
Abstract
Genes related to key cellular pathways are frequently altered in B cell ALL and are associated with poor survival especially in high-risk (HR) subgroups. We examined gene copy number abnormalities (CNA) in 101 Indian HR B cell ALL patients and their correlation with clinicopathological features by multiplex ligation-dependent probe amplification. Overall, CNA were detected in 59 (59%) cases, with 26, 10 and 23% of cases harboring 1, 2 or +3 CNA. CNA were more prevalent in BCR-ABL1 (60%), pediatric (64%) and high WCC (WBC count) (63%) patients. Frequent genes deletions included CDNK2A/B (26%), IKZF1 (25%), PAX5 (14%), JAK2 (7%), BTG1 (6%), RB1 (5%), EBF1 (4%), ETV6 (4%), while PAR1 region genes were predominantly duplicated (20%). EBF1 deletions selectively associated with adults, IKZF1 deletions occurred frequently in high WCC and BCR-ABL1 cases, while PAR1 region gains significantly associated with MLL-AF4 cases. IKZF1 haploinsufficiency group was predominant, especially in adults (65%), high WCC (60%) patients and BCR-ABL1-negative (78%) patients. Most cases harbored multiple concurrent CNA, with IKZF1 concomitantly occurring with CDNK2A/B, PAX5 and BTG1, while JAK2 occurred with CDNK2A/B and PAX5. Mutually exclusive CNA included ETV6 and IKZF1/RB1, and EBF1 and JAK2. Our results corroborate with global reports, aggregating molecular markers in Indian HR B-ALL cases. Integration of CNA data from rapid methods like MLPA, onto background of existing gold-standard methods detecting significant chromosomal abnormalities, provides a comprehensive genetic profile in B-ALL.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
B-lineage acute lymphoblastic leukemia (B-ALL) is a genetically heterogeneous disease, characterized by various recurrent (chromosomal and submicroscopic) alterations. Many of these alterations serve as diagnostic and prognostic factors routinely used in risk stratification. Patients with hypodiploidy, MLL-AF4 and BCR-ABL1 stratify into poor-outcome high-risk (HR) groups, while presence of hyperdiploidy and ETV6-RUNX1 implies good prognosis risk groups [1]. Yet, either several ALL patients, including those with HR features (older age, males, high WBC count), and relapse cases remain genetically undefined or disease etiology remains poorly understood [2]. Subsequently, genomic studies in B-ALL identified recurrent copy number abnormalities (CNA) targeting key genes in B-lymphoid development pathway (IKZF1, ETV6, PAX5, EBF1), cell cycle control, tumor suppression (RB1, CDKN2A/2B, BTG1), and cytokine receptors localized to PAR1 region (CRLF2, IL3RA, CSF2RA, P2RY8, SHOX) [3,4,5].
Recent studies showed diagnostic or prognostic relevance of some of these CNA, suggesting their potential use in risk stratification [6,7,8,9]. Notably, deletions of CDKN2A/B, CRLF2 and IKZF1 were associated with high-risk disease and poor outcome [10,11,12]. Moreover, some CNA represented cooperating aberrations that correlate with specific cytogenetic subtypes [6, 12, 13]. Like, IKZF1 deletions are hallmarks of multiple subtypes showing poor prognosis, including BCR-ABL1 cases [7, 8, 14]. In fact, IKZF1 deletions serve as prognostic markers in pediatric B-ALL and as strong predictors of relapse at diagnosis, conferring a threefold increased relapse risk in BCR-ABL1 ALL patients [15].
Compared to Western patients, the overall outcome in Indian B-ALL patients is poor, and one contributing factor includes higher frequency of poor-risk and fewer good-risk genetic subtypes seen in Indian B-ALL patients [16,17,18]. Testing for multiple mutations including point mutations, insertions/ deletions and translocations has become a standard of care in leukemias and in turn sheds light on the disease pathogenesis [19]. Use of MLPA, as an adjunct to cytogenetic analysis, provides a rapid, accurate, high-throughput and low-cost approach for uncovering molecular profile of multiple clinically relevant CNA in genes recurrently affected in B-lineage ALL. Therefore, the current study evaluated the frequency and type of CNA in commonly affected genes, and their association with clinicopathological features within high-risk B-ALL patients from India.
Materials and methods
Study subjects
The present study was performed at the R&D Division, SRL Ltd., India. High-risk (HR) B-lineage ALL cases (n = 101) were subjected to copy number analysis which were selected from the same study cohort which was reported earlier from our group [16]. Inclusion criteria involved cytogenetic, FISH or molecular evidence of unfavorable aberrations like hypodiploidy (n = 50), BCR-ABL1 (n = 48) and MLL-AF4 (n = 7), similar to previous HR cohorts [1, 20]. Table 1 summarizes the clinical and demographic data of cohort. Median age was 16 years (1–74) with 55 pediatric (<18 years) and 46 adult (≥18 years) patients. Median WCC (WBC count) was 39 × 109/L (0.2–468) with 61 low WCC and 40 high WCC cases. MLPA controls comprised healthy subjects with no previous or concurrent malignancy [21]. Treatment and outcome were not evaluated. Informed consent was obtained according to the Declaration of Helsinki, and study was approved by the institutional ethics committee.
Copy number abnormalities (CNA) analysis
Genomic DNA was isolated from peripheral blood or bone marrow using QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and quantified using NanoDrop Spectrophotometer (Thermo Scientific) for the presence of at least 5 ng/ul DNA. DNA integrity was further checked on 0.8% Agarose gel. Gene CNA were analyzed using MLPA kit (P335) (MRC-Holland, Amsterdam, Netherlands) as per protocol. This kit includes probes for EBF1 (5q33.3, ex 1, 10, 14, 16), IKZF1 (7p12.2, ex 1-8), PAX5 (9p13.2, ex 10, 8-5, 2, 1), CDKN2A/B (9p21.3, ex 2a, 5, 2), JAK2 (9p24.1, ex 23), ETV6 (12p13.2, ex 1-3, 5, 8), BTG1/BTG1 downstream region (12q21.33, ex 1-2), RB1 (13q14.2, ex 6, 14, 19, 24, 26) and PAR1 region genes (SHOX area, CRLF2, ex 4, CSF2RA, ex 10, IL3RA, ex 1, P2RY8, ex 2) (Xp22.33/Yp11.32). Complete details are available online (www.mrc-holland.com). The FAM-labeled PCR fragments were resolved by capillary electrophoresis on ABI 3500Dx Genetic Analyzer (Applied Biosystems, Foster City, CA), and peak intensities were analyzed using Coffalyser software (MRC-Holland). Relative copy number was obtained after (intra- and inter-sample) normalizations of peaks against internal reference probes and normal control DNA, respectively. As per manufacturer protocol, copy number ratio of each gene was interpreted as follows: <0.3, as homozygous deletion; ≥0.3 to <0.7, as heterozygous deletion; ≥0.7 to ≤1.3, as normal; >1.3 to <1.7, as heterozygous gain and ≥1.7, as homozygous gain. IKZF1 deletions were classified by their functional effects into 3 groups: “dominant negative group,” including exons 4–7 deletion; “haploinsufficiency group,” including whole gene deletions and deletions affecting exon 2; and “miscellaneous group,” representing all other deletions, as previously reported [14].
Statistical analysis
Routine statistical analysis, z-test of proportions and Fischer’s test were done using SPSS v20 (SPSS Inc., Chicago, IL), and two-sided P value <0.05 was considered significant.
Results
Distribution of copy number abnormalities (CNA) in HR cohort
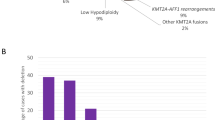
There was considerable heterogeneity seen in CNA distribution and number of CNA per case (Fig. 1). Overall, 59% (n = 59/101) cases showed CNA (i.e., deletion/gain in at least one gene). No CNA were observed in 41% (n = 42/101) cases, whereas 26% (n = 26), 10% (n = 10) and 23% (n = 23) of cases had 1, 2 or +3 CNA. Notably, CNA occurred at significantly higher frequencies overall (59%), and in BCR-ABL1 (60%, n = 29/48), pediatric (64%, n = 35/55) and high WCC (63%, n = 25/40) patients compared to those without CNA (all P < 0.05) (Fig. 1).

Distribution of copy number abnormalities (CNA) in HR cohort: a percentage of patients with and without CNA; b percentage of patients with increasing numbers of CNA
Gene CNA profile and correlation with clinicopathological features
As shown in Table 2, several gene copy number losses were more frequent than gains (P < 0.05). Frequent genes deletions included, CDNK2A/B (26%), IKZF1 (25%), PAX5 (14%), JAK2 (7%), BTG1 (6%), RB1 (5%), ETV6 (4%) and EBF1 (4%), while PAR1 region genes were chiefly duplicated (20 vs. 1%, P < 0.05) (Table 2).
Table 3 depicts correlation of gene CNA with clinicopathological features. EBF1 deletions exclusively associated with adults (9 vs. 0%, P = 0.02) while ETV6 and PAR1 deletions were absent in adults. Deletions in BTG1 (4 to 9%), RB1 (4 to 7%) and IKZF1 (20% to 30%) showed an increasing trend with age. No gender-related differences were seen. High WCC patients showed more deletions than low WCC cases, notably higher IKZF1 deletions (38 vs. 16%, P = 0.01). BCR-ABL1 cases showed more deletions than other subgroups, notably IKZF1 deletions (33 vs. 6%, P = 0.057). Interestingly, PAR1 region gains significantly associated with MLL-AF4 cases (71 vs. 16%, P = 0.0003), despite lower CNA levels seen in this group. Gains in other genes were too few for statistical analysis.
IKZF1 deletion subgroup profile
As seen in Table 4, IKZF1 deletions (n = 25 cases) were classified into three subgroups and correlated with clinical features. Among these, haploinsufficiency was most frequent (60%, n = 15, P < 0.001), involving deletions like Δ1–8 (n = 9) and Δ1–2, Δ2–3, Δ2–7, Δ2–8, Δ2–3, 5–8 and Δ1–3, 5–7 (n = 1 each). Dominant negative deletion Δ4–7 (28%, n = 7) and other miscellaneous deletions (12%, n = 3: Δ3–7, Δ4, 7 and Δ1, 4–7) occurred at lower frequencies.
Further, IKZF1 haploinsufficiency was significantly higher in BCR-ABL1-negative cases (78 vs. 22%), adult cases (65 vs. 35%), high WCC cases (60 vs. 40%), males (61 vs. 8%) and hypodiploid patients (64 vs. 9%) (all P < 0.03, Table 4).
Gene aberrations types, extent and combinations
As shown in Table 5, ample heterogeneity was seen in the type and extent of genetic change. All losses in EBF1, PAX5, ETV6 and JAK2 were heterozygous, while CDKN2A/B losses (46%) were mostly homozygous. Among IKZF1 deletions, 88% were heterozygous, 4% homozygous and rest 8% were mixed (P < 0.05). Likewise, BTG1 (67%) and RB1 (60%) deletions were chiefly heterozygous. All gains in JAK2, CDKN2A/B, ETV6 and BTG1 were heterozygous. Likewise, PAR1 (85%), EBF1 (75%) and PAX5 (71%) gains were chiefly heterozygous (Table 5).
Most cases harbored multiple concurrent CNA, and a total of 59 unique CNA profiles were observed (Table 5). Among these, profiles with single gene aberration (i.e., CNA=1, n = 26) involved: PAR1 (n = 9), IKZF1 (n = 7), CDNK2A/B (n = 5), RB1 (n = 2) and EBF1, PAX5, ETV6 (n = 1 each). Further, simultaneous aberrations in different genes were frequent (i.e., CNA>1, n = 33) and involved: IKZF1, CDKN2A/B, PAX5 (n = 4); IKZF1, CDKN2A/B, PAX5, BTG1 (n = 2); IKZF1, EBF1 (n = 2); IKZF1, PAR1 (n = 2); CDKN2A/B, JAK2, PAX5 (n = 2); CDKN2A/B, ETV6, BTG1 and PAR1 (n = 2), while other rare combinations were seen in single cases (n = 19). None of the cases with CNA in ETV6 showed concomitant CNA in IKZF1 or RB1, while cases with CNA in EBF1 lacked concomitant CNA in JAK2, indicating that these CNA were mutually exclusive.
Discussion
In the present study, several recurrent CNA were identified in genes related to leukemia and key cellular pathways by MLPA-based CNA analysis, consistent with previous studies [6, 12]. Even with a limited panel of tested genes, we detected significantly high levels of CNA (59–64%) in overall, pediatric, BCR-ABL1 and high WCC patients. These findings suggest aggressive disease biology and corroborate with 57–66% of CNA previously reported in pediatric, BCR-ABL1 and HR B-ALL patients [6, 11, 12, 14]. Our cohort presented fewer cases without CNA (41 vs. 65%) compared to the UKALLXII/ ECOG2993 trial [22] and more cases with ≥3 CNA (23 vs. 9–10%) than the ALL97/99 and UKALL2003 groups [6]. Further, a single gene CNA was noted in 26 (26%) cases, with the other 33 (33%) cases showing CNA in >1 gene, supporting the fact that B-ALL is a multistep process with several cooperating lesions.
Ample heterogeneity was observed in the frequency, extent and type of genetic change. Similar to previous CNA studies, losses were more frequent than gains in lymphoid development (IKZF1, PAX5, EBF1, ETV6), cell cycle control and tumor suppression genes (CDNK2A/B, BTG1, RB1), but not in cytokine receptor genes (PAR1 region) [13, 23]. Strikingly, many genes regulating B-lymphoid development are often affected in HR B-ALL and a greater number of lesions associate to poor outcome, suggesting that degree of “block” in this pathway contributes to both leukaemogenesis and treatment responsiveness [24].
In relation to individual abnormalities, CDKN2A/B deletions (26%) were most frequent. CDKN2A/B losses, reported earlier in 22–39% B-ALL cases [6, 25], are linked to poor prognosis in adult, BCR-ABL1-positive ALL, and rapid relapse in pediatric ALL [5, 10, 26]. CDKN2A/B code for tumor suppressors and cell cycle regulator proteins p16, p14 and p15, that inhibit cyclin-dependent kinases [26]. Similar to earlier reports, chiefly both A and B domains were deleted, probably leading to uncontrolled cell cycle progression [27]. Besides, cases with both mono- and biallelic deletions were noted, similar to prior reports [23, 28]. Further as noted earlier, CDKN2A/B deletions (9p21.3) co-occurred with abnormalities of 9p (like translocations and deletions) and loss of adjacent JAK2 (9p24.1) and PAX5 (9p13.2), suggesting they are common targets in B-ALL pathogenesis [10, 12, 29].
PAX5 showed 14% losses and fewer gains (7%), consistent with 10–25% PAX5 losses noted earlier in B-ALL [12, 13, 29]. Alternatively, HR-ALL studies using quantitative PCR or DNA chips report more PAX5 alterations (>30%), probably owing to differences between methods [11, 30,31,32]. PAX5 regulates B-lineage commitment by activating B-lineage-specific genes and repressing other-lineage genes; thus, its loss leads to B cell development blockage [30]. In pediatric ALL, CNA in PAX5 and IKZF1 alter expression levels and patterns [4, 11]. In adult B-ALL groups (GRAALL and GIMEMA), frequent PAX5 alterations bared no correlation with outcome [30, 32].
IKZF1 deletions (25%) varied largely in size and were frequent in our high WCC (38%) and BCR-ABL1 (33%) cases, suggesting a more aggressive disease. Earlier, 14–29% IKZF1 deletions were reported in pediatric, adult and HR B-ALL patients [6, 11, 12, 22, 33]. Further, prior correlations of IKZF1 losses with older age, high WCC, BCR-ABL1 and higher induction failure suggest resistance to standard therapy and need for intensive/alternate therapies [27, 33]. Globally, the independent and adverse impact of IKZF1 deletions in B-ALL is well documented [7, 9, 11, 14, 34]. They are preserved from diagnosis to relapse and maybe used to identify ALL-relapse and HR cases [31, 35]. IKZF1 deletions probably block B cell differentiation and activate stem cell gene expression program. Further, biallelic IKZF1 deletions, reported in 15–20% B-ALL, possibly provide survival advantage to leukemic cells [35, 36].
Classifying IKZF1 deletions by functional effects is vital in understanding their prognostic relevance. IKZF1 haploinsufficiency (60%) was more predominant than other groups, and significantly higher in our adults (65%), high WCC cases (60%) and BCR-ABL1-negative cases (78%). Likewise, IKZF1 haploinsufficiency was chiefly noted in 57–72% BCR-ABL1-negative, HR and adult B-ALL cases [11, 27, 36]. Haploinsufficiency deletions involving all exons or exon 2 result in reduced levels of ikaros protein, as loss of exon 2, harboring the translational start site, prevents IKZF1 translation, while exon 8 loss affects IKZF1 dimerization [36]. Dominant negative deletions involving exon 4–7 cause expression of dominant negative isoform Ik6, that lacks the N-terminal DNA binding zinc finger and shows oncogenic activity [35]. Recently, all IKZF1 deletion groups showed worse or equally poor prognosis in pediatric B-ALL [37].
The anti-proliferative gene, BTG1, was deleted in 6% cases and duplicated in 5% cases. Earlier, BTG1 deletions were noted in 6–10% B-ALL, often involving exon 2 and generating highly instable truncated BTG1 protein [11, 12, 38]. Further, BTG1 deletions are rare, but commonly occur with ETV6-RUNX1 and BCR-ABL1 cases [12, 38, 39], and show no correlation with B-ALL outcome [23].
The cell cycle regulator, RB1, only showed deletions (5%). Likewise, RB1 deletions were earlier reported in 6–11.3% B-ALL [11, 12], with a possible association with poor outcome noted in pediatric B-ALL [40].
EBF1, an essential transcription factor in B-lineage commitment, showed 4% heterozygous deletions only in adults, suggesting its lower incidence in pediatric cases. Likewise, IKZF1 and EBF1 deletions occur more frequently in NCI-HR cases that have higher age and WCC [12, 13]. Earlier, EBF1 deletions were noted in 2–7.7% B-ALL and associated with poor outcome [11, 12, 23]. Also, EBF1 deletions were more frequent in relapsed B-ALL than at diagnosis, indicating their possible role in disease recurrence [5].
The transcription factor ETV6, commonly involved in translocations of B-ALL and other leukemias, showed fewer gains (3%) and 4% heterozygous losses restricted to pediatric cases, suggesting their lower frequency in HR B-ALL cases. In agreement, ETV6 deletions, seen earlier in 8.2–22% of B-ALL, are frequent in good prognostic ETV6-RUNX1 and NCI-SR cases and show no impact on B-ALL outcome [6, 11,12,13].
Notably, gains in PAR1 region (at Xp22.33/Yp11.32) were significantly higher (20% overall and 71% in MLL-AF4 cases) than 7.4% PAR1 gains reported previously [27]. In contrast, prior HR-ALL studies mostly report recurrent PAR1 deletions or poor prognostic CRLF2 rearrangements, particularly with concomitant IKZF1 and JAK2 alterations [8, 23, 41]. PAR1 deletions (resulting in P2RY8-CRLF2 fusion) possibly cause CRLF2 over-expression, which activates JAK-STAT pathway and unchecked B cell proliferation [8, 23].
Most cases harbored multiple concurrent CNA, in addition to gross chromosomal alterations, as reported earlier [4, 27]. IKZF1 deletions frequently harbored CNA in CDNK2A/B, PAX5, BTG1, while JAK2 alterations co-occurred with CNA in CDNK2A/B and PAX5, suggesting these recurrent combinations as partially overlapping CNA in HR B-ALL. In contrast, CNA in ETV6 and IKZF1/RB1, and EBF1 and JAK2 were mutually exclusive.
As previously noted, the frequency and type of CNA strongly varied with B-ALL cytogenetic subgroups [12, 13]. Notably, BCR-ABL1 group harbored more distinct CNA, especially IKZF1, PAX5 and CDKN2A/B deletions, consistent with prior studies [4, 12, 31]. In contrast, MLL-AF4 cases had fewer CNA, corroborating with earlier reports [4, 22, 42]. This finding may be expected due to potency of this chromosomal abnormality to induce leukemia, requiring few cooperating genetic alterations [23, 24]. Among our hypodiploid cases with CNA (56%), 4% were near-haploid (NH, 24–29 chromosomes), 20% were low-hypodiploid (LH, 30–39 chromosomes) and 32% were medium-hypodiploid (MH, 40–45 chromosomes) cases. Agreeing with previous studies, reduced CNA found in hypodiploid group may likely be an artifact of analyzing CNA in context of ploidy changes, probably as neither the genes nor probes tested were located on the affected chromosomes [25]. Besides, prior reports suggest this group is genetically distinct, selectively harboring more RAS-activating and IKZF2- and IKZF3-inactivating mutations [43].
Summarily, HR B-ALL cases harbor multiple distinct CNA. Our results corroborate previous reports and emphasize screening of submicroscopic alterations as additional markers for risk stratification, especially in HR patients. Nonetheless, differences in frequency of abnormalities may be attributed to smaller size and shorter duration of our study compared to larger studies evaluating data over greater years.
References
Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. 2015;125:3977–87.
Loh ML, Mullighan CG. Advances in the genetics of high-risk childhood B-progenitor acute lymphoblastic leukemia and juvenile myelomonocytic leukemia: implications for therapy. Clin Cancer Res. 2012;18:2754–67.
Kuiper RP, Schoenmakers EFPM, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21:1258–66.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64.
Yang JJ, Bhojwani D, Yang W, Cai X, Stocco G, Crews K, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008;112:4178–83.
Moorman AV, Enshaei A, Schwab C, Wade R, Chilton L, Elliott A, et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124:1434–44.
Yamashita Y, Shimada A, Yamada T, Yamaji K, Hori T, Tsurusawa M, et al. IKZF1 and CRLF2 gene alterations correlate with poor prognosis in Japanese BCR-ABL1-negative high-risk B-cell precursor acute lymphoblastic leukemia. Pediatr Blood Cancer. 2013;60:1587–92.
Harvey RC, Mullighan CG, Chen I-M, Wharton W, Mikhail FM, Carroll AJ, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, hispanic/latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115:5312–21.
Dorge P, Meissner B, Zimmermann M, Moricke A, Schrauder A, Bouquin J-P, et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica. 2013;98:428–32.
Iacobucci I, Ferrari A, Lonetti A, Papayannidis C, Paoloni F, Trino S, et al. CDKN2A/B alterations impair prognosis in adult BCR-ABL1—positive acute lymphoblastic leukemia patients. Clin Cancer Res. 2011;17:7413–23.
Mullighan CG, Su X, Zhang J, Radtke I, Phillips LAA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–80.
Schwab CJ, Chilton L, Morrison H, Jones L, Al-Shehhi H, Erhorn A, et al. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica. 2013;98:1081–8.
Barbosa TC, Terra-Granado E, Quezado Magalhães IM, Neves GR, Gadelha A, Guedes Filho GE, et al. Frequency of copy number abnormalities in common genes associated with B-cell precursor acute lymphoblastic leukemia cytogenetic subtypes in Brazilian children. Cancer Genet. 2015;208:492–501.
van der Veer A, Zaliova M, Mottadelli F, De Lorenzo P, Te Kronnie G, Harrison CJ, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood. 2014;123:1691–8.
Waanders E, van der Velden VHJ, van der Schoot CE, van Leeuwen FN, van Reijmersdal SV, de Haas V, et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia. 2011;25:254–8.
Bhandari P, Ahmad F, Dalvi R, Koppaka N, Kokate P, Das BR, et al. Cytogenetic profile of de novo B lineage acute lymphoblastic leukemia: determination of frequency, distribution pattern and identification of rare and novel chromosomal aberrations in Indian patients. Asian Pac J Cancer Prev APJCP. 2015;16:7219–29.
Siraj AK, Kamat S, Gutiérrez MI, Banavali S, Timpson G, Sazawal S, et al. Frequencies of the major subgroups of precursor B-cell acute lymphoblastic leukemia in Indian children differ from the West. Leukemia. 2003;17:1192–3.
Sazawal S, Bhatia K, Gutierrez MI, Saxena R, Arya LS, Bhargava M. Paucity of TEL-AML 1 translocation, by multiplex RT-PCR, in B-lineage acute lymphoblastic leukemia (ALL) in Indian patients. Am J Hematol. 2004;76:80–2.
Konialis C, Savola S, Karapanou S, Markaki A, Karabela M, Polychronopoulou S, et al. Routine application of a novel MLPA-based first-line screening test uncovers clinically relevant copy number aberrations in haematological malignancies undetectable by conventional cytogenetics. Hematology. 2014;19:217–24.
Bhojwani D, Howard SC, Pui C-H. High-risk childhood acute lymphoblastic leukemia. Clin Lymphoma Myeloma. 2009;9:S222.
Bhandari P, Ahmad F, Mandava S, Das BR. Association of genetic variants in ARID5B, IKZF1 and CEBPE with risk of childhood de novo B-lineage acute lymphoblastic leukemia in India. Asian Pac J Cancer Prev APJCP. 2016;17:3989–95.
Marks DI, Moorman AV, Chilton L, Paietta E, Enshaie A, DeWald G, et al. The clinical characteristics, therapy and outcome of 85 adults with acute lymphoblastic leukemia and t(4;11)(q21;q23)/MLL-AFF1 prospectively treated in the UKALLXII/ECOG2993 trial. Haematologica. 2013;98:945–52.
Moorman AV, Schwab C, Ensor HM, Russell LJ, Morrison H, Jones L, et al. IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J Clin Oncol. 2012;30:3100–8.
Collins-Underwood JR, Mullighan CG. Genomic profiling of high-risk acute lymphoblastic leukemia. Leukemia. 2010;24:1676–85.
Schwab CJ, Jones LR, Morrison H, Ryan SL, Yigittop H, Schouten JP, et al. Evaluation of multiplex ligation-dependent probe amplification as a method for the detection of copy number abnormalities in B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2010;49:1104–13.
Kim M, Yim S-H, Cho N-S, Kang S-H, Ko D-H, Oh B, et al. Homozygous deletion of CDKN2A (p16, p14) and CDKN2B (p15) genes is a poor prognostic factor in adult but not in childhood B-lineage acute lymphoblastic leukemia: a comparative deletion and hypermethylation study. Cancer Genet Cytogenet. 2009;195:59–65.
Gupta SK, Bakhshi S, Kumar L, Kamal VK, Kumar R. Gene copy number alteration profile and its clinical correlation in B-cell acute lymphoblastic leukemia. Leuk Lymphoma. 2017;58:333–42.
Rand V, Parker H, Russell LJ, Schwab C, Ensor H, Irving J, et al. Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2011;117:6848–55.
Kim M, Choi JE, She CJ, Hwang SM, Shin HY, Ahn HS, et al. PAX5 deletion is common and concurrently occurs with CDKN2A deletion in B-lineage acute lymphoblastic leukemia. Blood Cells Mol Dis. 2011;47:62–6.
Familiades J, Bousquet M, Lafage-Pochitaloff M, Béné M-C, Beldjord K, De Vos J, et al. PAX5 mutations occur frequently in adult B-cell progenitor acute lymphoblastic leukemia and PAX5 haploinsufficiency is associated with BCR-ABL1 and TCF3-PBX1 fusion genes: a GRAALL study. Leukemia. 2009;23:1989–98.
Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–4.
Iacobucci I, Lonetti A, Paoloni F, Papayannidis C, Ferrari A, Storlazzi CT, et al. The PAX5 gene is frequently rearranged in BCR-ABL1-positive acute lymphoblastic leukemia but is not associated with outcome. A report on behalf of the GIMEMA Acute Leukemia Working Party. Haematologica. 2010;95:1683–90.
Safavi S, Hansson M, Karlsson K, Biloglav A, Johansson B, Paulsson K. Novel gene targets detected by genomic profiling in a consecutive series of 126 adults with acute lymphoblastic leukemia. Haematologica. 2015;100:55–61.
Asai D, Imamura T, Suenobu S, Saito A, Hasegawa D, Deguchi T, et al. IKZF1 deletion is associated with a poor outcome in pediatric B-cell precursor acute lymphoblastic leukemia in Japan. Cancer Med. 2013;2:412–9.
Iacobucci I, Storlazzi CT, Cilloni D, Lonetti A, Ottaviani E, Soverini S, et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell’Adulto Acute Leukemia Working Party (GIMEMA AL WP). Blood. 2009;114:2159–67.
Dupuis A, Gaub MP, Legrain M, Drenou B, Mauvieux L, Lutz P, et al. Biclonal and biallelic deletions occur in 20% of B-ALL cases with IKZF1 mutations. Leukemia. 2013;27:503–7.
Boer JM, van der Veer A, Rizopoulos D, Fiocco M, Sonneveld E, de Groot-Kruseman HA, et al. Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia. 2016;30:32–8.
Waanders E, Scheijen B, van der Meer LT, van Reijmersdal SV, van Emst L, Kroeze Y, et al. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet. 2012;8:e1002533.
Xie J, Wang Q, Wang Q, Yao H, Wen L, Ma L, et al. High frequency of BTG1 deletions in patients with BCR-ABL1—positive acute leukemia. Cancer Genet. 2014;207:226–30.
Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11:429–38.
Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–66.
Bardini M, Spinelli R, Bungaro S, Mangano E, Corral L, Cifola I, et al. DNA copy-number abnormalities do not occur in infant ALL with t(4;11)/MLL-AF4. Leukemia. 2010;24:169–76.
Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–52.
Acknowledgements
The authors are grateful to the management of SRL Ltd. for providing the necessary infrastructure facilities. No other research support is associated with this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The study was approved by the SRL-Ethics Committee and was in accordance with the Declaration of Helsinki. This article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all subjects as well as parents/guardians of minor subjects included in the study.
Rights and permissions
About this article

Cite this article
Bhandari, P., Ahmad, F. & Das, B.R. Molecular profiling of gene copy number abnormalities in key regulatory genes in high-risk B-lineage acute lymphoblastic leukemia: frequency and their association with clinicopathological findings in Indian patients. Med Oncol 34, 92 (2017). https://doi.org/10.1007/s12032-017-0940-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-017-0940-3