33]. For MAFF, moon et al. had reported that MAFF could regulate tumor invasion and metastasis, and MAFF is also an important mediator of gene activation and repression in response to microenvironment changes in oxygen [34]. The mitochondrial transporter SLC25A43 was also down-regulated after knockdown of ENO3 in CRC cells. The potential role of SLC25A43 in CRC progression and cancer metabolism deserve our further exploration.
Taken together, our findings illustrate that high ENO3 expression level predicts poor prognosis and promotes CRC aggressiveness by enhancing cell glycolysis. NAFT5, MAFF, and SLC25A43 were identified and validated as the potential downstream genes of ENO3. However, further experiments such as rescue experiments, RNA pull down are still required to validate the downstream molecular mechanisms of ENO3. Besides, considering the advantage in simulating tumor microenvironment, in vivo studies is needed to further explore the role of ENO3.
Abbreviations
- CRC:
-
Colorectal cancer
- ENO:
-
Enolase
- qRT-PCR:
-
Quantitative real-time PCR
- IHC:
-
Immunohistochemical
- DEGs:
-
Differentially expressed genes
- ROC:
-
Receiver operating characteristic curves
- AUC:
-
The area under the curve
- TCGA:
-
The cancer genome atlas
- GEO:
-
Gene expression omnibus
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33. https://doi.org/10.3322/caac.21654.
Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. https://doi.org/10.3322/caac.21660.
Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–7. https://doi.org/10.1016/j.cell.2008.08.021.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Hong J, Guo F, Lu SY, et al. F. nucleatum targets lncRNA ENO1-IT1 to promote glycolysis and oncogenesis in colorectal cancer. Gut. 2021;70:2123–37. https://doi.org/10.1136/gutjnl-2020-322780.
Wang Y, Lu JH, Wu QN, et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer. 2019;18:174. https://doi.org/10.1186/s12943-019-1105-0.
Yukimoto R, Nishida N, Hata T, et al. Specific activation of glycolytic enzyme enolase 2 in BRAF V600E-mutated colorectal cancer. Cancer Sci. 2021;112:2884–94. https://doi.org/10.1111/cas.14929.
Yang T, Shu X, Zhang HW, et al. Enolase 1 regulates stem cell-like properties in gastric cancer cells by stimulating glycolysis. Cell Death Dis. 2020;11:870. https://doi.org/10.1038/s41419-020-03087-4.
Czogalla B, Partenheimer A, Badmann S, et al. Nuclear Enolase-1/ MBP-1 expression and its association with the Wnt signaling in epithelial ovarian cancer. Transl Oncol. 2020;14: 100910. https://doi.org/10.1016/j.tranon.2020.100910.
Zhan P, Zhao S, Yan H, et al. Alpha-enolase promotes tumorigenesis and metastasis via regulating AMPK/mTOR pathway in colorectal cancer. Mol Carcinog. 2017;56:1427–37. https://doi.org/10.1002/mc.22603.
Xu CM, Luo YL, Li S, et al. Multifunctional neuron-specific enolase: its role in lung diseases. 2019. Biosci Rep. https://doi.org/10.1042/BSR20192732.
Zhu J, Wang S, Bai H, et al. Identification of five glycolysis-related gene signature and risk score model for colorectal cancer. Front Oncol. 2021;11: 588811. https://doi.org/10.3389/fonc.2021.588811.
Cui Z, Sun G, Bhandari R, et al. Comprehensive analysis of glycolysis-related genes for prognosis, immune features, and candidate drug development in colon cancer. Front Cell Dev Biol. 2021;9:684322. https://doi.org/10.3389/fcell.2021.684322.
Khoury T, Sait S, Hwang H, et al. Delay to formalin fixation effect on breast biomarkers. Mod Pathol. 2009;22:1457–67. https://doi.org/10.1038/modpathol.2009.117.
Pan X, Wu H, Chen G, Li W. Prognostic value of enolase gene family in colon cancer. Med Sci Monit. 2020;26: e922980. https://doi.org/10.12659/MSM.922980.
Hu T, Liu H, Liang Z, et al. Tumor-intrinsic CD47 signal regulates glycolysis and promotes colorectal cancer cell growth and metastasis. Theranostics. 2020;10:4056–72. https://doi.org/10.7150/thno.40860.
Kim J, Yu L, Chen W, et al. Wild-type p53 promotes cancer metabolic switch by inducing PUMA-dependent suppression of oxidative phosphorylation. Cancer Cell. 2019;35(191–203): e8. https://doi.org/10.1016/j.ccell.2018.12.012.
Weng ML, Chen WK, Chen XY, et al. Fasting inhibits aerobic glycolysis and proliferation in colorectal cancer via the Fdft1-mediated AKT/mTOR/HIF1alpha pathway suppression. Nat Commun. 2020;11:1869. https://doi.org/10.1038/s41467-020-15795-8.
Corbet C, Feron O. Cancer cell metabolism and mitochondria: nutrient plasticity for TCA cycle fueling. Biochim Biophys Acta Rev Cancer. 2017;1868:7–15. https://doi.org/10.1016/j.bbcan.2017.01.002.
Ren S, Liu J, Feng Y, et al. Knockdown of circDENND4C inhibits glycolysis, migration and invasion by up-regulating miR-200b/c in breast cancer under hypoxia. J Exp Clin Cancer Res. 2019;38:388. https://doi.org/10.1186/s13046-019-1398-2.
Ostergaard L, Tietze A, Nielsen T, et al. The relationship between tumor blood flow, angiogenesis, tumor hypoxia, and aerobic glycolysis. Cancer Res. 2013;73:5618–24. https://doi.org/10.1158/0008-5472.CAN-13-0964.
Shen C, Xuan B, Yan T, et al. m(6)A-dependent glycolysis enhances colorectal cancer progression. Mol Cancer. 2020;19:72. https://doi.org/10.1186/s12943-020-01190-w.
Boudon S, Ounaissi D, Viala D, et al. Label free shotgun proteomics for the identification of protein biomarkers for beef tenderness in muscle and plasma of heifers. J Proteomics. 2020;217: 103685. https://doi.org/10.1016/j.jprot.2020.103685.
Musumeci O, Brady S, Rodolico C, et al. Recurrent rhabdomyolysis due to muscle beta-enolase deficiency: very rare or underestimated? J Neurol. 2014;261:2424–8. https://doi.org/10.1007/s00415-014-7512-7.
Lu D, Xia Q, Yang Z, et al. ENO3 promoted the progression of NASH by negatively regulating ferroptosis via elevation of GPX4 expression and lipid accumulation. Ann Transl Med. 2021;9:661. https://doi.org/10.21037/atm-21-471.
Liu ZK, Zhang RY, Yong YL, et al. Identification of crucial genes based on expression profiles of hepatocellular carcinomas by bioinformatics analysis. PeerJ. 2019;7: e7436. https://doi.org/10.7717/peerj.7436.
Park C, Lee Y, Je S, et al. Overexpression and selective anticancer efficacy of ENO3 in STK11 mutant lung cancers. Mol Cells. 2019;42:804–9. https://doi.org/10.14348/molcells.2019.0099.
Cui H, Guo D, Zhang X, et al. ENO3 inhibits growth and metastasis of hepatocellular carcinoma via wnt/beta-catenin signaling pathway. Front Cell Dev Biol. 2021;9: 797102. https://doi.org/10.3389/fcell.2021.797102.
Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–14. https://doi.org/10.1016/j.tips.2012.01.005.
Zheng Y, Wu C, Yang J, et al. Insulin-like growth factor 1-induced enolase 2 deacetylation by HDAC3 promotes metastasis of pancreatic cancer. Signal Transduct Target Ther. 2020;5:53. https://doi.org/10.1038/s41392-020-0146-6.
Peng J, Liu F, Zheng H, Wu Q, Liu S. IncRNA ZFAS1 contributes to the radioresistance of nasopharyngeal carcinoma cells by sponging hsa-miR-7-5p to upregulate ENO2. Cell Cycle. 2021;20:126–41. https://doi.org/10.1080/15384101.2020.1864128.
Jiang Y, He R, Jiang Y, et al. Transcription factor NFAT5 contributes to the glycolytic phenotype rewiring and pancreatic cancer progression via transcription of PGK1. Cell Death Dis. 2019;10:948. https://doi.org/10.1038/s41419-019-2072-5.
Dou Y, Tian W, Wang H, Lv S. Circ_0001944 contributes to glycolysis and tumor growth by upregulating NFAT5 through acting as a decoy for miR-142-5p in non-small cell lung cancer. Cancer Manag Res. 2021;13:3775–87. https://doi.org/10.2147/CMAR.S302814.
Moon EJ, Mello SS, Li CG, et al. The HIF target MAFF promotes tumor invasion and metastasis through IL11 and STAT3 signaling. Nat Commun. 2021;12:4308. https://doi.org/10.1038/s41467-021-24631-6.
Funding
This work was financially supported by the National Foundation of Natural Science of China (82073229).
Author information
Authors and Affiliations
Contributions
JC, ZZ, and JN: Complete experiments and drafted the manuscript. JS, FJ, ZW: Provided clinical samples and CRC cell lines. MX and LW: Designed the study and revised the manuscript. All authors red and approved the submitted version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
None.
Ethics approval
The study protocol was approved by the Clinical Research Ethics Committee of the Second Affiliated Hospital of Zhejiang University and informed consent was obtained from all patients. Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
12032_2022_1676_MOESM6_ESM.pdf
Supplementary Figure S1. mRNA levels of ENO3 across TCGA cancers with CRC and normal samples. *p < 0.05, **p < 0.01, ***p < 0.001. Supplementary file6 (PDF 322 kb)
12032_2022_1676_MOESM7_ESM.pdf
Supplementary Figure S2. ENO3 is upregulated in CRC tissues. (A) ENO3 mRNA expression levels in 14 pairs of human primary CRC tissues and normal mucosa. (B) ENO3 protein expression in 5 pairs of human primary CRC tumor tissues and adjacent normal tissues was assessed by western blot. *p < 0.05. Supplementary file7 (PDF 326 kb)
Rights and permissions
About this article

Cite this article
Chen, J., Zhang, Z., Ni, J. et al. ENO3 promotes colorectal cancer progression by enhancing cell glycolysis. Med Oncol 39, 80 (2022). https://doi.org/10.1007/s12032-022-01676-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-022-01676-1