
Abstract
Although the causes of prostate cancer (PCa) and benign prostatic hyperplasia (BPH) are not known, the role of oxidative stress, aging, and diet are suspected to increase the incidence of prostate complications. The cholesterol oxidation derivative (oxysterol) 27-hydroxycholesterol (27-OHC) is the most prevalent cholesterol metabolite in the blood. As aging, oxidative stress, and hypercholesterolemia are associated with increased risk of PCa and BPH, and because 27-OHC levels are also increased with aging, hypercholesterolemia, and oxidative stress, determining the role of 27-OHC in the progression of PCas and BPH is warranted. In this study, we determined the effect of 27-OHC in human prostate epithelial cells RWPE-1. We found that 27-OHC stimulates proliferation and increases androgen receptor (AR) transcriptional activity. 27-OHC also increased prostate-specific antigen expression and enhanced AR binding to the androgen response element compared to controls. Silencing AR expression with siRNA markedly reduced the 27-OHC-induced proliferation. Furthermore, 27-OHC blocked docetaxel-induced apoptosis. Altogether, our results suggest that 27-OHC may play an important role in PCa and BPH progression by promoting proliferation and suppressing apoptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prostate health is an area of growing concern. Over 14 % of men will develop prostate cancer (PCa), and greater than 70 % of men will develop benign prostatic hyperplasia (BPH) by the age of 70 [1, 2]. The etiologies of PCa and BPH are unknown, but environmental factors including diet are suggested to play a role in the progression of these pathologies. Aging, genetic susceptibility, obesity, low physical activity, androgens, and inflammatory conditions are also all associated with both PCa and BPH [3, 4].
Cholesterol involvement in PCa has been suspected for over a century when it was found to accumulate in PCa cells [5]. More recent studies also demonstrated a positive correlation between high plasma cholesterol and high-grade PCa [6]. Moreover, some studies reported a reduction in the risk of high-grade PCa [7, 8] and BPH [9] with the cholesterol-lowering statins. However, the efficacy of lowering cholesterol levels currently not proven and whether increased cholesterol levels are associated or causal factors of PCa and BPH is still unknown. Several lines of evidence suggest that cholesterol oxidation products, not cholesterol per se, may increase the risk of PCa and BPH risk. 27-Hydroxycholesterol (27-OHC) is the main oxysterol in the circulation, and its levels can be increased by aging, oxidative stress, and hypercholesterolemia [10, 11], factors that are all suspected to increase the risk of PCa and BPH. There are reasons to believe that 27-OHC is deleterious as this oxysterol has been shown to act as a selective estrogen receptor modulator (SERM) [12, 13] and a liver X receptor (LXR) ligand [14]. Both estrogen receptors (ER) and LXR are involved in steroid signaling pathways and influence inflammation, cell proliferation, and many other metabolic processes. However, the extent to which 27-OHC also regulates androgen receptors (AR) which play a central role in the pathogenesis of BPH and PCa is yet to be determined. In this study, we determined the effects of 27-OHC on cell proliferation and the role of AR in 27-OHC-induced cell proliferation.
We found that 27-OHC increases cell proliferation and AR transcriptional activity. We also demonstrate that 27-OHC enhances AR binding to the prostate-specific antigen (PSA) promoter and increases PSA expression. To investigate whether AR is required in 27-OHC-induced cell proliferation, we silenced AR gene expression using siRNA and found that 27-OHC-induced cell proliferation is AR-dependent. Additionally, to determine whether 27-OHC contributes to apoptosis resistance, we treated cells with 27-OHC and docetaxel and found that 27-OHC inhibits the pro-apoptotic effects of docetaxel by visualizing nuclear fragmentation using TUNEL assay.
Methods
Reagents
27-OHC was purchased from Santa Cruz Biotechnologies (Dallas, TX), docetaxel and fulvestrant from Cayman Chemicals (Ann Arbor, MI), β-estradiol from Sigma-Aldrich (St. Louis, MO), and the reporter constructs encoding androgen receptor response elements conjugated to the firefly luciferase gene from SA Biosciences (Valencia, CA). All cell culture reagents, with the exception of fetal bovine serum (FBS; Atlanta Biologicals; Flowery Branch, GA), were from Invitrogen. Human RWPE-1 cells were purchased from ATCC (Manassas, VA). Concentrations of solvent in treatments were less than 0.1 %.
Cell culture
Non-tumorigenic human prostate epithelial RWPE-1 cells were maintained in keratinocyte serum-free medium (Invitrogen; Carlsbad, CA) supplemented with 0.05 mg/ml BPE and 5 ng/ml EGF. Cells were supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin (Sigma; St. Louis, MO) and cultured at 5 % CO2 and 37 °C. Cells were treated with vehicle (ethanol in media), 0.1 or 1 μM of 27-OHC. Stock solutions of 27-OHC were prepared in 100 % ethanol and stored at −80 °C. 27-OHC stock solution was dissolved in appropriate volumes of media to prepare the working solutions of 0.1 or 1 μM. Our study was approved by the Institutional Biosafety Committee of the School of Medicine at the University of North Dakota.
Cell proliferation assays
To determine the effects of 27-OHC on cell proliferation, we treated cells with 27-OHC at concentrations that have been shown to promote breast cancer progression [10, 15, 16]. Proliferation assays were conducted on black 96-well plates using CyQUANT Direct Cell Proliferation Assay (Invitrogen; Carlsbad, CA) which quantifies cell number using DNA content and membrane integrity. Cells seeded at 5 × 103 cells/well were treated with vehicle 0.1 or 1 μM of 27-OHC and incubated for 48 h. Cells were then stained as per the manufacturer’s protocol and read using Spectra MAX GEMINI EM (Molecular Devices; Sunnyvale, CA).
Metabolic activity assay
Cell metabolic activity was quantified by measurement of the reduction in MTS to formazan product using CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega; Madison, WI) according to the manufacturer’s protocol. The assay of the formation of formazan was performed by measuring absorbance change using a microplate reader (Spectromax plus; Molecular Devices; Sunnyvale, CA) 48 h after treatments.
Dual luciferase assays
Dual luciferase assays were conducted on white 96-well plates using the Dual Luciferase Reporter Assay System (Promega; Madison, WI). Cells were incubated for 18 h with transfection-ready AR response element conjugated with firefly luciferase construct and constitutively expressing Renilla luciferase construct using Lipofectamine 2000 (Invitrogen; Carlsbad, CA) as per the manufacturer’s protocol. Following transfection, cells were treated with 0 (vehicle), 0.1 or 1 μM 27-OHC for 24 h. After 24 h of treatment, cells were lysed and measured in relative luciferase units (RLU) as per the manufacturer’s protocol. Firefly luciferase readings were normalized against constitutive Renilla luciferase readings.
Chromatin immunoprecipitation (ChIP) analysis
ChIP analysis was performed to evaluate the extent of AR binding to the DNA elements in the ARE regions, respectively, using SimpleChIP™ Enzymatic Chromatic IP kit (Cell Signaling; Beverly, MA). Briefly, cells from each treatment group (1 × 107 cells) were washed with PBS, trypsinized, and centrifuged at 5000g. The pellet containing the cells was further washed with PBS and cross-linked using 37 % formaldehyde for 15 min followed by the addition of glycine solution to cease the cross-linking reaction. The cells were washed with 4× volumes of 1× PBS and centrifuged at ~300g for 5 min. The pellet was re-suspended and incubated for 10 min in 5 ml of cell lysis buffer containing DTT and protease and phosphatase inhibitors. The cross-linked chromatin from each sample was apportioned into three equal parts. One-third of the cross-linked chromatin was set aside as “input.” One-third of the cross-linked chromatin from each sample was incubated with 5 µg of AR rabbit antibody (Active Motif), while the remaining one-third of the cross-linked chromatin from each sample was incubated with 5 µg of normal rabbit IgG to serve as negative control. The cross-linked chromatin samples were incubated overnight at 4 °C with their respective antibodies. The DNA–protein complexes were collected using Protein G agarose beads. The samples were incubated with 2 µl of proteinase K for 2 h at 65 °C. The crude DNA extract was eluted and then washed several times with wash buffer containing ethanol followed by purification with the DNA spin columns. The pure DNA was eluted out of the DNA spin columns using DNA elution buffer. The relative abundance of the AR antibody-precipitated chromatin containing the AR binding site in the ARE region was determined by qPCR using an SYBR Green Mastermix kit following the manufacturer’s instructions (Invitrogen; Carlsbad, CA). ARE sequence on the PSA promoter F: 5′-TCTGCCTTTGTCCCCTAGAT-3′ and R: 5′-AACCTTCATTCCCCAGGACT-3′ [17]. The amplification was performed using Step ONE plus PCR Detection System (Invitrogen; Carlsbad, CA). The fold enrichment was calculated using the ΔΔC t method which normalizes ChIP C t values of each sample to the % input and background.
Western blot analysis
Treated cells were washed with PBS, trypsinized, and centrifuged at 5000g. The pellet was washed with PBS and homogenized in M-PER tissue protein extraction reagent (Thermo Scientific; Waltham, MA) supplemented with protease and phosphatase inhibitors. Denatured proteins (5 µg) were separated in 10 or 12.5 % SDS-PAGE gels, transferred to a PVDF membrane (Millipore), and incubated with antibodies to PSA (1:1000, Santa Cruz; Dallas, TX), or AR (1:1000, Santa Cruz; Dallas, TX). β-actin was used as a gel-loading control. The blots were developed with enhanced chemiluminescence (ECL Clarity kit, Bio-Rad). Bands were visualized on a polyvinylidene difluoride membrane and analyzed by LabWorks 4.5 software on a UVP Bioimaging System. Quantification of results was performed by densitometry and the results analyzed as total integrated densitometric values (arbitrary units).
Small interfering RNA
The cells were transfected with AR siRNA using Lipofectamine 2000 (Invitrogen; Carlsbad, CA) and incubated for 24 h, followed by their respective treatments. siRNA to AR sense and antisense strands were: 5′-CGGGAAGUUUAGAGAGCUATT-3′, 5′UAGCUCUCUAAACUUCCCGTG-3′ (Hs_AR_5, SA Biosciences; Valencia, CA).
TUNEL assay
The TUNEL assay was performed using DeadEnd Fluorometric TUNEL assay (Promega; Valencia, CA) for detection of apoptosis. The TUNEL staining was performed according to manufacturer’s instructions. Cells were permeabilized with Triton-X, washed with PBS, and incubated with terminal deoxynucleotidyl transferase, fluorescein-12-dUTP. The fluorescein-12-dUTP-labeled DNA was then visualized directly by fluorescence microscopy. DAPI was used as counter-stain for staining the nucleus. Slides were visualized using DMI 6000 (Leica Microsystems; Buffalo Grove, IL).
Statistical analysis
The significance of differences was assessed by unpaired t test and one-way analysis of variance (one-way ANOVA) followed by Tukey’s post hoc test. Statistical analysis was performed with GraphPad Prism software 4.01. Quantitative data for experimental analysis are presented as mean values ± SEM with unit value assigned to control and the magnitude of differences among the samples being expressed relative to the unit value of control.
Results
The oxysterol 27-OHC increases cell proliferation
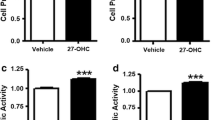
We found that 27-OHC increases proliferation by ~60 % at 0.1 µM and ~40 % at 1.0 µM (Fig. 1a). To confirm our results, we performed MTS assay to assess the mitochondrial activity of cells. We found that 27-OHC (0.1 and 1 µM) also increased the metabolic activity of the cells (Fig. 1b). These data suggest that 27-OHC induces cell proliferation in prostate epithelial cells.

27-OHC induces proliferation in prostate cells. Cell proliferation assay in RWPE-1 (a) cells demonstrates a significant increase in proliferation in the presence of 27-OHC (0.1 and 1 μM). MTS assay shows a significant increase in cell metabolic activity in the presence of 27-OHC in RWPE-1 (b). Readings were recorded 48 h after treatment with 27-OHC. Data are expressed as mean ± SEM ***p < 0.001 and **p < 0.01 versus controls, ## p < 0.01 and ### p < 0.001 versus 0.1 μM 27-OHC treatment
The oxysterol 27-OHC increases AR transcriptional activity
We treated cells with 27-OHC and measured the transcriptional activity of AR via ARE-tagged luciferase reporter. We found that AR transcriptional activity was significantly increased after 27-OHC treatment (Fig. 2). This suggests that 27-OHC regulates AR transcriptional activity.

27-OHC increases AR transcriptional activity; luciferase reporter assay shows an increase in AR transcriptional activity in the presence of 27-OHC (0.1 and 1 μM) in RWPE-1 cells. Readings were recorded 24 h after treatment. Data are expressed as mean ± SEM **p < 0.01 versus controls
The oxysterol 27-OHC increases PSA expression and AR binding to the PSA promoter
Using western blot analyses, we showed elevated levels of PSA in 27-OHC-treated cells. PSA levels significantly increased with 0.1 and 1.0 µM of 27-OHC (Fig. 3a, b). To determine whether the elevated levels of PSA were due to the increase in AR transcriptional activity, we performed a ChIP assay to analyze AR binding to the androgen receptor element (ARE) on the PSA promoter. We found an increase in AR binding to the ARE of the PSA promoter by ~17-fold when treated with 0.1 µM of 27-OHC and ~11-fold when treated with 1 µM of 27-OHC (Fig. 3c). These data suggest that 27-OHC enhances AR-mediated PSA expression.

27-OHC increases PSA protein levels via AR representative western blot (a) and densitometric analysis (b) showing a significant increase in PSA protein levels in RWPE-1 cells treated with 27-OHC (0.1 and 1 μM) for 48 h. ChIP analysis of AR binding to the ARE on the PSA promoter shows an increase in AR binding in the presence of 27-OHC that is higher with 0.1 than 1.0 μM concentrations (c). Data are expressed as mean ± SEM ***p < 0.001 and **p < 0.01 versus controls, ## p < 0.01, and # p < 0.05 versus 1 µM 27-OHC treatment
The oxysterol 27-OHC-induced cell proliferation is dependent on AR
We found that siRNA to AR reduced the protein levels of AR in untreated cells (Fig. 4a) and diminished cell proliferation in 27-OHC-treated cells (Fig. 4b). This result suggests that 27-OHC-induced cell proliferation involves AR. As 27-OHC does not directly bind to AR (data not shown) and is an ER modulator [12, 16, 18], it was important to determine the role of ER in prostate cell proliferation. We found that fulvestrant, an ER inhibitor, when concomitantly treated with 27-OHC, attenuated 27-OHC-induced cell proliferation (Fig. 4c). This result suggests that ER may function in concert with AR to induce 27-OHC-induced cell proliferation.

27-OHC-induced cell proliferation is AR-dependent; western blot analysis a shows the efficiency of siRNA to AR with protein levels markedly lower in RWPE-1 cells treated with siRNA to AR than with scrambled siRNA. Cell proliferation assay b demonstrates that 27-OHC (0.1 and 1.0 µM) increases cell proliferation, and co-treatment with siRNA to AR markedly reduces proliferation. The assay was performed following a 24-h incubation of RWPE-1 cells with the siRNAs on a 96-well plate with their respective treatments. Cell proliferation assay c demonstrates that 27-OHC-induced cell proliferation is attenuated when treated with 10 µM fulvestrant. Readings were recorded 48 h after the treatments. Data are expressed as mean ± SEM ***p < 0.001 versus vehicle only, ## p < 0.01 versus 0.1 µM 27-OHC only, ### p < 0.001 versus 0.1 or 1 µM 27-OHC only treatment
27-OHC suppressed docetaxel-induced apoptosis
We found that docetaxel induced apoptosis and 27-OHC (0.1 or 1.0 µM) markedly diminished docetaxel-induced apoptosis (Fig. 5). These data suggest that 27-OHC inhibits docetaxel-induced apoptosis.

27-OHC opposes docetaxel-induced apoptosis. Confocal images panels (a) and quantitation (b) for TUNEL assay in RWPE-1 cells treated with 27-OHC and/or docetaxel for 24 h. Docetaxel (0.1 µM) induced a marked increase in the apoptotic cells (green) and treatment with 27-OHC at both 0.1 and 1.0 µM diminished apoptosis induced by docetaxel. Data are expressed as mean ± SEM ***p < 0.001, **p < 0.01 and *p < 0.05 versus controls, ### p < 0.001 versus docetaxel only treatment. Bar 100 µM
Discussion
This study was designed to determine the effects of 27-OHC on prostate cell proliferation and apoptosis. We also determined the involvement of AR in cell proliferation induced by 27-OHC. We demonstrate that 27-OHC increases proliferation and activates AR transcriptional activity in the normal prostate cells RWPE-1. We further demonstrate that 27-OHC-induced cell proliferation is AR-dependent. Additionally, we show that 27-OHC opposes apoptotic cell death induced by docetaxel. Our data suggest that 27-OHC increases proliferation in normal epithelial prostate cells in an AR-dependent manner.
27-OHC, the most abundant cholesterol metabolite in the circulatory system, is synthesized by the cytochrome P-450 enzyme, sterol 27 hydroxylase (CYP27A1), in the inner mitochondrial membrane of the liver and is metabolized by CYP7B1 enzyme [14, 19]. The main source of 27-OHC levels in the human body emanates from oxidation of cholesterol. As cholesterol levels increase following intake of diets rich in cholesterol or overproduction of endogenous cholesterol, production of 27-OHC increases. Patients with hypercholesterolemia were reported to have high levels of 27-OHC in their blood [10, 11, 18]. Additionally, 27-OHC levels in the plasma have been shown to increase with age in males but not in females [20]. Also, males are known to have higher basal levels of 27-OHC in plasma than females [20, 21]. 27-OHC is also known to activate or inhibit nuclear receptors depending on the target tissue. For instance, 27-OHC inhibits ER signaling in the vasculature [22] and bone [23]; however, it activates ER signaling in breast [10, 15, 16].
To the best of our knowledge, there has been no established link between 27-OHC, PCa, and BPH. In this study, we report an increase in cell proliferation in prostate cells in the presence of 27-OHC at concentrations below (0.1 µM) or above (1.0 µM) the physiological concentrations [24, 25]. Total 27-OHC (esterified + non-esterified) is found in the human blood plasma ranging from 0.2 to 0.6 µM in a healthy individual [11, 26]. Also, it has been shown that the levels of non-esterified 27-OHC, which is the most biologically active form of 27-OHC [27, 28], is reported to be less than 20 % of total 27-OHC in the human body [20, 29, 30]. In disease conditions, such as cancer and neurodegenerative diseases, macrophages migrate to affected areas releasing excess 27-OHC in the surrounding tissue [10, 30, 31]. We also found that fulvestrant, an ER inhibitor, attenuated 27-OHC-induced cell proliferation in both concentrations to statistically same levels. Overall, our data suggest that 27-OHC may play an important role in prostate cell proliferation, which may result in the progression of BPH and/or PCa.
Androgen receptor (AR) plays an important role in prostate growth and development. There is a positive relationship between AR transcriptional activity and PCa progression [32, 33]. Over 80 % of PCa patients respond to anti-androgens or androgen deprivation therapy that targets and inhibits the AR activity [32, 34]. Additionally, androgens are also involved in BPH pathogenesis via AR [35, 36]. However, the effects of cholesterol metabolites including the oxysterol 27-OHC on the AR signaling pathway remain to be determined.
The AR signaling pathway plays a critical role in the development and progression of PCa [32, 33, 36]. AR signaling is chiefly targeted in the context of PCa using androgen ablation therapies [32, 33]. As we found that AR transcriptional activity was increased with 27-OHC treatment, we determined the expression levels of PSA, a well-known downstream target of AR, in these cells [37]. PSA is a serine protease which is produced by prostate epithelial cells and PCa cells. It is used as a serum biomarker to monitor PCa progression in patients [38]. Subsequently, we found that levels of PSA were increased. Also, using ChIP assay, we demonstrate that the binding of AR to the PSA promoter in the presence of 27-OHC is increased. We also show that 27-OHC-induced proliferation is AR-dependent. When AR gene expression was silenced, 27-OHC was unable to induce an increase in proliferation. As there was no additive effect in proliferation when cells were treated simultaneously with 27-OHC and E2 and because the ER selective inhibitor (Fulvestrant) prevented the 27-OHC-induced cell proliferation in RWPE-1 cells, we suggest the existence of an ER–AR crosstalk in the presence of 27-OHC since we show that the AR knockdown also reduces 27-OHC-induced proliferation.
Since 27-OHC induces AR transcriptional activity and 27-OHC levels increase with age [20], 27-OHC may play a role in castrate-resistant prostate cancer. However, further studies are needed to evaluate the role of 27-OHC in castrate-resistant PCa.
Apoptosis is an important process that keeps the number of cells dividing under tight control [39]. To assess the role of 27-OHC in regulating apoptosis, we utilized a pro-apoptotic drug, docetaxel [40], a current chemotherapeutic drug of choice for advanced PCa [41, 42]. To further understand whether 27-OHC plays a potential role in chemotherapeutic resistance, we treated cells with docetaxel and 27-OHC. We found that 27-OHC attenuated the pro-apoptotic effects of docetaxel. Docetaxel is clinically used to battle metastatic PCa [43] and is also known experimentally to have pro-apoptotic properties in non-tumorigenic RWPE-1 cells [44]. To better understand the role of 27-OHC in PCa, further in vivo studies are warranted. These results warrant further investigation to understand the anti-apoptotic role of 27-OHC in chemotherapeutic resistance to PCa and BPH.
In summary, we demonstrate that 27-OHC induces an increase in proliferation in normal prostatic epithelial cells in an AR-dependent manner. We also report for the first time the docetaxel-resistant role of 27-OHC in epithelial cells. Our study provides a novel insight into the molecular mechanisms of 27-OHC and its role in modulating AR signaling pathway that is tightly linked to cell proliferation which is associated with PCa and BPH. Further studies are warranted to delineate the 27-OHC activated AR signaling pathway which may lead to unraveling novel therapeutic avenues for PCa and BPH.
References
Guess HA, Arrighi HM, Metter EJ, Fozard JL. Cumulative prevalence of prostatism matches the autopsy prevalence of benign prostatic hyperplasia. Prostate. 1990;17:241–6.
Delongchamps NB, Singh A, Haas GP. The role of prevalence in the diagnosis of prostate cancer. Cancer Control. 2006;13:158–68.
Chokkalingam AP, Nyrén O, Johansson J-E, Gridley G, McLaughlin JK, Adami H-O, et al. Prostate carcinoma risk subsequent to diagnosis of benign prostatic hyperplasia: a population-based cohort study in Sweden. Cancer. 2003;98:1727–34.
Ørsted DD, Bojesen SE. The link between benign prostatic hyperplasia and prostate cancer. Nat Rev Urol. 2013;10:49–54.
White CP. On the occurrence of crystals in tumours. J Pathol Bacteriol. 1909;13:3–10.
Mondul AM, Clipp SL, Helzlsouer KJ, Platz EA. Association between plasma total cholesterol concentration and incident prostate cancer in the CLUE II cohort. Cancer Causes Control. 2010;21:61–8.
Breau RH, Karnes RJ, Jacobson DJ, McGree ME, Jacobsen SJ, Nehra A, et al. The association between statin use and the diagnosis of prostate cancer in a population based cohort. J Urol. 2010;184:494–9.
Papadopoulos G, Delakas D, Nakopoulou L, Kassimatis T. Statins and prostate cancer: molecular and clinical aspects. Eur J Cancer. 2011;47:819–30.
Lee SH, Park TJ, Bae MH, Choi SH, Cho YS, Joo KJ, et al. Impact of treatment with statins on prostate-specific antigen and prostate volume in patients with benign prostatic hyperplasia. Korean J Urol. 2013;54:750–5.
Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342:1094–8.
Hirayama T, Mizokami Y, Honda A, Homma Y, Ikegami T, Saito Y, et al. Serum concentration of 27-hydroxycholesterol predicts the effects of high-cholesterol diet on plasma LDL cholesterol level. Hepatol Res. 2009;39:149–56.
Umetani M, Shaul PW. 27-Hydroxycholesterol: the first identified endogenous SERM. Trends Endocrinol Metab. 2011;22:130–5.
DuSell CD, Umetani M, Shaul PW, Mangelsdorf DJ, McDonnell DP. 27-Hydroxycholesterol is an endogenous selective estrogen receptor modulator. Mol Endocrinol. 2008;22:65–77.
Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, et al. 27-Hydroxycholesterol is an endogenous ligand for liver X receptor in cholesterol-loaded cells. J Biol Chem. 2001;276:38378–87.
Cruz P, Torres C, Ramírez ME, Epuñán MJ, Valladares LE, Sierralta WD. Proliferation of human mammary cancer cells exposed to 27-hydroxycholesterol. Exp Ther Med. 2010;1:531–6.
Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS, et al. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Rep. 2013;5:637–45.
Horie-Inoue K, Bono H, Okazaki Y, Inoue S. Identification and functional analysis of consensus androgen response elements in human prostate cancer cells. Biochem Biophys Res Commun. 2004;325:1312–7.
Nelson ER, DuSell CD, Wang X, Howe MK, Evans G, Michalek RD, et al. The oxysterol, 27-hydroxycholesterol, links cholesterol metabolism to bone homeostasis through its actions on the estrogen and liver X receptors. Endocrinology. 2011;152:4691–705.
Ma D, Liu W, Wang Y. ApoA-I or ABCA1 expression suppresses fatty acid synthesis by reducing 27-hydroxycholesterol levels. Biochimie. 2014;103:101–8.
Burkard I, von Eckardstein A, Waeber G, Vollenweider P, Rentsch KM. Lipoprotein distribution and biological variation of 24S- and 27-hydroxycholesterol in healthy volunteers. Atherosclerosis. 2007;194:71–8.
Dzeletovic S, Breuer O, Lund E, Diczfalusy U. Determination of cholesterol oxidation products in human plasma by isotope dilution-mass spectrometry. Anal Biochem. 1995;225:73–80.
Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, et al. 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat Med. 2007;13:1185–92.
DuSell CD, Nelson ER, Wang X, Abdo J, Mödder UI, Umetani M, et al. The endogenous selective estrogen receptor modulator 27-hydroxycholesterol is a negative regulator of bone homeostasis. Endocrinology. 2010;151:3675–85.
Marwarha G, Ghribi O. Does the oxysterol 27-hydroxycholesterol underlie Alzheimer’s disease-Parkinson’s disease overlap? Exp Gerontol. 2015;68:13–8.
Brown AJ, Jessup W. Oxysterols and atherosclerosis. Atherosclerosis. 1999;142:1–28.
Schüle R, Siddique T, Deng H-X, Yang Y, Donkervoort S, Hansson M, et al. Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J Lipid Res. 2010;51:819–23.
Meaney S, Lütjohann D, Diczfalusy U, Björkhem I. Formation of oxysterols from different pools of cholesterol as studied by stable isotope technique: cerebral origin of most circulating 24S-hydroxycholesterol in rats, but not in mice. Biochim Biophys Acta. 2000;1486:293–8.
Meaney S, Hassan M, Sakinis A, Lütjohann D, von Bergmann K, Wennmalm A, et al. Evidence that the major oxysterols in human circulation originate from distinct pools of cholesterol: a stable isotope study. J Lipid Res. 2001;42:70–8.
Ramirez DMO, Andersson S, Russell DW. Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J Comp Neurol. 2008;507:1676–93.
Bandaru VVR, Haughey NJ. Quantitative detection of free 24S-hydroxycholesterol, and 27-hydroxycholesterol from human serum. BMC Neurosci. 2014;15:137.
Cruz P, Epuñán MJ, Ramírez ME, Torres CG, Valladares LE, Sierralta WD. 27-hydroxycholesterol and the expression of three estrogen-sensitive proteins in MCF7 cells. Oncol Rep. 2012;28:992–8.
Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308.
Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinog. 2011;10:20.
Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10:981–91.
Lu T, Lin W-J, Izumi K, Wang X, Xu D, Fang L-Y, et al. Targeting androgen receptor to suppress macrophage-induced EMT and benign prostatic hyperplasia (BPH) development. Mol Endocrinol. 2012;26:1707–15.
Izumi K, Mizokami A, Lin W-J, Lai K-P, Chang C. Androgen receptor roles in the development of benign prostate hyperplasia. Am J Pathol. 2013;182:1942–9.
Saxena P, Trerotola M, Wang T, Li J, Sayeed A, Vanoudenhove J, et al. PSA regulates androgen receptor expression in prostate cancer cells. Prostate. 2012;72:769–76.
Kim J, Coetzee GA. Prostate specific antigen gene regulation by androgen receptor. J Cell Biochem. 2004;93:233–41.
Hipfner DR, Cohen SM. Connecting proliferation and apoptosis in development and disease. Nat Rev Mol Cell Biol. 2004;5:805–15.
Mhaidat NM, Thorne RF, Zhang XD, Hersey P. Regulation of docetaxel-induced apoptosis of human melanoma cells by different isoforms of protein kinase C. Mol Cancer Res. 2007;5:1073–81.
Petrylak DP. The treatment of hormone-refractory prostate cancer: docetaxel and beyond. Rev Urol. 2006;8(Suppl 2):S48–55.
Kellokumpu-Lehtinen P-L, Harmenberg U, Joensuu T, McDermott R, Hervonen P, Ginman C, et al. 2-Weekly versus 3-weekly docetaxel to treat castration-resistant advanced prostate cancer: a randomised, phase 3 trial. Lancet Oncol. 2013;14:117–24.
McKeage K, Keam SJ. Docetaxel in hormone-refractory metastatic prostate cancer. Drugs. 2005;65:2287–94 (discussion 2295–7).
Karanika S, Karantanos T, Kurosaka S, Wang J, Hirayama T, Yang G, et al. GLIPR1-ΔTM synergizes with docetaxel in cell death and suppresses resistance to docetaxel in prostate cancer cells. Mol Cancer. 2015;14:122.
Acknowledgments
This work was supported by University of North Dakota School of Medicine seed grant to Othman Ghribi. The funding source had no involvement in the study design, collection, analysis or interpretation of data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Additional information
Disclaimer: This material is the result of work supported with resources and the use of facilities at the Fargo VA Medical Center. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Rights and permissions
About this article

Cite this article
Raza, S., Meyer, M., Schommer, J. et al. 27-Hydroxycholesterol stimulates cell proliferation and resistance to docetaxel-induced apoptosis in prostate epithelial cells. Med Oncol 33, 12 (2016). https://doi.org/10.1007/s12032-015-0725-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-015-0725-5