
Abstract
Accumulating evidence have suggested that some microRNAs are aberrantly expressed in prostate cancer. In our previous work, we had identified a panel of four differentially expressed microRNAs in prostate cancer. In the present study, we have investigated common molecular targets of this panel of miRNAs (DEMs) and key hub genes that can serve as potential candidate biomarkers in the pathogenesis and progression of prostate cancer. A joint bioinformatics approach was employed to identify differentially expressed genes (DEGs) in prostate cancer. Gene enrichment analysis followed by the protein–protein interaction (PPI) network construction and selection of hub genes was further performed using String and Cytoscape, respectively. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of the identified hub genes was conducted using the Database for Annotation, Visualization and Integrated Discovery (DAVID) tool. In total, 496 genes were identified to be common targets of DEMs in prostate cancer and 13 key hub genes were identified from three modules of the PPI network of the DEGs. Further top five genes viz Rhoa, PI3KCA, CDC42, MAPK3, TP53 were used for Enrichment analysis which revealed their association with vital cellular and functional pathways in prostate cancer indicating their potential as candidate biomarkers in prostate cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prostate cancer is one of the leading causes of cancer-related mortality in men with estimated number of 164,690 new cases and 29,430 deaths, respectively, in the United States according to 2018 statistics [1]. A large number of factors have been associated with the patient survival including the stage of cancer at the time of diagnosis, and thus early detection may aid in better prognosis. Various clinical parameters such as pathological biopsy, prostate-specific antigen (PSA) value, histopathological scores (Gleason score) and imaging diagnostics allows detection of the disease, however, they lack in specificity and sensitivity [2]. First, PSA as a prognostic marker has detrimental effects as it is not tumor specific [3] and screens indolent tumors leading to unnecessary consecutive biopsies [4]. Although the combination of PSA and digital rectal examination allow certain risk stratification, however, it is associated with unavoidable risk of misclassification and thus fails in assisting the most beneficial treatment option [5]. Many attempts have been made in the identification of common molecular signatures that can serve as reliable biomarkers and help in decision-making in critical scenarios and one such approach is microRNAs as blood-based non-invasive biomarkers for cancer.
MicroRNAs (miRNAs) are highly conserved, endogenous, single-stranded, non-protein coding oligonucleotides [6]. They play an important role in the regulation of gene expression at post-transcriptional and post-translational level [7]. MiRNAs bind to the 3′-untranslated region of the target mRNAs and depending on the level of complementarity between the target mRNA and miRNA it either leads to transcriptional repression or translation inhibition of the targets [8] and thus it regulates multiple signaling pathways [9]. MiRNAs are reported to regulate various important metabolic and cellular pathways such as proliferation, migration, invasion, apoptosis, cell cycle, differentiation, survival and are reported to be aberrantly expressed in various cancers including prostate cancer [10, 11]. MiRNAs can be isolated from body fluids in stable condition and can be detected in small volumes with high specificity and sensitivity [12,13,14]. All these factors make miRNAs potential putative biomarker which can be of great clinical significance.
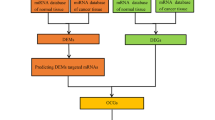
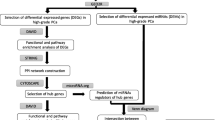
Recently, we had identified a panel of four most consistently reported dysregulated miRNAs in prostate cancer [15]. In the present study, we have used a joint bioinformatics approach to analyze this panel of differentially expressed miRNAs (DEMs) to identify key genes that are differentially expressed (DEGs) in PCa. Subsequently, functional and pathway enrichment analysis was performed on the identified DEGs, and their potential function and signaling pathways regulated by them in PCa were identified. Further protein–protein interaction (PPI) was constructed, and key hub genes and modules were identified using various plug-ins in Cytoscape (Fig. 1). The present study aimed to evaluate the underlying molecular and biological pathways driven by this panel of signature miRNAs in PCa and to identify certain hub genes that can serve as potential candidate diagnostic and prognostic biomarkers for PCa.

Flowchart of the methodology used in the study
Materials and methods
Identification of DEMs
A systematical literature search was carried out using PubMed and Google Scholar and the keywords used were: microRNA, miRNA, and prostate cancer. Relevant full text and peer-reviewed research articles written in English were considered. A comprehensive list of differentially expressed microRNAs (miRNAs) (DEMs) in prostate cancer was prepared followed by identification of a panel of four consistently reported dysregulated miRNAs in prostate cancer namely, miR-141, miR-375, miR-221 and miR-21 as per our previously published paper [15].
Target prediction of DEMs in PCa
Targets of the DEMs in PCa were retrieved using in silico tools and databases such as miRDB (http://www.mirdb.org/) [16], miRTarbase (http://mirtarbase.mbc.nctu.edu.tw/php/index.php) [17] and miRwalk (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/) [18]. These databases were used as they comprise of the experimentally validated miRNA targets and are the most frequently updated ones. A comprehensive list of target genes, regulated by this panel of DEMs was prepared followed by duplicate removal. Followed by that an overlapping analysis was carried out using a Venn diagram and a list of hub genes targeted by this panel of DEMs in PCa was identified.
Protein–protein interaction (PPI) network and module analysis
The protein–protein interaction (PPI) network analysis of the identified hub genes was constructed using the Search Tool for the Retrieval of Interacting Genes (STRING; https://string-db.org/cgi/network.pl) [19] database. An interaction score of ≥ 0.7 (high confidence) was considered the threshold value. The resulting PPI network was then visualized using Cytoscape software (version 3.7.0; https://cytoscape.org/) [20]. The Molecular Complex Detection (MCODE version 1.5.1) application (https://cytoscape.org/apps/mcode) [21] was used to perform module analysis. A degree cutoff = 2, node density cutoff = 0.2, node score cutoff = 0.2, K-core = 2 and max depth = 100 was considered as the threshold. Further hub gene analysis was carried out using cytoHubba (version 0.1; https://cytoscape.org/apps/cytohubba) [22] application of Cytoscape where degree ≥ 10 was used as the threshold.
Gene, function and pathway enrichment analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID 6.8, https://david.ncifcrf.gov/) [23] was used to perform Gene Ontology (GO) annotation analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG, https://www.genome.jp/kegg/) [24] pathway analysis of the hub genes identified by cytoHubba (version 0.1; https://cytoscape.org/apps/cytohubba) application of Cytoscape. GeneCodis (http://genecodis.cnb.csic.es/analysis) [25] and Panther (http://www.pantherdb.org/) [26] was used for the functional enrichment analysis. The GO of the targets of identified DEMs were further categorized into biological functions (BF), molecular functions (MF) and cellular components (CC). p > 0.05 was used as the threshold value for considering the results to be statistically significant.
Results
Identification of differentially expressed microRNAs (DEMs)
In our previous publication, we had identified a panel of four dysregulated signature miRNAs (DEMs) in PCa from the literature which were reported by almost all authors to be differentially expressed in PCa irrespective of the profiling platform. This panel of signature DEMs was selected for further analysis using bioinformatics [15].
Identification of differentially expressed genes (DEGs)
A total of 1132, 497, 824 and 1218 genes were identified to be targets of miR-141, miR-375, miR-221 and miR-21 respectively in PCa after duplicates removal. Overlapping analysis using Venn diagram identified a total of 496 genes to be common targets of this panel of signature DEMs (Fig. 2).

Venn diagram showing common genes targeted by the panel of differentially expressed miRNAs (DEMs) in prostate cancer. DEMs shared by at least three datasets were extracted to identify target genes
PPI network construction and module analysis
To analyze the interaction between the identified common targets of the panel of signature DEMs, the protein–protein interaction (PPI) network was constructed using STRING. The PPI network of the identified targets of the panel of signature DEMs consisted of 485 nodes and 349 edges with a confidence score of ≥ 0.7 (Fig. 3). The network was retrieved from STRING and visualized using Cytoscape software version 3.7.0. A total of 13 hub genes were selected from the PPI network with a degree ≥ 10 (Fig. 4). The hub genes include Ras homolog family member A (RHOA), Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA), Tumor protein p53 (TP53), cell division cycle 42 (CDC42), Mitogen-activated protein kinase 3 (MAPK3), V-myc myelocytomatosis viral oncogene homolog (avian) (MYC), Catenin (cadherin-associated protein), beta 1, (CTNNB1), Heat shock protein 90 kDa alpha (cytosolic), class A member 1; (HSP90AA1), Platelet-activating factor acetylhydrolase 1b, regulatory subunit 1 (PAFAH1B1), Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide (YWHAZ), Janus kinase 2 (JAK2), V-erb-b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma derived oncogene homolog (avian) (ERBB2), Integrin, beta 1 (fibronectin receptor, beta polypeptide, antigen CD29 includes MDF2, MSK12) (ITGB1) (Supplementary Table 1). According to the degree of importance, three significant modules were screened from the PPI network complex using MCODE application. Module 1 consisted of 7 nodes and 21 edges; Module 2 consisted of 6 nodes and 15 edges and Module 3 consisted of 10 nodes and 23 edges (Fig. 5a–c). Further, the PPI network of the identified hub genes consisted of 18 nodes and 59 edges with a confidence score of ≥ 0.7 (Fig. 6).

Protein–protein interactions (PPI) of the differentially expressed genes (DEGs) in prostate cancer. Colored lines indicate the type of interaction evidence between nodes

Top 13 identified hub genes in prostate cancer

The modules of the PPI network identified using the MCode plug-in of Cytoscape: a module 1, b module 2 and c module 3

Protein–protein interactions (PPI) of the identified hub genes using Cytohubba plug-in of Cytoscape. Colored lines indicate the type of interaction evidence between nodes
Gene, function and pathway enrichment analysis
To gain a better understanding of the identified 13 hub genes, GO and KEGG pathway enrichment analysis was performed using DAVID. The enriched GO terms were categorized in molecular functions (MF), biological functions (BF), and cellular components (CC) (Supplementary Table 2). The analysis revealed that majority of the genes in the MF category were mainly enriched in ‘protein binding,’ ‘enzyme binding,’ ‘ATP binding,’ ‘receptor binding, ‘ion binding’ and ‘kinase binding.’ In the BF category, majority of the genes were associated with ‘intracellular signal transduction,’ ‘organ development,’ ‘wound healing,’ ‘cell surface receptor signaling pathway,’ ‘regulation of cellular component organization.’ In addition, the GO CC enrichment analysis revealed that the majority of the genes in this category were enriched in ‘cytosol,’ ‘extracellular exosome,’ and ‘plasma membrane part.’
Further, the KEGG pathway enrichment analysis revealed that the genes were primarily enriched in ‘Pathways in cancer,’ ‘PI3K-Akt signaling pathway’, ‘Prostate cancer,’ ‘MicroRNAs in cancer’ and ‘Wnt signaling pathway.’ These results demonstrated that these genes are significantly enriched in the hallmarks of cancer.
Discussion
Prostate cancer (PCa) remains one of the major medical burden associated with mortality in men worldwide and the mortality associated with metastatic PCa is even higher [27]. Research into the molecular mechanisms might shed light in the identification of biomarkers for prostate cancer therapeutics. In our recently published work, we had identified a panel of four microRNAs (miRNAs) that are consistently reported by various authors to be differentially expressed (DEMs) in PCa irrespective of the profiling platform and sample size being recruited. In the current study, we retrieved the gene targets of this panel of DEMs using insilico tools. The protein–protein interaction (PPI) was constructed and was viewed using Cytoscape.
The PPI network analysis was followed by analysis of hub genes using Cytohubba plug-in of Cytoscape that identified 13 hub genes with ≥ 10°. The functional enrichment analysis of the identified hub genes further revealed their involvement in various molecular functions such as protein binding, receptor binding, kinase binding thus highlighting their role in regulating hallmarks of cancer [28]. Further, the biological pathway enrichment analysis showed that these identified hub genes contribute in intracellular signal transduction, organ development, wound healing, cell surface receptor signaling pathway indicating the involvement of these hub genes in tumorigenesis. The pathway analysis using KEGG shed light into some of the important signaling pathways being enriched by these hub genes namely pathways in cancer, PI3K–Akt signaling pathway, prostate cancer, MicroRNAs in cancer and Wnt signaling pathway.
Out of the 13 hub genes identified by the Cytohubba plug-in of Cytoscape, the top five important hub genes identified were Rhoa, PI3KCA, CDC42, MAPK3, and TP53. These genes are reported to regulate various vital molecular and cellular function in various carcinomas including prostate cancer.
Ras homolog gene family member (RhoA) is a member of the Ras homology family of small GTPases [29] and is reported to be upregulated in a variety of cancers, particularly epithelial cancers and has a role to play in the progression of cancer [30]. It plays a vital role in the regulation of the actin and microtubule cytoskeleton [31] and influences many cellular functions such as proliferation, migration, invasion, polarity, motility, adhesion and cell cycle progression [32]. In case of colorectal cancer inactivation of RhoA leads to increased proliferation and migration by activating Wnt/β catenin pathway and subsequently metastasis, acting as a tumor suppressor gene [33] and is associated with poor prognosis in colorectal cancer [34]. However, in case of gastric cancer RhoA is reported to be overexpressed and is associated with the progression of the cancer acting as an oncogene and silencing of its expression led to apoptosis induction [35] and reduced cell migration and invasion [36] and is also found to regulate epithelial-mesenchymal transition (EMT) in gastric cancer [37]. In prostate cancer, RhoA facilitates invasiveness of PCa cells and also induces androgen-receptor-mediated apoptosis of PCa cells [38]. In our analysis, RhoA was identified as the top hub gene with 21° and was found to be involved in Module 1 and Module 2. Further functional enrichment analysis revealed the involvement of RhoA in wound healing, cell cycle, cell activation, regulation of cell–cell adhesion, pathways in cancer and Wnt signaling pathway highlighting its role in regulating cancer metastasis.
PIK3CA encodes for phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (p110α) which is the catalytic subunit of the enzyme phosphatidylinositol 3-kinase (PI3K) and regulates the activation of AKT [39]. PIK3CA mutations are reported in various adenocarcinoma such as breast, colon, lung that results in the constitutive activation of p110α leading to the activation of AKT1 and its downstream component mammalian target of rapamycin (mTOR) and Cyclin-dependent kinase inhibitors [40,41,42,43]. In the case of prostate cancer upregulated expression of PIK3CA is reported to regulate proliferation, migration and invasion of PCa cells in vitro and is associated with PCa progression [44, 45]. Activation of PI3K can be a consequence of loss of function of phosphatase and tensin homologue (PTEN) reported in various cancers. PI3K–Akt–mTOR pathway plays a critical role in regulating metabolism, proliferation, cell motility and apoptosis [46]. In our analysis, PIK3CA is identified as a hub gene with 19° in the PPI network and as a core gene of Module 3. Further enrichment analysis revealed the association of PIK3CA with ATP binding, protein kinase activation, organ development, mTOR signaling pathway, apoptosis, PI3K–Akt signaling pathway suggesting PIK3CA to be a potential target for PCa therapeutics.
The tumor protein p53 (TP53) is a tumor suppressor gene, and its mutation is reported in almost all types of cancer [47,48,49]. The TP53 mutation is associated with various hallmarks of cancer such as proliferation, increased migration, invasion, resistance to apoptosis, anchorage-independent growth, increased colony formation, angiogenesis and cell survival [50] and it serves as prognostic and predictive markers of cancer depending on the cancer type and status [51]. In our analysis TP53 was identified to be a hub gene with 19° in the PPI network and is found to be core gene of Module 3 and enriched in enzyme binding, ATP binding, protein kinase activity, protein phosphatase binding, wound healing, negative regulation of cell differentiation, cell cycle, pathways in cancer, MAPK signaling pathway and apoptosis, suggesting its association with prostate cancer progression.
Cell division control protein 42 homologue (CDC42) is a member of Rho GTPase family and reported to regulate various cellular processes governing cancer progression such as cell division, cellular transformation, cell invasion, enzyme activity, membrane trafficking and cell polarity thereby promoting migration of cells [52]. The upregulated expression of Cdc42 is reported in various cancers and is associated with tumorigenesis, invasion, and metastasis [53, 54]. For instance, in the case of cervical cancer, the expression of Cdc42 correlates positively with the grade of cervical lesions and its overexpression leads to migration and invasion by pseudopodia formation [55]. Similarly in the case of gastric cancer, inhibiting the expression of Cdc42 induced cell cycle arrest with simultaneous downregulation of MMP9 expression resulting in reduced proliferation, migration and invasion of gastric cancer cells [56]. In the case of esophageal cancer, knockdown of CXCR4 resulted in downregulated expression of Cdc42 resulting in reduced proliferation, migration, invasion of esophageal cells and suppression of tumor growth in xenograft models [57]. In case of prostate cancer also, Cdc42 regulates motility and invasive potential of PCa cells by actin-cytoskeleton rearrangement [58] also it is reported that activation of Cdc42-associated tyrosine kinase Ack1leads to accelerated prostate xenograft tumor growth in mice, poor prognosis, increased cell motility, invasion and metastasis [59]. In our analysis, we found that CDC42 is identified as one of the hub gene with 17° in the PPI network and is found to be a core element of the Module 2. Further biological and functional enrichment analysis demonstrated its association with protein binding, enzyme binding, protein kinase binding, receptor binding, regulation of cell–cell adhesion, pathways in cancer and MAPK signaling pathway.
Mitogen-activated protein kinase (MAPK) plays an important role in converting extracellular signals into cellular response [60] and regulate various cellular functions of the cell such as gene expression, differentiation, proliferation, migration, apoptosis [61] and are also required for the induction of epithelial-mesenchymal transition (EMT) [62]. The MAPK signaling pathway is reported to be one of the most common dysregulated pathways in many cancers. MAPK3 was identified as a hub gene with 17° in the PPI network, and as a core gene of Module 3. The enrichment analysis demonstrated that MAPK3 is associated with protein binding, enzyme binding, ion binding, intracellular signal transduction, cell activation, cell surface receptor signaling pathway revealing that MAPK3 may be involved in the progression of prostate cancer.
In conclusion, 496 genes were found to be differentially expressed in prostate cancer (DEGs) that are common targets of the identified panel of differentially expressed miRNAs (DEMs) in prostate cancer. In addition, 13 key hub genes were identified from the PPI network analysis of the DEGs which can serve as potential candidate biomarkers in the pathogenesis and progression of prostate cancer. The hub genes and the identified panel of signature miRNAs may serve as novel biomarkers in prostate cancer management. Additionally, biological and functional enrichment analysis showed that these identified hub genes played a key role in major cellular and physiological functions such as proliferation, cell activation, wound healing, apoptosis, enzyme binding, protein kinase binding, Wnt regulation. Further, the DEMs-DEGs network was constructed that shed further light into the molecular mechanisms of prostate cancer metastasis. However, further studies are required to validate and confirm our findings.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30.
Velonas V, Woo H, Remedios C, Assinder S. Current status of biomarkers for prostate cancer. Int J Mol Sci. 2013;14(6):11034–60.
Semjonow A, Brandt B, Oberpenning F, Roth S, Hertle L. Discordance of assay methods creates pitfalls for the interpretation of prostate-specific antigen values. Prostate. 1996;29(S7):3–16.
Filella X, Foj L, Augé JM, Molina R, Alcover J. Clinical utility of % p2PSA and prostate health index in the detection of prostate cancer. Clin Chem Lab Med (CCLM). 2014;52(9):1347–55.
Thompson IM, Pauler DK, Goodman PJ, Tangen CM, Lucia MS, Parnes HL, Crowley JJ. Prevalence of prostate cancer among men with a prostate-specific antigen level ≤ 4.0 ng per milliliter. N Eng. J Med. 2004;350(22):2239–46.
Wahid F, Shehzad A, Khan T, Kim YY. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. BBA Mol Cell Res. 2010;1803(11):1231–43.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Rothschild SI. MicroRNA therapies in cancer. Mol Cell Ther. 2014;2(1):7.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33.
MacFarlane LA, Murphy RP. MicroRNA: biogenesis, function and role in cancer. Curr Genomics. 2010;11(7):537–61.
Singh AN, Sharma N. In silico Meta-Analysis of Circulatory microRNAs in Prostate Cancer. J Anal Oncol. 2017;6(2):107–16.
Kim YK. Extracellular microRNAs as biomarkers in human disease. Chonnam Med J. 2015;51(2):51–7.
Sita-Lumsden A, Dart DA, Waxman J, Bevan CL. Circulating microRNAs as potential new biomarkers for prostate cancer. Br J Cancer. 2013;108(10):1925.
Schwarzenbach H, Nishida N, Calin GA, Pantel K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat Rev Clin Oncol. 2014;11(3):145.
Sharma N, Baruah MM. The microRNA signatures: aberrantly expressed miRNAs in prostate cancer. Clin Transl Oncol. 2018. https://doi.org/10.1007/s12094-018-1910-8.
Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2014;43(D1):D146–52.
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Tsai TR. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2015;44(D1):D239–47.
Sticht C, De La Torre C, Parveen A, Gretz N. miRWalk: an online resource for prediction of microRNA binding sites. PLoS ONE. 2018;13(10):e0206239.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Jensen LJ. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016;45:D362–8.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4(1):2.
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(4):S11.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4(1):44.
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa MKEGG. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2009;38(suppl_1):D355–60.
Nogales-Cadenas R, Carmona-Saez P, Vazquez M, Vicente C, Yang X, Tirado F, Pascual-Montano A. GeneCodis: interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res. 2009;37(suppl_2):W317–22.
Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2016;45(D1):D183–9.
Patil N, Gaitonde K. Clinical perspective of prostate cancer. Top Magn Reson Imaging. 2016;25(3):103–8.
Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res. 2017;7(5):1016.
Gilbert-Ross M, Marcus AI, Zhou W. RhoA, a novel tumor suppressor or oncogene as a therapeutic target. Genes Dis. 2015;2(1):2.
Orgaz JL, Herraiz C, Sanz-Moreno V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases. 2014;5(4):e29019.
Hall A. Rho family gtpases. Biochem Soc Trans. 2012;40(6):1378–82.
Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69.
Dopeso H, Rodrigues P, Bilic J, Bazzocco S, Cartón-García F, Macaya I, Martínez-Barriocanal Á. Mechanisms of inactivation of the tumour suppressor gene RHOA in colorectal cancer. Br J Cancer. 2018;118(1):106.
Jeong D, Park S, Kim H, Kim CJ, Ahn TS, Bae SB, Kwon HY. RhoA is associated with invasion and poor prognosis in colorectal cancer. Int J Oncol. 2016;48(2):714–22.
Song L, Guo Y, Xu B. Expressions of Ras Homolog Gene family, member A (RhoA) and cyclooxygenase-2 (COX-2) proteins in early gastric cancer and their role in the development of gastric cancer. Med Sci Monit. 2017;23:2979–84.
Yoon JH, Choi WS, Kim O, Choi BJ, Nam SW, Lee JY, Park WS. Gastrokine 1 inhibits gastric cancer cell migration and invasion by downregulating RhoA expression. Gastric Cancer. 2017;20(2):274–85.
Li H, Wang Z, Zhang W, Qian K, Xu W, Zhang S. Fbxw7 regulates tumor apoptosis, growth arrest and the epithelial-to-mesenchymal transition in part through the RhoA signaling pathway in gastric cancer. Cancer Lett. 2016;370(1):39–55.
Liu K, Li X, Wang J, Wang Y, Dong H, Li J. Genetic variants in RhoA and ROCK1 genes are associated with the development, progression and prognosis of prostate cancer. Oncotarget. 2017;8(12):19298.
Jason SL, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143(17):3050–60.
Moynahan ME, Chen D, He W, Sung P, Samoila A, You D, Baselga J. Correlation between PIK3CA mutations in cell-free DNA and everolimus efficacy in HR+, HER2− advanced breast cancer: results from BOLERO-2. Br J Cancer. 2017;116(6):726–30.
Bonetti LR, Barresi V, Bettelli S, Caprera C, Manfredini S, Maiorana A. Analysis of KRAS, NRAS, PIK3CA, and BRAF mutational profile in poorly differentiated clusters of KRAS-mutated colon cancer. Hum Pathol. 2017;62:91–8.
Green S, Trejo CL, McMahon M. PIK3CA(H1047R) accelerates and enhances KRAS(G12D)-driven lung tumorigenesis. Cancer Res. 2015;75(24):5378–91.
Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14(5):381–95.
Zhang S, Cai J, Xie W, Luo H, Yang F. miR 202 suppresses prostate cancer growth and metastasis by targeting PIK3CA. Exp Ther Med. 2018;16(2):1499–504.
Pearson HB, Li J, Meniel VS, Fennell CM, Waring P, Montgomery KG, Cullinane C. Identification of Pik3ca mutation as a genetic driver of prostate cancer that cooperates with Pten loss to accelerate progression and castration-resistant growth. Cancer Discov. 2018;8(6):764–79.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–19.
Kandioler D, Mittlböck M, Kappel S, Puhalla H, Herbst F, Langner C, Hofbauer F. TP53 mutational status and prediction of benefit from adjuvant 5-fluorouracil in stage III colon cancer patients. EBioMedicine. 2015;2(8):825–30.
Scheel A, Bellile E, McHugh JB, Walline HM, Prince ME, Urba S, Bradford C. Classification of TP53 mutations and HPV predict survival in advanced larynx cancer. Laryngoscope. 2016;126(9):E292–9.
Gao W, Jin J, Yin J, Land S, Gaither-Davis A, Christie N, Keohavong P. KRAS and TP53 mutations in bronchoscopy samples from former lung cancer patients. Mol Carcinog. 2017;56(2):381–8.
Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25(3):304–17.
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008.
Humphries-Bickley T, Castillo-Pichardo L, Hernandez-O-Farrill E, Borrero-Garcia LD, Forestier-Roman I, Gerena Y, Vlaar CP. Characterization of a dual Rac/Cdc42 inhibitor MBQ-167 in metastatic cancer. Mol Cancer Ther. 2017;16(5):805–18.
Ellenbroek SI, Collard JG. Rho GTPases: functions and association with cancer. Clin Exp Metastasis. 2007;24(8):657–72.
Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582(14):2093–101.
Ye H, Zhang Y, Geng L, Li Z. Cdc42 expression in cervical cancer and its effects on cervical tumor invasion and migration. Int J Oncol. 2015;46(2):757–63.
Du DS, Yang XZ, Wang Q, Dai WJ, Kuai WX, Liu YL, Tang XJ. Effects of CDC42 on the proliferation and invasion of gastric cancer cells. Mol Med Rep. 2016;13(1):550–4.
Guo J, Yu X, Gu J, Lin Z, Zhao G, Xu F, Ge D. Regulation of CXCR57/AKT-signaling-induced cell invasion and tumor metastasis by RhoA, Rac-1, and Cdc42 in human esophageal cancer. Tumor Biol. 2016;37(5):6371–8.
Guo Y, Zhang Z, Wei H, Wang J, Lv J, Zhang K, Wang Q. Cytotoxic necrotizing factor 1 promotes prostate cancer progression through activating the Cdc42–PAK1 axis. J Pathol. 2017;243(2):208–19.
Mahajan NP, Liu Y, Majumder S, Warren MR, Parker CE, Mohler JL, Whang YE. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci USA. 2007;104(20):8438–43.
Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12(1):9–18.
Imajo M, Tsuchiya Y, Nishida E. Regulatory mechanisms and functions of MAP kinase signaling pathways. IUBMB Life. 2006;58(5–6):312–7.
Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, Nakamori S, Mori M. Epithelial–mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010;101(2):293–9.
Acknowledgements
This work was supported by Grants from Symbiosis Centre for Research and Innovation (SCRI) and Symbiosis School of Biological Sciences (SSBS), Symbiosis International (Deemed University), Lavale, Pune, India. Ms. Meghna M. Baruah receive Senior Research Fellowship from Symbiosis Centre for Research and Innovation (SCRI), Symbiosis International (Deemed University), Lavale, Pune, India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Baruah, M.M., Sharma, N. In silico identification of key genes and signaling pathways targeted by a panel of signature microRNAs in prostate cancer. Med Oncol 36, 43 (2019). https://doi.org/10.1007/s12032-019-1268-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-019-1268-y