
Abstract
The destruction of the blood-brain barrier (BBB) contributes to a spectrum of neurological diseases such as stroke, and the hyperpermeability of endothelial cells is one of the characters of stroke, which is possibly exacerbated after reperfusion. However, the underlying mechanisms involving hyperpermeability after reperfusion between the endothelial cells remain poorly understood. Therefore, in the present study, the human microvascular endothelial cells (HBMECs) were exposed to oxygen-glucose deprivation/reperfusion (OGD/R) to mimic ischemic stroke condition in vitro with the aim to investigate the potential mechanisms induced by OGD/R. The permeability of cultured HBMECs was measured using FITC-labeled dextran in a Transwell system and transendothelial electrical resistance (TEER), while the RhoA activity was detected by pull-down assay. In addition, the phosphorylation of MYPT1, which reflects the activation of ROCK and the internalization of VE-cadherin, was detected by Western blot. It showed that OGD/R treatment significantly increased the permeability of HBMEC monolayers and facilitated the internalization of VE-cadherin in HBMEC monolayers. Pull-down assay showed that RhoA activation was obviously enhanced after OGD/R treatment, while RhoA and ROCK inhibitor significantly reversed OGD/R-induced HBMEC monolayers hyperpermeability and the internalization of VE-cadherin. Meanwhile, the knockdown assay showed that RhoA small interfering RNA (siRNA) led to similar effects. The inactivation of the downstream effector protein ROCK was also examined. Intriguingly, ROCK2 rather than ROCK1 exerted its adverse effects on HBMEC monolayer integrity, since ROCK2 knockdown markedly reverses the injury of OGD/R in HBMEC monolayers. In conclusion, the present study provides evidence that OGD/R may induce HBMEC monolayer hyperpermeability via RhoA/ROCK2-mediated VE-cadherin internalization, which may provide an impetus for the development of therapeutics targeting BBB damage in ischemic stroke.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The blood-brain barrier (BBB) dysfunction contributes to occurrence and development of many neurological disorders (Brailoiu et al. 2018), including the cerebral edema in stroke (Kassner and Merali 2015; Prakash and Carmichael 2015; Xiang et al. 2017). In the acute and the subacute phase of stroke, the impairment of BBB is marked by the decreased expression of junction proteins and increased permeability, which may last for a few days. BBB dysfunction leads to severe clinical consequences of stroke such as hemorrhagic transformation (HT) and motor and cognitive function deficits (Khatri et al. 2012).
Endothelial cells (ECs) of the microvasculature represent a critical site of barrier regulation (Taddei et al. 2008). Endothelial cells are tightly connected to each other via interactions of adherens junction (AJ) proteins, such as VE-cadherin (Vandenbroucke et al. 2008; Stamatovic et al. 2016). VE-cadherin is a calcium-dependent cell-cell adhesion molecule and is the endothelial-specific transmembrane component that localizes exclusively at endothelial cell-cell contacts. It maintains endothelial permeability and vessel integrity by interactions with catenins, including β-catenin and p120-catenin (Harris and Nelson 2010). Though the barrier function of the endothelium is supported by multiple intercellular adhesion systems, disruption of VE-cadherin is sufficient to disrupt other intercellular junctions. Evidence has shown that blockage of VE-cadherin with the specific antibodies significantly increases vascular permeability (Semina et al. 2014). Moreover, recent studies have revealed that impaired vascular barrier function is accompanied by VE-cadherin decreased and enhanced internalization in neuroinflammation (Li et al. 2012, 2018). Therefore, due to the crucial roles of VE-cadherin in responsible for the opening and closing of the endothelial barrier, it might be a critical therapeutic target for ECs permeability changes.
RhoA, a small GTPases of the Rho protein family, is demonstrated to be the key regulator of actin cytoskeleton and cell junction (Heasman and Ridley 2008; Huang et al. 2015). The activation of RhoA disturbs endothelial barrier function and leads to the impairment of cell adhesion junction. Rho-associated protein kinase (ROCK) is the effector protein of small Rho GTPases, including two subtypes ROCK1 and ROCK2. RhoA/ROCK signaling pathway exerts important roles in regulating multiple intracellular activities, including cell contraction, migration, axon elongation, cell adhesion, and differentiation. It has indicated that Y27632 and Fasudil, the ROCK inhibitors, improve endothelial function, increase cerebral blood flow, and protect against focal cerebral ischemia (Rikitake et al. 2005). Additionally, loss of RhoA shows decreased phosphorylation of Myosin Light Chain and increases VE-cadherin expression at cell-cell contacts after thrombin stimulation (MCA et al. 2017). Based on the evidence aforementioned, we hypothesized that RhoA/ROCK signaling pathway might be involved in the OGD/R-induced endothelial cell hyperpermeability.
Therefore, the main goal of the present study was attempted to investigate the HBMECs hyperpermeability after OGD/R, meanwhile, the potential mechanism involving RhoA/ROCK signaling pathway in this process was also fully discussed.
Material and Methods
Reagents and Antibodies
Anti VE-cadherin was purchased from Abcam (Cambridge, MA), RhoA antibody, phosphorylation of MYPT1 antibody, and GAPDH antibody were all from Cell Signaling Technology (Danvers, MA), and C3-transferase was from Cytoskeletion (Danvers, CO). Y-27632 was from Calbiochem (San Diego, CA). All other chemicals were of the best available quality, usually analytical grade.
HBMEC Monolayer Permeability Assay
Primary HBMECs were obtained from Cell Systems Corporation (ACBRI376, Kirkland, WA), cultured in complete growth media EBM-2 (Lonza, Walkersville, MD) with 10% FBS in a humidified 5% CO2 incubator at 37 °C. The cells were seeded on the inner surface of collagen-coated transwell inserts (6.5-mm diameter, 0.4-μm pore size polycarbonate filter; Corning, NY), which were placed in wells of a 24-well plate. When the monolayer of cells was confluent, confirmed by ensuring that it is impermeable to media, the cells were serum starved for 8 h with EBM-2 media without growth supplement before OGD/R treatment. The control group is the monolayer culture without OGD/R treatment. For OGD/R treatment, EBM-2 media were replaced with DMEM without glucose, and the cells were then put in a specialized, humidified chamber (Heidolph, incubator 1000, Brinkmann Instruments, Westbury, NY) at 37 °C, which contained an anaerobic gas mixture (90% N2, 5% H2, and 5% CO2). After 4-h incubation, the cultures were removed from the anaerobic chamber, and the OGD medium was replaced with EBM-2 complete medium. Then the cells were allowed to recover in a regular incubator for 20 h (reoxygenation) before permeability measurement. For the C3 transferase (RhoA inhibitor) treated group, C3 transferase was added to the medium (final concentration of 1 μg/ml) before OGD/R. For the Y27632 (ROCK inhibitor) treated group, Y27632 was added to the medium (final concentration 10 μM) before OGD/R. After 20-h reoxygenation, media in both upper and lower chambers were removed and replaced with fresh media without supplement. Permeability was measured by adding 0.1 mg/ml of fluorescein isothiocyanate (FITC)-labeled dextran (MW, 70,000 Da; Sigma, St. Louis, MO) to the upper chamber, with a lower compartment containing fresh serum-free media. After incubation for 20 min, 100 μl of the sample from the lower compartment was measured for fluorescence at excitation 490 nm and emission 520 nm. All independent experiments were performed in triplicate.
Transendothelial Electrical Resistance
The method is based on an established process to monitor transendothelial electrical resistance of cultured HBMEC monolayers by an EndOhm-6 chamber and an EVOM resistance meter (Millicell, World Precision Instruments, Sarasota, FL) following the manufacturer instructions. Briefly, the transwells containing cultured endothelial cell monolayers were transferred into the EndOhm-6 chamber containing 0.1 M KCl. The culture media within the transwells was also replaced with 0.1 M KCl. EndOhm cap was then inserted on the top of the chamber and transwell and then connected with the chamber using a connector cable, and resistance was then measured using the EVOM resistance meter. A blank transwell containing 0.1 M KCl without any cells was used as blank control.
Western Blot
HBMECs were harvested and lysed with RIPA buffer. The cell extracts were subjected to western blot analysis as previously described. The trypsinization assay, which was performed as previously described, was used to distinguish cell surface and intracellular pools of VE-cadherin (Xiao et al. 2003). In brief, HBMECs treated with different conditions were rinsed and then incubated in trypsin-EDTA to remove cell surface VE-cadherin. Then, the cells were collected and lysed with RIPA buffer for western blot assay. As a negative control, cells were harvested in parallel using RIPA buffer without trypsinization.
GTP-RhoA Pull-Down Assay
Cells were stimulated and washed with ice-cold 1× PBS and lysed in lysis buffer A (25 mM Tris pH 7.5, 150 mM NaCl, 1% NP-40, 5 mM MgCl2, 5% glycerol and protease inhibitors including 1 mM PMSF, 1 μg/ml of each leupeptin, aprotinin, and pepstatin A). Lysates were clarified by centrifugation and equalized for total volume and protein concentration. After incubation with GST-Rhotekin-Rho binding domain (RBD) beads, washing three times with ice-cold lysis buffer B (50 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5 mM MgCl2, 100 μM orthovanadate, with protease inhibitors), the bound fraction (active RhoA-GTP) was separated on SDS-PAGE. Total RhoA levels were similarly analyzed using a reserved aliquot of whole cell lysate. Active RhoA and total RhoA were analyzed by western blotting with an anti-RhoA antibody. The relative amount of active RhoA was determined by taking the ratio of RhoA sedimented by GST-RBD beads (active RhoA) divided by the amount of total RhoA in the whole cell lysate. The results were quantified using Photoshop 7.01 software.
Small Interfering RNA-Mediated Gene Silencing
RhoA, ROCK1, ROCK2 siRNA, and control siRNA were obtained from Santa Cruz Biotechnology. siRNA transfection was conducted following the manufacturer’s instruction. Briefly, HBMECs were cultured on six-well plates or transwells for fusion. siRNA duplex and siRNA Transfection Reagent (Santa Cruz) were separately diluted with Transfection Medium (Santa Cruz) and then mixed together and incubated at room temperature for 40 min. The cells were washed once with siRNA Transfection Medium. Then it is incubated with mixed siRNA and siRNA Transfection Reagent for 5 h at 37 °C. The cells on six-well plates were collected for Western Blot to detect protein level, and cells in transwells underwent OGD/R treatment and tested for permeability.
Statistical Analysis
Data were analyzed with SPSS 17.0 software. Experimental results were presented as mean ± SD. Multiple comparisons were evaluated by one-way ANOVA followed by a Student-Newman-Keuls post hoc test. p < 0.05 was considered statistically significant.
Results
OGD/R Induces HBMEC Monolayer Hyperpermeability
Since the endothelial monolayer permeability is closely associated with cerebral edema, the first step was to examine whether OGD/R treatment contributed to endothelial monolayer permeability. The cultured HBMEC monolayers in transwells were subjected to 4 h of OGD followed by 20 h of reoxygenation, and then the monolayer permeability was evaluated by the FITC-dextran (4, 10, 70 kDa); assay was then measured. As shown in Fig. 1a, the mean percentage of 4, 10, and 70 kDa FITC-dextran migrated through the HBMEC monolayer are 18.1%, 17.3%, and 15.2% in the control group, respectively. Furthermore, there was no significant difference among the three groups. OGD/R treatment significantly increased the mean percentage of 4, 10, and 70 kDa FITC-dextran migrated through the HBMEC monolayer. As shown in Fig. 1b, OGD/R treatment significantly increased the permeability of HBMEC monolayers, manifested as more than twofold number of cell fluorescence was detected in the OGD/R treatment group when compared with the control group (p < 0.01). In the following experiment, we chose 70 kDa FITC-dextran as the indicator of the permeability of HBMEC monolayers. To further corroborate the finds that OGD/R-mediated monolayer hyperpermeability aforementioned, the EndOhm chamber and EVOM resistance meter was used and the TEER in the two groups was detected. In line with the results in Fig. 1a, OGD/R significantly decreased the TEER of cultured HBMEC monolayers compared to that in the control group (Fig. 1c, p < 0.01), suggesting that OGD/R induces monolayer hyperpermeability.

OGD/R induces HBMEC monolayers hyperpermeability. Cultured HBMECs in transwells were subjected to 4-h OGD followed by 20-h reoxygenation. The integrity of HBMEC monolayers was measured by FITC-dextran permeability and TEER. a The percentage of FITC-dextran (4, 10, 70 kDa) in the upper chamber and in the bottom chamber. b The amount of FITC-dextran (4, 10, 70 kDa) diffused to the bottom chamber was analyzed. c HBMEC monolayers were treated as indicated and the electrical resistance was measured. Cells without treatment belonged to the control group. Data are expressed as means ± SD of three independent experiments (*p < 0.01 vs. the control group. **p < 0.01 vs. the control group)
OGD/R Activates RhoA/ROCK Signaling Pathway and Facilitates Internalization of VE-Cadherin in HBMEC Monolayers
Having determined that OGD/R-induced monolayer hyperpermeability, we then sought to examine whether RhoA/ROCK signaling pathway was involved in this process. As shown in Fig. 2a and b, OGD/R treatment significantly increased RhoA activity (p < 0.05) and MYPT1 phosphorylation (p < 0.01), indicating an activation of RhoA/ROCK signaling pathway. We also monitored another key protein VE-cadherin after OGD/R treatment, because of its critical roles in maintaining cell membrane integrity (Benn et al. 2016). Therefore, the trypsinization and Western blot assays were performed to explore the expression of internalized VE-cadherin in HBMECs. As shown in Fig. 2c, OGD/R markedly enhanced the intracellular VE-cadherin accumulation in HBMECs (p < 0.05) and decreased the total VE-cadherin level (p < 0.01).

OGD/R activates RhoA/ROCK signaling pathway and causes internalization of VE-cadherin in HBMEC monolayers. a The activation of RhoA was tested by pull-down assay. The active RhoA protein (RhoA-GTP) level was normalized to the total RhoA. Cells without treatment belonged to the control group. b Effect of OGD/R on MYPT1 phosphorylation at Thr850. The protein levels were quantified by densitometric analysis, normalized to GAPDH. c After treatment with or without OGD/R, trypsinized and non-trypsinized HBMECs were lysed, and the proteins were analyzed by Western Blot. Quantification of intracellular VE-cadherin was shown. Data are expressed as means ± SD of three independent experiments (*p < 0.05 vs. the control group. **p < 0.01 vs. the control group)
Inhibition of RhoA/ROCK Signaling Pathway Ameliorated OGD/R-Induced HBMEC Monolayers Hyperpermeability
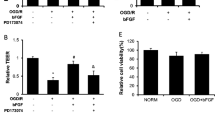
To further support the function of the RhoA/ROCK signaling in OGD/R-induced HBMEC monolayer hyperpermeability, C3 transferase (1 μg/ml), the RhoA inhibitor, and Y27632 (10 μM), the blocker of ROCK was used in the present work. It showed that pretreatment of Y27632 obviously reduced the permeability of HBMEC monolayers compared to that in the OGD/R group (p < 0.05). Meanwhile, with the transendothelial electrical resistance assay, a similar result observed that administration of C3 transferase significantly reduced the transendothelial electrical resistance, suggesting the impairment of OGD/R on the HBMEC monolayers (Fig. 3a and b, p < 0.05). Additionally, OGD/R-induced hyperpermeability was further strengthened by the observation that both of the inhibitors substantially attenuated OGD/R-induced VE-cadherin internalization compared with OGD/R treatment alone (Fig. 3c, p < 0.05). These results, taken together, indicated that the RhoA/ROCK signaling pathway exerts important roles in OGD/R-induced hyperpermeability.

Inhibition of RhoA/ROCK signaling pathway protects against OGD/R-induced HBMEC monolayers hyperpermeability. HBMEC monolayers were subjected to OGD/R in the presence or absence of C3 transferase (1 μg/ml) for 30 min or Y27632 (10 μM) for 30 min. Then, the integrity of monolayers was measured by FITC-dextran permeability and TEER. a The amount of FITC-dextran diffused to the bottom chamber was analyzed. b HBMEC monolayers were treated as indicated and the electrical resistance was measured. c After treatment with or without OGD/R, trypsinized and non-trypsinized HBMECs were lysed, and the proteins were analyzed by western blot. Quantification of intracellular VE-cadherin was shown. Data are expressed as means ± SD of three independent experiments (*p < 0.05 vs. the control group. **p < 0.01 vs. the control group. #p < 0.05 vs. the group treated with OGD/R)
RhoA Suppression Obviously Inhibited OGD/R-Induced HBMEC Monolayers Hyperpermeability and Internalized VE-Cadherin
Having determined the roles of RhoA/ROCK signaling pathway in OGD/R-induced HBMEC monolayers hyperpermeability via the pharmacological way, the siRNA technology was exploited to further assess the RhoA/ROCK signaling pathway in OGD/R-induced HBMEC monolayers impairment (Fig. 4). As depicted in Fig. 4a, RhoA siRNA effectively knockdown RhoA expression, which contributed to obvious decreased FITC-dextran leakage and increased TEER of HBMEC monolayers (Fig. 4b and c, p < 0.05); in accordance with these investigation aforementioned, internalization of VE-cadherin was obviously decreased after RhoA knockdown when compared with the OGD/R treated group (Fig. 4d, p < 0.05).

RhoA depletion ameliorates HBMEC monolayers hyperpermeability and internalization of VE-cadherin induced by OGD/R. a HMVECs were transferred RhoA siRNA, then the knockdown efficiency was confirmed with Western Blot. b Permeability of HMVEC monolayers was measured by 70 kDa weight FITC-dextran. c HBMEC monolayers’ electrical resistance was measured. d After treatment with or without OGD/R, trypsinized and non-trypsinized HBMECs were lysed, and the proteins were analyzed by Western Blot. Quantification of intracellular VE-cadherin was shown. Data are expressed as means ± SD of three independent experiments (*p < 0.05 vs. the control group. #p < 0.05 vs. the group treated with OGD/R)
ROCK2 Suppression Inhibits OGD/R-Induced HBMEC Monolayers Hyperpermeability and Internalized VE-Cadherin
Since the ROCK inhibitor (Y27632) is non-selective and cannot distinguish between the two ROCK isoforms, then we further knockdown ROCK1 and ROCK2 by performing siRNA technology. It showed that ROCK1 knockdown did not obviously affect the FITC-dextran leakage and TEER (Fig. 5b and c, p > 0.05). However, ROCK2 depletion markedly retarded OGD/R-induced increase of FITC-dextran leakage and reduction of TEER in HBMEC monolayers, accompany by decrease of internalization of VE-cadherin after OGD/R (Fig. 6b and c, p < 0.05); these results demonstrated that ROCK2, rather than ROCK1, was involved in OGD/R-mediated monolayer damage.

ROCK1 depletion has no effect on HBMEC monolayers hyperpermeability and internalization of VE-cadherin after OGD/R. a HMVECs were transferred ROCK1 siRNA, then the knockdown efficiencies were confirmed with Western Blot. b Permeability of HMVEC monolayers was measured by 70 kDa weight FITC-dextran. c HBMEC monolayers’ electrical resistance was measured. d After treatment with or without OGD/R, trypsinized and non-trypsinized HBMECs were lysed, and the proteins were analyzed by Western Blot. Quantification of intracellular VE-cadherin was shown. Data are expressed as means ± SD of three independent experiments (*p < 0.05 vs. the control group. #p < 0.05 vs. the group treated with OGD/R)

ROCK2 depletion ameliorates HBMEC monolayers hyperpermeability and internalization of VE-cadherin induced by OGD/R. a HMVECs were transferred ROCK2 siRNA, then the knockdown efficiencies were confirmed with Western Blot. b Permeability of HMVEC monolayers was measured by 70 kDa weight FITC-dextran. c HBMEC monolayers’ electrical resistance was measured. d After treatment with or without OGD/R, trypsinized and non-trypsinized HBMECs were lysed, and the proteins were analyzed by Western Blot. Quantification of intracellular VE-cadherin was shown. Data are expressed as means ± SD of three independent experiments (*p < 0.05 vs. the control group. #p < 0.05 vs. the group treated with OGD/R)
Discussion
Ischemic stroke is a reduction or cessation of blood flow to the brain, which deprives the cerebral tissue of important nutrients and oxygen as well as the removal of metabolic waste products. The fibrinolytic treatment of ischemic stroke or successful recanalization restores the oxygenation but initiates secondary local inflammation after reperfusion which in turn exacerbates cerebral tissue injury, the so-called reperfusion injury (I/R) (Asaithambi et al. 2014). Recent advances in treatments (i.e., endovascular thrombectomy and tissue plasminogen activator (t-PA)) that target the stroke-causing blood clot, while improving overall stroke mortality rates, have had much less of an impact on overall stroke morbidity. This may in part be attributed to the lack of therapeutics targeting reperfusion-induced injury after the blood clot has been removed, which, if left unchecked, can expand injury from its core into the surrounding at-risk tissue (penumbra) (Edwards and Bix 2019). I/R injury causes widespread inflammation, reactive oxidation, excitotoxicity, and cell-specific dysregulation of metabolic processes, leading to cerebrovascular hyperpermeability (Mark and Davis 2002), which may further lead to the destruction of the BBB, and thereby finalized brain vasogenic edema. Therefore, suppression of BBB damage induced by I/R will be beneficial to prevent ischemic stroke and improve prognosis (Heye et al. 2014; Shi et al. 2017). In the current study, we highlighted the potential molecular mechanisms underlying BBB damage in OGD/R, and we found that in the OGD/R cell model, the activation of the RhoA/ROCK2 signaling pathway significantly enhanced OGD/R-induced BBB damage and upregulation of the internalization of VE-cadherin.
Brain microvessel endothelial cells (BMECs) form a metabolic and physical barrier, separating the periphery from the brain to maintain cerebral homeostasis. Endothelial barrier dysfunction is one of the major pathological characteristics for BBB disruption after stroke (Jin et al. 2010; Heye et al. 2014). Hypoxia-reoxygenation induces endothelial cell hyperpermeability and edema formation, which presents a major impediment for the recovery of the organ (Aslam et al. 2013). In line with these pathological characteristics, our results in the cell OGD/R model showed that OGD/R leads to HBMEC monolayers hyperpermeability.
RhoA, one subtype of the Rho family, is an important regulator of endothelial actin cytoskeleton dynamics and AJs, which plays a crucial role in the maintenance of the endothelial barrier. Hypoxia-reoxygenation induces loss of endothelial barrier function and edema formation accompanied by a rise in RhoA/ROCK signaling (Aslam et al. 2013). Luo SY reported that inhibition RhoA/ROCK pathway had a potent protective effect on myocardial I/R injury in rats (Luo et al. 2016). These results strongly supported our results that OGD/R caused a pronounced activation of RhoA, which was accompanied by enhanced phosphorylation of MYPT1 at Thr850 (ROCK site). Based on this observation, we hypothesized that inhibition of RhoA/ROCK signaling would protect against reoxygenation-induced BBB hyperpermeability.
Using the pharmacological assay, C3 transferase or Y27632 inhibited the RhoA/ROCK signaling pathway, respectively. The results showed that inhibition of either RhoA with C3 transferase or ROCK with Y27632 significantly abrogated OGD/R-induced hyperpermeability. These data indicated that the activated RhoA/ROCK pathway is involved in OGD/R-induced endothelial monolayer permeability.
The adherence junction proteins are known to be vital to vascular barrier function; however, little is known about the role of VE-cadherin in the context of the pathophysiology of stroke-induced BBB breakdown and injury. Recent studies indicate that some pools of cadherin on the cell surface are endocytosed and recycled back to the plasma membrane and enhanced internalization of VE-cadherin leads to vascular dysfunction (Gavard 2014), suggesting that cadherin endocytosis is an important regulatory mechanism that controls cadherin cell surface levels and perhaps regulates overall levels of cadherin expression. The extracellular portion of these adhesive proteins can be hydrolyzed into the serum, which may be able to indicate the occurrence and development of the disease. It has been reported that the expression level of serum VE-cadherin in patients with acute myocardial infarction is increased and the degree of elevation is related to the severity of the disease (Soeki et al. 2004). Serum VE-cadherin may also be a biological indicator for assessing the blood-brain barrier after stroke. Therefore, the present study aimed to evaluate the interactions between RhoA/ROCK pathway and internalization of VE-cadherin. Our data showed that OGD/R caused a strong activation of RhoA, accompanied by enhanced internalization of VE-cadherin and decreased expression of VE-cadherin in cell-cell junction. Intriguingly, inhibition of RhoA or ROCK significantly abrogated OGD/R-induced internalization of VE-cadherin. These results indicated that activated RhoA/ROCK pathway is involved in OGD/R-induced endothelial monolayer permeability and increase internalization of VE-cadherin (Fig. 7).

Hypothetical model of OGD/R-induced endothelial cell hyperpermeability. OGD/R activates RhoA/ROCK signaling pathway and facilitates internalization of VE-cadherin in HBMEC monolayers, inducing monolayer hyperpermeability. The inactivation of RhoA/Rock signaling pathway shows anti-permeability activity through downregulation of internalization of VE-cadherin
To confirm that the RhoA activity mediated the OGD/R-induced HBMEC monolayers hyperpermeability, we successfully suppressed RhoA by performing siRNA technology and confirmed the effects of RhoA suppression on HBMEC monolayers. The results showed that RhoA suppression significantly inhibited OGD/R-induced HBMEC monolayers hyperpermeability and increased the expression of VE-cadherin. These results are consistent with those of the application of RhoA inhibitor, indicating that OGD/R-induced barrier dysfunction and the expression of VE-cadherin through RhoA activation.
Concerning the tissue distribution of Rho-kinases, it was reported that ROCK I was highly expressed in non-neuronal tissues such as the liver and lungs, whereas ROCK2 was predominantly expressed in the brain and muscles (Nakamura et al. 2001). We further suppressed ROCK1 and ROCK2 by performing siRNA technology respectively. The data show that the loss of ROCK2, but not ROCK1, led to enhanced endothelial function. Similar to our results, it reported that ROCK2 is the main isoform in the brain, with significant expression in many cell types of the neurovascular unit that is relevant to stroke severity. These data indicate that inhibition of ROCK2 partially protects against hypoxia-reoxygenation-induced endothelial hyperpermeability.
There are two limitations in our study. First, the astrocyte-endothelium co-culture system or neuron-astrocyte-endothelium co-culture system is often used as the model of blood-brain barrier (BBB) in vitro (Wu et al. 2018). However, siRNA transfection of RhoA, ROCK1, and ROCK2 in the model of endothelium cells, astrocytes, or neuron needs different experimental conditions. Thus, we chose the model of monoculture of brain endothelial cell without any other cell type of neurovascular unit. Ysrayl BB also used the HBMECs as the model of BBB in vitro in his recently published article (Ysrayl et al. 2019). In our future studies, we should build a more suitable model of BBB to mimic its function and structure form. Second, this is only an in vitro study to demonstrate the effect of RhoA/ROCK and VE-cadherin on OGD/R-induced HBMEC monolayers permeability. We should use the cerebral ischemia animal stroke models to investigate the effect of RhoA/ROCK signaling pathway on BBB dysfunction.
In summary, in this study, we clearly demonstrated that the RhoA/ROCK2 pathway was involved in OGD/R-induced HBMEC monolayers hyperpermeability through regulating the internalization of VE-cadherin. This mechanism probably also contributes to the neuroprotective effects after ischemic stroke. Thus, inactivation of RhoA/ROCK2/VE-cadherin may potentially develop therapeutic strategies to mitigate the clinical consequences of ischemic stroke.
References
Asaithambi G, Tong X, George MG, Tsai AW, Peacock JM, Luepker RV, Lakshminarayan K (2014) Acute stroke reperfusion therapy trends in the expanded treatment window era. J Stroke Cerebrovasc Dis 23:2316–2321
Aslam M, Schluter KD, Rohrbach S, Rafiq A, Nazli S, Piper HM, Noll T, Schulz R, Gündüz D (2013) Hypoxia-reoxygenation-induced endothelial barrier failure: role of RhoA, Rac1 and myosin light chain kinase. J Physiol 591:461–473
Benn A, Bredow C, Casanova I, Vukičević S, Knaus P (2016) VE-cadherin facilitates BMP-induced endothelial cell permeability and signaling. J Cell Sci 129:206–218
Brailoiu E, Barlow CL, Ramirez SH, Abood ME, Brailoiu GC (2018) Effects of platelet-activating factor on brain microvascular endothelial cells. Neuroscience 377:105–113
Edwards D, Bix GJ (2019) Roles of blood-brain barrier integrins and extracellular matrix in stroke. Am J Physiol Cell Physiol 316:252–263
Gavard J (2014) Endothelial permeability and VE-cadherin: a wacky comradeship. Cell Adhes Migr 8:158–164
Harris ES, Nelson WJ (2010) VE-cadherin: at the front, center, and sides of endothelial cell organization and function. Curr Opin Cell Biol 22:651–658
Heasman SJ, Ridley AJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9:690–701
Heye AK, Culling RD, Valdés HMC, Thrippleton MJ, Wardlaw JM (2014) Assessment of blood-brain barrier disruption using dynamic contrast-enhanced MRI. A systematic review. Neuroimage Clin 6:262–274
Huang Y, Tan Q, Chen R, Cao B, Li W (2015) Sevoflurane prevents lipopolysaccharide-induced barrier dysfunction in human lung microvascular endothelial cells: Rho-mediated alterations of VE-cadherin. Biochem Biophys Res Commun 468:119–124
Jin R, Yang G, Li G (2010) Molecular insights and therapeutic targets for blood-brain barrier disruption in ischemic stroke: critical role of matrix metalloproteinases and tissue-type plasminogen activator. Neurobiol Dis 38:376–385
Kassner A, Merali Z (2015) Assessment of blood-brain barrier disruption in stroke. Stroke 46:3310–3315
Khatri R, McKinney AM, Swenson B, Janardhan V (2012) Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology 79:52–57
Li R, Ren M, Chen N, Luo M, Zhang Z, Wu J (2012) Vitronectin increases vascular permeability by promoting VE-cadherin internalization at cell junctions. PLoS One 7:e37195
Li W, Chen Z, Chin I, Chen Z, Dai H (2018) The role of VE-cadherin in blood-brain barrier integrity under central nervous system pathological conditions. Curr Neuropharmacol 16:1375–1384
Luo SY, Chen S, Qin YD, Chen ZW (2016) Urotensin-II receptor antagonist SB-710411 protects rat heart against ischemia-reperfusion injury via RhoA/ROCK pathway. PLoS One 11:e0146094
Mark KS, Davis TP (2002) Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol 282:1485–1494
MCA P, van Bezu JSM, van Nieuw Amerongen GP, van Hinsbergh VWM, Hordijk PL (2017) RhoA, RhoB and RhoC differentially regulate endothelial barrier function. Small GTPases:1–19
Nakamura M, Nagano T, Chikama T, Nishida T (2001) Role of the small GTP-binding protein rho in epithelial cell migration in the rabbit cornea. Invest Ophthalmol Vis Sci 42:941–947
Prakash R, Carmichael ST (2015) Blood-brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Curr Opin Neurol 28:556–564
Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, Moskowitz MA, Liao JK (2005) Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 36:2251–2257
Semina EV, Rubina KA, Sysoeva VY, Rutkevich PN, Kashirina NM, Tkachuk VA (2014) Novel mechanism regulating endothelial permeability via T-cadherin-dependent VE-cadherin phosphorylation and clathrin-mediated endocytosis. Mol Cell Biochem 387:39–53
Shi Y, Jiang X, Zhang L, Pu H, Hu X, Zhang W, Cai W, Gao Y, Leak RK, Keep RF, Bennett MV, Chen J (2017) Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood-brain barrier disruption after ischemic brain injury. Proc Natl Acad Sci U S A 114:1243–1252
Soeki T, Tamura Y, Shinohara H, Sakabe K, Onose Y, Fukuda N (2004) Elevated concentration of soluble vascular endothelial cadherin is associated with coronary atherosclerosis. Circ J 68:1–5
Stamatovic SM, Johnson AM, Keep RF, Andjelkovic AV (2016) Junctional proteins of the blood-brain barrier: new insights into function and dysfunction. Tissue Barriers 4:e1154641
Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E (2008) Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol 10:923–934
Vandenbroucke E, Mehta D, Minshall R, Malik AB (2008) Regulation of endothelial junctional permeability. Ann N Y Acad Sci 1123:134–145
Wu L, Ye Z, Pan Y, Li X, Fu X, Zhang B, Li Y, Lin W, Li X, Gao Q (2018) Vascular endothelial growth factor aggravates cerebral ischemia and reperfusion-induced blood-brain-barrier disruption through regulating LOC102640519/HOXC13/ZO-1 signaling. Exp Cell Res 369:275–283
Xiang J, Routhe LJ, Wilkinson DA, Hua Y, Moos T, Xi G, Keep RF (2017) The choroid plexus as a site of damage in hemorrhagic and ischemic stroke and its role in responding to injury. Fluids Barriers CNS 14:8
Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, Kowalczyk AP (2003) Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol 163:535–545
Ysrayl BB, Balasubramaniam M, Albert I, Villalta F, Pandhare J, Dash C (2019) A novel role of prolidase in cocaine-mediated breach in the barrier of brain microvascular endothelial cells. Sci Rep 9:2567
Acknowledgments
We thank Professor Gui bo (Department of Anesthesiology, 1st Affiliated Hospital of Nanjing Medical University, China) for his selfless help in the research process.
Funding
This work was supported by the National Natural Science Foundation of China (81701872) and by a grant from the Nanjing Medical University Science and Technology Development Fund Project (2015NJMUZD010).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, J., Sun, L., Ding, Gb. et al. Oxygen-Glucose Deprivation/Reoxygenation Induces Human Brain Microvascular Endothelial Cell Hyperpermeability Via VE-Cadherin Internalization: Roles of RhoA/ROCK2. J Mol Neurosci 69, 49–59 (2019). https://doi.org/10.1007/s12031-019-01326-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-019-01326-8