
Abstract
Cell therapy and stem cell transplantation strategies have provided potential therapeutic approaches for the treatment of neurological disorders. Adipose-derived mesenchymal stem cells (ADMSCs) are abundant adult stem cells with low immunogenicity, which can be used for allogeneic cell replacement therapies. Differentiation of ADMSCs into acetylcholine-secreting motoneurons (MNs) is a promising treatment for MN diseases, such as spinal muscular atrophy (SMA), which is associated with the level of SMN1 gene expression. The SMN2 gene plays an important role in MN disorders, as it can somewhat compensate for the lack of SMN1 expression in SMA patients. Although the differentiation potential of ADMSCs into MNs has been previously established, overexpression of SMN2 gene in a shorter period with a longer survival has yet to be elucidated. Ponasterone A (PNA), an ecdysteroid hormone activating the PI3K/Akt pathway, was studied as a new steroid to promote SMN2 overexpression in MNs differentiated from ADMSCs. After induction with retinoic acid, sonic hedgehog, forskolin, and PNA, MN phenotypes were differentiated from ADMSCs, and immunochemical staining, specific for β-tubulin, neuron-specific enolase, and choline acetyltransferase, was performed. Also, the results of real-time PCR assay indicated nestin, Pax6, Nkx2.2, Hb9, Olig2, and SMN2 expression in the differentiated cells. After 2 weeks of treatment, cultures supplemented with PNA showed a longer survival and a 1.2-fold increase in the expression of SMN2 (an overall 5.6-fold increase; *P ≤ 0.05), as confirmed by the Western blot analysis. The PNA treatment increased the levels of ChAT, Isl1, Hb9, and Nkx2 expression in MN-like cells. Our findings highlight the role of PNA in the upregulation of SMN2 genes from MSC-derived MN-like cells, which may serve as a potential candidate in cellular therapy for SMA patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Motoneurons (MNs) can be found in the spinal cord and brain with organized patterns. They are connected to muscles for controlling muscular activities as voluntary actions (Du et al. 2015) and transmit electrochemical signals from the central nervous system to all parts of the body (Bertini and Mercuri 2018). The inactivating mutations in the survival motor neuron (SMN) gene lead to decreased SMN protein expression (Lefebvre et al. 1995) and selective death of MNs (Hao le et al. 2012). This phenomenon causes disorders, such as spinal muscular atrophy (SMA), which is dependent on the level of SMN gene expression in MNs (Zerres et al. 1997).
SMA patients can be divided into different groups according to the age of disease onset (Kostova et al. 2007), which is directly related to the copy number of SMN1 gene or expression level of SMN2 gene (Zheleznyakova et al. 2011). In type-I SMA (Werdnig-Hoffmann disease), there is a homozygous deletion of SMN1 gene (De Sanctis et al. 2018). The homozygous absence of SMN1 gene is the primary cause of SMA, while its severity is determined by the level of SMN2 gene expression. One copy of SMN2 gene produces about 10% of full-length SMN protein (FL-SMN) identical to the SMN1 gene, as SMN2 transcripts are spliced due to a silent mutation in exon 7 (Brichta et al. 2003). A small portion of SMN2 transcripts is not spliced alternatively and produces a very small amount of functional FL-SMN, compensating for the lack of SMN1 gene expression (Lorson et al. 2010).
The mechanisms which can regulate SMN2 splicing and expression have been extensively studied in the literature (Groen et al. 2018), and the important role of increased SMN2 expression as a potential therapy for SMA has been suggested. The overexpression of SMN2 gene improves the neuromuscular function and MN survival (Turner et al. 2014). Therefore, the increased expression of SMN2 gene in an ordinary cellular system can lead to the application of modified MNs in clinical trials for SMA patients to inhibit disease progression or even reduce the severity of SMA complications.
Mesenchymal stromal cells (MSCs) are promising targets in cell and gene therapies because of their extensive self-renewal and multipotential capacity in regeneration of multiple tissues and neurogenesis in particular (Rahmani et al. 2013; Noureddini et al. 2012; Verdi et al. 2014a, b; Bahrami et al. 2017). Considerable progress has been made in the past decade in differentiating human stem cells into MNs by using a cocktail of different growth factors. However, the percentage of stem cells becoming MNs ranges from less than 0.5 to more than 50% (Thonhoff et al. 2009), and the underlying molecular mechanisms controlling the fate of MNs remain largely instinctive.
Although adipose-derived MSCs (ADMSCs) have been successfully differentiated into MNs (Liqing et al. 2011; Darvishi et al. 2017a), changes in the expression of SMN2 gene for achieving a higher SMN2 yield have not been described yet. In this study, we focused on a steroid hormone, ponasterone A (PNA), as an ecdysone analog and a novel approach facilitating MN formation. We also found that it can induce SMN2 gene upregulation in differentiated MNs. Ecdysteroids, as the most abundant steroids, show very low toxicity in human cells (LD50 > 6 g/kg). They are not hypertensive, and in spite of their anabolic action, they have neither androgenic nor estrogenic effects (Lafont and Dinan 2003).
Binding of PNA to its receptor (EcR) initiates a transcriptional cascade, which triggers many cellular responses (Riddiford et al. 2000). It can potentiate the activation of PI3-k/Akt pathway, which can ultimately interfere with cell growth and survival (Constantino et al. 2001). The PI3K/Akt pathway modulates the decision of human neural stem cells (hNSCs) to become MNs both in vitro and after transplantation into the spinal cord (Ojeda et al. 2011). Inhibition of Fas ligand and TNF-related apoptosis by PNA occurs at the level of caspase-8 activation (Oehme et al. 2006). Animal studies show that ecdysteroids can induce acetylcholine esterase in the brain (Catalan et al. 1984). Also, several activities of ecdysteroids, such as neurotransmitter synthesis/degradation, neuroprotection, and neuromodulation, have been described (Catalan et al. 1984; Okada et al. 1998; Tsujiyama et al. 1995).
Since SMN2 gene is neuroprotective in the spinal cord and can delay the presentation of symptoms in SMA patients, we aimed to introduce a method by using PNA for efficient differentiation of human ADMSCs into MNs, overexpressing SMN2 genes with a longer survival. This study was retrieved from our stem cell clinical trial on SMA patients.
Materials and Methods
Isolation, Culture, and Expansion of ADMSCs
The fat tissue was obtained after proper consenting process and approvals from Tehran University of Medical Sciences ethics committee. The tissue derived from normal volunteers aged between 18 and 70 years via abdominal lipoaspiration process. The subcutaneous abdominal fat tissue stromal cells were isolated by collagenase extraction as described previously (Amirkhani et al. 2016). Furthermore, these cells were expanded and characterized for the presence of MSC markers CD73, CD90, CD105, and CD146 per ISCT (International Society for Cellular Therapy) criteria and were found more than 95% positive (Dominici et al. 2006). The whole cell suspension was incubated for 30 min at 4 °C with FcR-blocking reagent and a primary antibody solution containing magnetic microbeads conjugated against CD105 (cat no. 130–094-926, Miltenyi Biotech, Germany). Washed cells were passed through a magnetic separator. CD105+ cells were recovered by eluting the column with phosphate buffer saline (cat no. 12579099, PBS, Gibco; UK) containing 0.5% bovine serum albumin (cat no. 9048-46-8, BSA, Gibco; UK). The pure population of ADMSCs was cultured in low glucose Dulbecco modified Eagle medium (cat no. 11530596, DMEM, Gibco, UK) supplemented with 10% fetal bovine serum (cat no. 11563397, FBS, Gibco, UK) and 50 units per ml penicillin plus 50 mg/ml streptomycin (cat no. 15070063, Pen/Strep, Gibco; UK). The harvested cells were plated in 25-cm2 flasks (CLS430372, Corning; Germany) and maintained at 37 °C in a 5% CO2 incubator with 95% humidity. Them changed every 3 days, and adherent cells were serially passaged at 80–90% confluence. The ADMSCs were confirmed for their capacity to differentiate toward the adipogenic, osteogenic, and chondrogenic lineages as described previously (Verdi et al. 2014b).
Motoneuron Differentiation Process
Our method was based on the (1) embryoid body (EB) induction and then (2) neurosphere formation followed by (3) neural rosette formation and finally (4) motoneurons maturation (Fig. 1). In passage 2, we expanded ADMSCs in DMEM: F12 (cat no. 11530566, Gibco; UK), 10% FBS (Gibco; UK), and 2 ng/ml basic fibroblast growth factor (cat no. 11123149001, bFGF, Sigma, USA). These flasks were harvested at 80–90% of confluence. To form the cell aggregates, 8 × 105 ADMSCs were plated in each well of low-adherent six-well plates (CLS3471-24EA, Corning; Germany) to obtain the EB-like aggregates. The EB-like aggregates formed on day 3 and then the media changed to the neurosphere medium that consists of 5 ng/ml bFGF and 1× sodium-pyruvate (cat no. 12539059, Gibco; UK). On day 7, when the cells formed neurosphere-like structures in suspension, we plated them on laminin-coated flasks (Cat No. 354533, Corning; Germany). Also, we have separated few neurosphere-like structures for complementary characterization studies. The remaining spheres were inserted into the 0.1% laminin-coated T25 flasks. The cultures were incubated at 37 °C for 2 h. The neural rosette formation was taking place in DMEM: F12, 10% FBS, 2% B27 (cat no. 21103-049, Invitrogen, USA), 10 ng/ml fibroblast growth factor 2 (FGF2, SRP4037, Sigma Aldrich, USA), 250 μM isobutylmethylxanthine (IBMX, cat no. 28822-58-4, Cayman Chemical, USA), and 1 mM β-mercaptoethanol (cat no. 60-24-2, Sigma, USA). On day 3 after plating, the medium was then substituted by DMEM: F12 containing 0.01 ng/ml Retinoic acid (RA, cat no. 302-79-4, Sigma, USA) and 5 μM Forskolin (66575-29-9, F3917, Sigma, UK). After 4 days, we changed the medium to the MN-maturation media consisting of DMEM: F12, 0.01 ng/ml retinoic acid, 10 ng/ml bFGF, 100 ng/ml Sonic Hedgehog (SHH, 1845SH, R&D systems, USA), and 50 ng/ml PNA (13408–56-5, Biomol, Germany) to increase the expression of SMN2 gene. The RA and SHH molecules are necessary to regulate the expression of the homeodomain and basic helix-loop-helix transcription factors. The cells were harvested at each stage and stored at − 20 °C. A group of ADMSCs cultured on the laminin-coated flask without differentiation factors served as the control. The medium was changed every 3 or 4 days due to the culture confluence.

Scheme for generating motoneurons from human ADMSCs. The ADMSCs cultured on laminin-coated flask before induction and maturation of MNs. Different combinations of signaling factors were added at the beginning of each stage of differentiation. (MN: motor neuron; DMEM: Dulbecco modified Eagle medium; FBS: fetal bovine serum; bFGF: basic fibroblast growth factor; RA: retinoic acid; Shh: Sonic HedgeHog; LM: laminin; PNA: ponasterone A; IBMX: isobutylmethylxanthine; BME: β-mercaptoethanol)
Immunocytochemistry Analysis
Cells were washed once with cold PBS, fixed with 4% paraformaldehyde (CAS. 30525-89-4, Merck; Germany) for 10 min, permeabilized with 0.1% Triton X-100 (9002-93-1, Sigma, USA) in PBS, and blocked with BSA. Primary antibodies of Nestin (1:200; Cat No. AB5922, Chemicon, USA), β-III-tubulin (1:400, Cat No. 05-559, Chemicon, USA), choline acetyltransferase (ChAT, 1:250, Cat No. AB144P, Chemicon, USA), and neuron-specific enolase (NSE, 1:100, cat no. AB53025, Abcam, Cambridge, UK) were incubated with samples overnight at 4 °C, followed by fluorescein isothiocyanate (FITC) or Alexa Fluor 568-conjugated secondary antibodies (1:1000, Cat No. 565786, BD PharmingenTM; Canada). Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole; CAS. 28,718–90-3, Sigma, USA). Primary antibodies were omitted for control. Images were collected with a Olympus fluorescent microscope (Olympus, Japan).
Real-Time PCR Analysis
The RNA samples were isolated from cell pellets at each stage of the differentiation process using an RNA isolation kit (RNeasy MicroKit, Cat No. 74004, Qiagen, USA) as described previously (Shoae-Hassani et al. 2017). cDNA was synthesized using cDNA synthesis kit (Superscript II First-strand cDNA synthesis kit, cat no. 18064014, Invitrogen, USA). The PCR mix contains cDNA sample, by mixing with 10 μl of SYBER Green master mix (cat no. 4309155, 2×, Applied Biosystems, USA), 0.5 μM of each forward and reverse primers (Table 1), and nuclease-free water in a total volume of 25 μl. The comparative Ct method, 2 − ∆∆Ct, was used for relative gene expression analysis. Cycling parameters were as follows: denaturation at 94 °C for 45 s, annealing for 45 s, and elongation at 72 °C for 1 min, for 30 cycles and the final extension at 72 °C for 10 min on Step One ABI Thermal Cycler (Applied Biosystems, USA).
Western Blot Analysis
The total protein was extracted from ADMSCs and differentiated cell lysates in all groups using RIPA lysis buffer [50 mM Tris, pH 8.0, 150 mM NaCl, (cat no. 79993, Sigma, USA) 1 mM EDTA (cat no. E9884, Sigma, USA), 1% sodium deoxycholate (cat no. D6750, Sigma, USA), 0.1% SDS (cat no. L3771, Sigma; USA)] and analyzed in 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described previously (Chen et al. 2015). After SDS-PAGE, the proteins transferred onto nitrocellulose papers (cat no. 77010, Thermo Fisher Scientific, USA). These membranes were blocked with 5% skimmed milk for 1 h. The membranes were exposed to the primary antibody (1:500, MAB.E230, Merck-Millipore, USA) for 3 h, and then they were washed three times for 10 min with TBST. In the next step, the membranes incubated for 1 h with the HRP-conjugated secondary antibody (Cat No. A9917, Sigma, USA). The membrane was washed 5 times with Tris buffer, and then specific bands were quantified using a ChemiImager System (Alpha Innotech Corporation). Anti-훽 actin antibody (cat no. Ab8227, Abcam; UK) was used as an internal control. The primary antibodies used were anti-SMN2 (cat no. 610647, BD; Canada, 1:5000 dilution) and anti-actin (cat no. 130301, Santa Cruz Biotechnology, Inc., USA, 1:3000 dilution). The secondary antibody used was horse anti-mouse IgG-HRP (Cat No. 31181, Invitrogen, USA; 1:5000 dilution).
Cell Viability Analysis
The 3-(4,5-dimethyl-thiazolyl-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Cat No. M2128, Sigma, USA) assay was carried out to measure the metabolic activity of the ADMSC culture in differentiation media with or without PNA. We added 500 μl of 5 mg/ml MTT solution to each well of 24-well plate and then incubated cultures at 37 °C for 4 h. The medium was removed, the formazan crystals were dissolved in dimethyl sulfoxide (DMSO, Cat No. D8418, Sigma, USA), and the absorbance at 570 nm was measured using an ELISA reader (Beckman Coulter DTX880, MA, USA). Also, survival of the MNs after differentiation in the presence or absence of PNA was measured by the percentage of originally plated cells every 24 h for 7 days.
Statistical Analysis
Data are presented as mean ± standard deviation (n = 3). Statistical analysis was carried out using one-way analysis of variance (ANOVA), and P value less than 0.05 was considered statistically significant.
Results
Morphological Characteristics of ADMSCs
ADMSCs were harvested by collagenase digestion as described earlier. Flow cytometry analysis after MACS purification showed that CD105+, CD90+, and CD146+ markers were expressed more than 99% in human ADMSCs, and these cells were negative for hematopoietic, endothelial, and progenitor CD34/CD45 markers (Fig. 2a). The expanded cells fulfilled the criteria for their markers, set by the International Society for Cellular Therapy (ISCT) (Dominici et al. 2006). These cells were spindle-shaped and had a fibroblast-like morphology as shown in Fig. 2b. The ADMSCs could adopt adipogenic (Fig. 2c), osteogenic (Fig. 2d), and chondrogenic (Fig. 2e) phenotypes. The undifferentiated cells were negative for staining, as well.

Flow cytometry analysis of CD105+ cell populations derived from ADMSCs and their differentiation. ADMSCs processed by a magnetic cell sorter to isolate CD105+ cells. The Y-axis is the number of cells analyzed in gated 104 cells and the X-axis represents staining intensity. After isolation by MACS, more than 99.99% of the cells were CD105+, 99.95% CD90+, 99.93% CD146+ and they were negative for the CD34 hematopoietic marker (a). ADMSCs culture in DMEM low glucose medium after 1 week without induction (b). Oil Red staining shows adipogenic differentiation of neutral lipids in the differentiated cells (c). Alizarin Red staining pointed out Osteogenesis of ADMSCs (d). Chondrogenic differentiation was demonstrated by Toluidine Blue staining (e). Scale bar = 20 μm
Neuronal Phenotypes of Treated ADMSCs
Human ADMSCs (Fig. 3a, b), treated by the MN culture medium, showed dramatic changes in cell shape from a flat cell body to a round cell body with neurite-like processes, similar to embryoid body-like aggregates (Fig. 3c), neurosphere-like structures (Fig. 3d), and finally mature MN-like cells (Fig. 3e, f). On day 7, the number of shiny cells increased in the culture, and their structure became more distinct after 24 h, forming neural rosettes (Fig. 3c). These cells showed a typical neuronal morphology with extended neurites, a shining nucleus, and a retracted cytoplasm. Neurospheres were formed from EBs as compact bodies in comparison with their mother cells during 3 days (Fig. 3d). MN-like cells were induced in the presence or absence of PNA in the induction media (Fig. 3e, f).

Human ADMSC culture and differentiation to MNs. ADMSCs 1 day after culture in passage 1 (a). ADMSCs first day of culture in the second passage (P2) (b). Embryoid body formation after 4 days of culture in differentiation medium (c). Neurosphere-derived from EB, one week after induction (d). Terminal differentiation of neurospheres into MN-like cells treated with the PNA (e) and without the PNA (f). Scale bar = 10 μm
Immunocytochemistry of PNA-, RA-, and Shh-Induced MN Differentiation
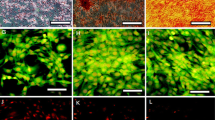
Nestin, as a neural progenitor cell marker, was detected at a high positive rate after differentiation (Fig. 4a). After placing the ADMSC culture in the differentiation medium for 5 days, the cytoplasm of many ADMSCs was retracted toward the cell nuclei, producing contracted cell bodies with extended neurites. These bodies became more spherical and showed a neuronal cell shape. Also, the immunocytochemistry results showed that the differentiated cells were positive for β-III-tubulin (Fig. 4b). These neuron-like cells continued to develop, and axon outgrowth was confirmed after 10 days (Fig. 4c). Simultaneously, neuron-like cells began to express NSE (Fig. 4d) and ChAT (Fig. 4e, f), which are mature MN markers expressed abundantly in the nucleus and proximal axons. Also, β-III-tubulin (Fig. 4g) continued to be expressed after 14 days. All these characteristics of differentiated ADMSCs are similar to human MNs.

Expression of neuronal cell markers on the MNs differentiated from ADMSCs. Immunostaining of nestin (a), and β-III tubulin (b) after 3 days of culture. Expression of β-III tubulin continued on the day 7 and neurosphere formation (c). Expression of neuron-specific enolase (NSE) (d) and ChAT (e) in MN-like cells differentiated from ADMSCs treated by MN induction media supplemented with PNA. The merged image of NSE/ChAT expression (f). Expression of β-III tubulin continued on the day 14 and MN-like cells maturation (g). The green color indicates fluorescein isothiocyanate (FITC) staining, the red color indicates Alexa-fluor, and the blue one indicates 4,6-diamidino-2-phenylindole (DAPI) staining. Scale bar = 20 μm
Real-Time PCR (RT-PCR) Assay
The RT-PCR assay was performed on PNA-treated (Fig. 5a) and PNA-untreated (Fig. 5b) samples on days 0, 1, 5, 10, and 15 of differentiation (Fig. 5a, b). The undifferentiated ADMSCs only expressed Pax6 as the spinal cord progenitor marker. When RA and Shh were added to the induction medium, the cells expressed Olig2 and Nkx2.2 genes, as markers of ventral-neural spinal cord progenitors. Also, Nkx2.2 is involved in the morphogenesis of the central nervous system. On day 5, Pax6, Olig2, and Nkx2.2 genes were continuously expressed. On the other hand, the expression of Hb9, as a mature MN marker, was very low from day 5. On day 15, the differentiated cells were positive for Olig2 and Hb9 but negative for nestin and Pax6. The expression of nestin decreased in the last stages of differentiation (Fig. 5a).

Regulation of MN markers and SMN2 expression. The expression of neuronal genes in different stages of the differentiation process after treatment with/without PNA (a). Except for nestin and pax6, all other neuronal markers expressed up to 15 days in both treatment groups (*p ≤ 0.05; *p ≤ 0.005). RT-PCR shows 5.6-fold higher expression of the SMN2 gene after PNA treatment (p ≤ 0.05). The data were obtained from three different and independent experiments (b). Western blot assay of SMN2 protein in the treated and untreated groups (c). The data are representative of at least three independent experiments. GAPDH was used as an internal control
On the other hand, the expression of ChAT on the final days increased remarkably, compared to days 5 and 10. Overexpression of this MN-specific gene was evident on the final days. PNA treatment showed ChAT and Isl1 overexpression in the differentiated cells (Fig. 5a). The expression levels of ChAT and Isl1 were respectively 71 and 76% higher in PNA treatment, compared to only RA and Shh treatments (P > 0.05). According to our results, expression of ChAT, Hb9, and Isl1 genes increased significantly during differentiation (P ≤ 0.05), and PNA overexpressed these markers. Also, expression of nestin and Pax6 was downregulated following PNA induction (Fig. 5a). These results revealed that treatment with PNA could improve efficient differentiation of ADMSCs into MN-like cells with overexpression of key MN factors.
Effects of PNA Treatment on SMN2 Expression
RT-PCR Assay
After treatment of ADMSC cultures with the MN induction media in the presence or absence of PNA, RT-PCR assay was carried out under quantitative conditions, using primers at exons 6 and 8 of SMN2. The FL-SMN2 and Δ7SMN2 transcripts, as well as their ratios, were calculated relative to GAPDH as the internal control. Our findings clearly showed that PNA treatment results in a 5.6-fold increase (P ≤ 0.05) in FL-SMN2 mRNA expression (Fig. 5c). PNA-untreated cells expressed SMN2 4.8 times more than ADMSCs (P ≤ 0.05; Fig. 5c). Also, there was a 1.2-fold increase in PNA overexpression of SMN2 genes in comparison with PNA-untreated cultures (P ≤ 0.05; Fig. 5c). Also, a 3.4-fold increase was observed in the Δ7SMN2 ratio (P ≤ 0.05; Fig. 5c).
Western Blot Analysis
The SMN2 protein expression was measured in MN-like cells, differentiated with or without PNA. The Western blot analysis using high-resolving SDS gel indicated very slight expression of SMN2 genes in ADMSCs (Fig. 5c). The SMN2 genes were expressed in mature MN-like cells, differentiated from ADMSCs without PNA induction (Fig. 5c); this finding confirms our RT-PCR results. The PNA-treated MNs even expressed more SMN2 genes, compared to PNA-untreated MN-like cells (Fig. 5c). These results confirm the expression of SMN2 genes at translational levels.
Viability of Cells
The MTT assay was performed to investigate the viability of cells, cultured in different stages at 1, 3, 5, 7, and 14 days after induction with differential media (Fig. 1). ADMSCs, which were cultured in DMEM without any induction factors, showed higher viability than cells cultured in the complex media (Fig. 6a; P ≤ 0.05). On days 5, 7, and 14, the viability of cells continued to decrease step by step as a consequence of adding factors, such as RA, forskolin, and Shh (P ≤ 0.05; Fig. 6a). On the other hand, PNA significantly increased the MN survival (P ≤ 0.05). Survival of MN-like cells in the presence or absence of PNA was measured by the percentage of originally plated cells every 24 h for 7 days (Fig. 6b). Based on the findings, the survival rate of differentiated MN-like cells in PNA-treated cells was 16% higher than that of PNA-untreated cells after the seventh day (P > 0.05).

Changes in the MNs cell viability after differentiation process. MTT shows the MN-like cells viability rate in comparison with the undifferentiated ADMSCs. There was a significant difference in the cell survival time between the treatment and control groups. Results are expressed based on the percentage of survival in the control group (mean S.E.M., *p ≤ 0.05). a The viability of two groups of MN-like cells measured by the percentage of originally plated cells with/without PNA treatment that shows the protective effect of PNA in the cell survival. PNA increased differentiated MN-like cell survival by 16% after 1 week (*p ≤ 0.05; **p ≤ 0.005) (b). The data were obtained from three different and independent experiments
Discussion
The main goal of this study was to evaluate the potential of PNA in the upregulation of SMN2 gene in MN-like cells differentiated from ADMSCs. Also, we confirmed the stimulation of Shh signaling pathway (Ebrahimi-Barough et al. 2017; Oh et al. 2009) by PNA to promote efficient differentiation of ADMSCs to MN-like cells.
Stem cell transplantation is a promising approach for improving the condition of patients with MN diseases, such as SMA, as it results in cell replacement or activation of molecular mechanisms, which promote endogenous MN function and neuroprotective activities against MN degeneration. Recently, several therapies have been developed for SMA, including gene therapy using antisense oligonucleotides (Spinraza or nusinersen) (Maruyama et al. 2018); however, there are still questions about their ease of delivery and cost-effectiveness (Seo et al. 2013; Richardson 2018). Cellular therapies may compensate for the lack of SMN gene expression in these patients.
In 2009, the California Stem Cell Institute (USA) announced a pre-investigational new drug (IND) meeting with the US Food and Drug Administration (FDA) for a phase I/II trial to inject human-grade MNs in SMA patients (Lunn et al. 2011). MNs are proposed to reduce SMA symptoms through cell replacement and muscle innervation. It is assumed that neuronal support, combined with the overexpression of SMN2 genes, may offer an effective strategy for SMA treatment, as four SMN2 copies are associated with a mild SMA phenotype (Wirth et al. 2006), and people with five or more copies may remain clinically asymptomatic (Sproule and Kaufmann 2010).
Adult stem cells are multipotent cells, derived from various adult tissues, such as adipose tissues. ADMSCs are suitable candidates for cell therapies owing to their intrinsic potential for self-renewal and differentiation into mesodermal and non-mesodermal lineages (Amirkhani et al. 2016). They exhibit low immunogenicity, which can be applied in allogeneic cell replacement therapies. Also, they can be proper candidates for investigating MNs in in vitro models. A number of protocols have been developed, including various steps for differentiation of stem cells into MNs. However, these protocols have not focused on the amount of SMN gene expression in differentiated MNs.
RA, in combination with Shh, is commonly used for the induction of transcription factors in MN differentiation (Wu et al. 2012). In our study, PNA was used to upregulate SMN gene expression (Fig. 5c), but we found that it could accelerate the differentiation process and increase MN markers, as well (Fig. 3e, f). In 2013, Liu et al. induced the differentiation of umbilical cord MSCs into MN-like cells at a very low efficiency (Liu et al. 2013). Additionally, in 2015, Joghataei et al. differentiated Wharton’s jelly-derived and chorion-derived MSCs into MN-like cells in 2D and 3D cultures (Bagher et al. 2015; Faghihi et al. 2015). Darvishi et al. also reported the differentiation of neural stem cell-derived ADMSCs into MN-like cells (Darvishi et al. 2017b).
The discussed studies reported differences in the timing and efficiency of MSC differentiation into MNs. These differences were attributed to variations in the main principles of the protocols (e.g., concentration and time of adding specific factors), besides minor differences in factors, such as cellular density or precise medium composition in stem cell cultures. Moreover, stem cells from each individual have unique characteristics with respect to the secretion profile and terminal differentiation capacity. Our differentiation protocol with PNA only took 3 weeks to produce mature MN-like cells from ADMSCs (Fig. 3e, f), which also influenced SMN2 gene expression (Fig. 5b, c).
The PI3K/Akt signaling pathway is a signal transduction pathway, and its activation is crucial to protecting neurons against apoptosis (Guo et al. 2007). In our experiment, PNA promoted MN differentiation and survival by protecting MNs against apoptosis; it also upregulated SMN2 genes. The mechanism of PNA in the activation of gene transcription may be related to the activation of endogenous PI3K/Akt pathway. Previously, Ashcroft et al. demonstrated that inducible activation of endogenous PI3K results in efficient nerve growth factor-mediated survival and neurite outgrowth in induced pluripotent stem cell (iPSC)-derived MNs (Ashcroft et al. 1999).
In addition, López-Carballo et al. suggested that the PI3K/Akt signaling pathway is important in the regulation of RA-induced neuronal survival of human cells (López-Carballo et al. 2002). In another study, Zhang et al. showed that the PI3K/Akt signaling pathway contributed to neuronal survival and neurite outgrowth, induced by methyl 3,4-dihydroxybenzoate (Zhang et al. 2015). Furthermore, Allodi et al. stated that IGF-2 treatment activated the PI3K/Akt signaling pathway in MNs, as evidenced by the increased level of phosphorylated Akt and glycogen synthase kinase-3β (GSK3β) phosphorylation, which inhibits GSK3 activity (Allodi et al. 2016). This finding is in agreement with a study by Yang et al. in 2013, which identified a GSK inhibitor as a survival factor for iPSC-derived MNs (Yang et al. 2013). Also, PI3K/Akt signaling is related to MAPK/ERK signaling pathways (Zhou et al. 2015).
Interactions between the PI3K/Akt and MAPK/ERK1/2 pathways may occur at different stages of cell cycle, producing a variety of consequences. In some cases, the ERK1/2 pathway is inhibited by Akt via direct phosphorylation and inhibition at the Raf-1 node (Zhou et al. 2015). Based on some reports, the MEK/ERK pathway potentiates growth factor-induced Akt phosphorylation, and ERK activation results in the attenuation of Akt phosphorylation (Hayashi et al. 2008). In addition, it was revealed that PI3-kinase activity is required for enhanced Akt phosphorylation, induced by the inhibition of ERK pathway (Hayashi et al. 2008).
Although AKT and ERK signaling pathways are often simultaneously activated in response to growth factors and hormones (Rodriguez-Viciana et al. 1997), their putative negative cross-talks lead to the control of neuron survival (Subramanian et al. 2005). In such cases, activation of the AKT pathway induces ERK pathway inhibition. It is interesting to note the role of ERK pathway in MN function (Hollis et al. 2009). The ERK pathway, which is constitutively overactivated in the spinal cord of SMA mouse models (Biondi et al. 2010), plays a repressive role in SMN2 gene transcription (Branchu et al. 2013).
Moreover, ERK inhibition in SMA myotubes leads to an increase in SMN production, suggesting the repressive role of the ERK pathway in SMN2 gene transcription (Also-Rallo et al. 2011). Therefore, PNA can affect SMN expression via inhibiting ERK signals through its PI3 kinase activity. Strategies, which upregulate SMN2 transcripts, can stabilize neuromuscular junctions in SMA patients and preclinical animal models.
In 2008, Gavrilina et al. revealed that the SMN transgene could correct the phenotype of SMN-deficient mice (Gavrilina et al. 2008) and even increase survival (Le et al. 2000; Monani et al. 2000). In our differentiation process, the immunocytochemistry and RT-PCR data represented the highest expression of differentiation markers of MNs in cultured cells with PNA. The heterogeneity of cell population in terms of nestin expression (Fig. 4a) decreased and disappeared upon differentiation through formation of spheroids on laminin-coated flasks (Fig. 5a), which are normally used to enhance neuronal cell attachment.
Cellular aggregation also induces ectoderm-specific genes, such as β-III tubulins, which separate neurons from glial cells, with continuous expression after 2 weeks of differentiation (Fig. 4b, c, g). Also, choline acetyltransferase, as an MN-specific marker, converts acetyl-CoA to choline. Considering its great importance, expression of ChAT was evaluated via both immunochemistry staining (Fig. 4e, f) and RT-PCR assay (Fig. 5a). The PCR cycles were terminated before saturation of amplification and normalized to GAPDH mRNA expression. The ChAT mRNA expression of ADMSCs was not significant, whereas it increased about twofold after differentiation to MNs (P ≤ 0.05; Fig. 5a). Also, it was overexpressed by 3.8-folds (P ≤ 0.05) when treated with the medium-containing PNA (Fig. 5b).
We also evaluated the expression of neuron-specific enolase (NSE), a marker of MN, which shows the regeneration of neuronal cells (Fig. 4d, f). NSE was expressed from the second week of differentiation. It may show neurotrophic function, as it controls neuronal survival, differentiation, and neurite regeneration via activation of PI3K and MAPK signaling pathways (Haque et al. 2018).
Optimization of MN differentiation depends on the development of effective methods for MN quantification in cultures. In 2013, Amoroso et al. analyzed Hb9 and ISL1 gene expression in developing human spinal cords (Liqing et al. 2011). The Hb9 and ISL1 genes were expressed only in subsets of spinal MNs. To prevent underestimation of MN formation, combined expression of both markers was measured; therefore, we used these two markers, as well. Hb9 gene is an essential factor in the development of MNs (Liqing et al. 2011). Hb9 was not expressed in the control or ADMSC group. Despite the efficient differentiation of MN-like cells, the resulting neuron-like cells express HB9 and Isl1 genes in a pattern similar to that observed in human MNs in vivo (Brichta et al. 2003).
Our cell viability MTT assay and direct observation of plated cells showed that the differentiation process decreased cell survival by up to 50% (*P ≤ 0.05) after 14 days of culturing (Fig. 6a). In the differentiation groups, survival of ADMSCs cultured with PNA was 16% higher than that of cells cultured without PNA (P ≤ 0.05). A PNA concentration range of 5–500 ng/mL was used to find the optimal dose of this steroid for the MN-like cell differentiation process. Finally, 50 ng/mL of PNA was confirmed as the optimal dose in our experiments. This concentration of PNA increased neuron-like cell viability, as shown in Fig. 6b (*P ≤ 0.05). Overall, antiapoptotic genes, activated after PNA administration, might protect MNs against cell death.
Conclusion
The results of this study showed that PNA can be used for differentiation of MSCs into MN phenotypes. Expression of MN markers, such as ChAT, Isl-1, and NSE genes, at transcriptional and translational levels showed that PNA could promote efficient differentiation of MSCs into MN-like cells with upregulation of SMN2 genes. This finding introduces a novel strategy for deriving MNs from MSCs for modeling research, preclinical applications, and human clinical trials in near future.
References
Allodi I, Comley L, Nichterwitz S, Nizzardo M, Simone C, Benitez JA, Cao M, Corti S, Hedlund E (2016) Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci Rep 6:25960
Also-Rallo E, Alías L, Martínez-Hernández R, Caselles L, Barceló MJ, Baiget M, Bernal S, Tizzano EF (2011) Treatment of spinal muscular atrophy cells with drugs that upregulate SMN expression reveals inter- and intra-patient variability. Eur J Hum Genet 19(10):1059–1065
Amirkhani MA, Mohseni R, Soleimani M, Shoae-Hassani A, Nilforoushzadeh MA (2016) A rapid sonication based method for preparation of stromal vascular fraction and mesenchymal stem cells from fat tissue. Bioimpacts 6(2):99–104
Ashcroft M, Stephens RM, Hallberg B, Downward J, Kaplan DR (1999) The selective and inducible activation of endogenous PI3-kinase in PC12 cells results in efficient NGF-mediated survival but defective neurite outgrowth. Oncogene 18:4586–4597
Bagher Z et al (2015) Differentiation of Wharton’s jelly-derived mesenchymal stem cells into motor neuron-like cells on three-dimensional collagen-grafted nanofibers. Mol Neurobiol 53:2397–2408
Bahrami N, Bayat M, Mohamadnia A, Khakbiz M, Yazdankhah M, Ai J, Ebrahimi-Barough S (2017) Purmorphamine as a Shh signaling activator small molecule promotes motor neuron differentiation of mesenchymal stem cells cultured on nanofibrous PCL scaffold. Mol Neurobiol 54(7):5668–5675
Bertini E, Mercuri E (2018) Motor neuron disease: a prospective natural history study of type 1 spinal muscular atrophy. Nat Rev Neurol 14(4):197–198
Biondi O, Branchu J, Sanchez G, Lancelin C, Deforges S, Lopes P, Pariset C, Lecolle S, Cote J, Chanoine C, Charbonnier F (2010) In vivo NMDA receptor activation accelerates motor unit maturation, protects spinal motor neurons, and enhances SMN2 gene expression in severe spinal muscular atrophy mice. J Neurosci 30(34):11288–11299
Branchu J, Biondi O, Chali F, Collin T, Leroy F, Mamchaoui K, Makoukji J, Pariset C, Lopes P, Massaad C, Chanoine C, Charbonnier F (2013) Shift from extracellular signal-regulated kinase to AKT/cAMP response element-binding protein pathway increases survival-motor-neuron expression in spinal-muscular-atrophy-like mice and patient cells. J Neurosci 33(10):4280–4294
Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B (2003) Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet 12(19):2481–2489
Catalan RE, Aragones MD, Godoy JE, Martinez AM (1984) Ecdysterone induces acetylcholinesterase in mammalian brain. Comp Biochem Physiol C 78(1):193–195
Chen YC, Chang JG, Jong YJ, Liu TY, Yuo CY (2015) High expression level of Tra2-beta1 is responsible for increased SMN2 exon 7 inclusion in the testis of SMA mice. PLoS One 10(3):e0120721
Constantino S, Santos R, Gisselbrecht S, Gouilleux F (2001) The ecdysteroid inducible gene expression system: unexpected effects of muristerone A and ponasterone A on cytokine signalling in mammalian cells. Eur Cytokine Netw 12:365–367
Darvishi M, Tiraihi T, Mesbah-Namin SA, Delshad AR, Taheri T (2017a) Motor neuron transdifferentiation of neural stem cell from adipose-derived stem cell characterized by differential gene expression. Cell Mol Neurobiol 37(2):275–289
Darvishi M et al (2017b) Motor neuron transdifferentiation of neural stem cell from adipose-derived stem cell characterized by differential gene expression. Cellular and Molecular Neurobiology 37(2):275–289
De Sanctis R et al (2018) Clinical phenotypes and trajectories of disease progression in type 1 spinal muscular atrophy. Neuromuscul Disord 28(1):24–28
Dominici M, le Blanc K, Mueller I, Slaper-Cortenbach I, Marini FC, Krause DS, Deans RJ, Keating A, Prockop DJ, Horwitz EM (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8(4):315–317
Du ZW et al (2015) Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat Commun 6:6626
Ebrahimi-Barough S, Hoveizi E, Yazdankhah M, Ai J, Khakbiz M, Faghihi F, Tajerian R, Bayat N (2017) Inhibitor of PI3K/Akt signaling pathway small molecule promotes motor neuron differentiation of human endometrial stem cells cultured on electrospun biocomposite polycaprolactone/collagen scaffolds. Mol Neurobiol 54(4):2547–2554
Faghihi F, Mirzaei E, Ai J, Lotfi A, Sayahpour FA, Barough SE, Joghataei MT (2016) Differentiation potential of human chorion-derived mesenchymal stem cells into motor neuron-like cells in two- and threedimensional culture systems. Mol Neurobiol 53(3):1862–1872. https://doi.org/10.1007/s12035-015-9129-y
Gavrilina TO, McGovern VL, Workman E, Crawford TO, Gogliotti RG, DiDonato CJ, Monani UR, Morris GE, Burghes AHM (2008) Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum Mol Genet 17:1063–1075
Guo JS, Zeng YS, Li HB, Huang WL, Liu RY, Li XB, Ding Y, Wu LZ, Cai DZ (2007) Cotransplant of neural stem cells and NT-3 gene modified Schwann cells promote the recovery of transected spinal cord injury. Spinal Cord 45(1):15–24
Hao le T et al (2012) Survival motor neuron affects plastin 3 protein levels leading to motor defects. J Neurosci 32(15):5074–5084
Haque A, Polcyn R, Matzelle D, Banik NL (2018) New insights into the role of Neuron-Specific Enolase in neuro-inflammation, neurodegeneration, and neuroprotection. Brain Sci 18;8(2). https://doi.org/10.3390/brainsci8020033
Hayashi H, Tsuchiya Y, Nakayama K, Satoh T, Nishida E (2008) Down-regulation of the PI3-kinase/Akt pathway by ERK MAP kinase in growth factor signaling. Genes Cells 13(9):941–947
Hollis ER, Jamshidi P, Low K, Blesch A, Tuszynski MH (2009) Induction of corticospinal regeneration by lentiviral trkB-induced Erk activation. Proc Natl Acad Sci U S A 106(17):7215–7220
Groen JNE et al (2018) Temporal and tissue-specific variability of SMN protein levels in mouse models of spinal muscular atrophy. Hum Mol Genet 27(16):2851–2862
Kostova FV, Williams VC, Heemskerk J, Iannaccone S, DiDonato C, Swoboda K, Maria BL (2007) Spinal muscular atrophy: classification, diagnosis, management, pathogenesis, and future research directions. J Child Neurol 22(8):926–945
Lafont R, Dinan L (2003) Practical uses for ecdysteroids in mammals including humans: an update. J Insect Sci 3:7
Le TT et al (2000) The survival motor neuron (SMN) protein: effect of exon loss and mutation on protein localization. Neurogenetics 3(1):7–16
Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80(1):155–165
Liqing Y, Jia G, Jiqing C, Ran G, Fei C, Jie K, Yanyun W, Cheng Z (2011) Directed differentiation of motor neuron cell-like cells from human adipose-derived stem cells in vitro. Neuroreport 22(8):370–373
Liu X, Li D, Jiang D, Fang Y (2013) Acetylcholine secretion by motor neuron-like cells from umbilical cord mesenchymal stem cells. Neural Regen Res 8:2086–2092
López-Carballo G, Moreno L, Masiá S, Pérez P, Barettino D (2002) Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway by retinoic acid is required for neural differentiation of SH-SY5Y human neuroblastoma cells. J Biol Chem 277:25297–25304
Lorson CL, Rindt H, Shababi M (2010) Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet 19(R1):R111–R118
Lunn JS, Sakowski SA, Federici T, Glass JD, Boulis NM, Feldman EL (2011) Stem cell technology for the study and treatment of motor neuron diseases. Regen Med 6(2):201–213
Maruyama R, Touznik A, Yokota T (2018) Evaluation of exon inclusion induced by splice switching antisense oligonucleotides in SMA patient fibroblasts. J Vis Exp 11(135). https://doi.org/10.3791/57530
Monani UR, Coovert DD, Burghes AH (2000) Animal models of spinal muscular atrophy. Hum Mol Genet 9(16):2451–2457
Noureddini M, Verdi J, Mortazavi-Tabatabaei SA, Sharif S, Azimi A, Keyhanvar P, Shoae-Hassani A (2012) Human endometrial stem cell neurogenesis in response to NGF and bFGF. Cell Biol Int 36(10):961–966
Oehme I, Bosser S, Zornig M (2006) Agonists of an ecdysone-inducible mammalian expression system inhibit Fas Ligand- and TRAIL-induced apoptosis in the human colon carcinoma cell line RKO. Cell Death Differ 13(2):189–201
Oh S, Huang X, Liu J, Litingtung Y, Chiang C (2009) Shh and Gli3 activities are required for timely generation of motor neuron progenitors. Dev Biol 331(2):261–269
Ojeda L, Gao J, Hooten KG, Wang E, Thonhoff JR, Dunn TJ, Gao T, Wu P (2011) Critical role of PI3K/Akt/GSK3beta in motoneuron specification from human neural stem cells in response to FGF2 and EGF. PLoS One 6(8):e23414
Okada M, Ishihara K, Sasa M, Izumi R, Yajin K, Harada Y (1998) Enhancement of GABA-mediated inhibition of rat medial vestibular nucleus neurons by the neurosteroid 20-hydroxyecdysone. Acta Otolaryngol 118(1):11–16
Rahmani A et al (2013) Neurogenesis and increase in differentiated neural cell survival via phosphorylation of Akt1 after fluoxetine treatment of stem cells. Biomed Res Int 2013:582526
Richardson RC (2018) Cost-effectiveness of Nusinersen for spinal muscular atrophy. JAMA Pediatr 172:701
Riddiford LM, Cherbas P, Truman JW (2000) Ecdysone receptors and their biological actions. Vitam Horm 60:1–73
Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J (1997) Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell 89(3):457–467
Seo J, Howell MD, Singh NN, Singh RN (2013) Spinal muscular atrophy: an update on therapeutic progress. Biochim Biophys Acta 1832(12):2180–2190
Shoae-Hassani A, Behfar M, Mortazavi-Tabatabaei SA, Ai J, Mohseni R, Hamidieh AA (2017) Natural killer cells from the subcutaneous adipose tissue underexpress the NKp30 and NKp44 in obese persons and are less active against major histocompatibility complex class I non-expressing neoplastic cells. Front Immunol 8:1486
Sproule DM, Kaufmann P (2010) Therapeutic developments in spinal muscular atrophy. Ther Adv Neurol Disord 3(3):173–185
Subramanian C, Opipari AW, Castle VP, Kwok RPS (2005) Histone deacetylase inhibition induces apoptosis in neuroblastoma. Cell Cycle 4(12):1741–1743
Thonhoff JR, Ojeda L, Wu P (2009) Stem cell-derived motor neurons: applications and challenges in amyotrophic lateral sclerosis. Curr Stem Cell Res Ther 4(3):178–199
Tsujiyama S, Ujihara H, Ishihara K, Sasa M (1995) Potentiation of GABA-induced inhibition by 20-hydroxyecdysone, a neurosteroid, in cultured rat cortical neurons. Jpn J Pharmacol 68(1):133–136
Turner BJ, Alfazema N, Sheean RK, Sleigh JN, Davies KE, Horne MK, Talbot K (2014) Overexpression of survival motor neuron improves neuromuscular function and motor neuron survival in mutant SOD1 mice. Neurobiol Aging 35(4):906–915
Verdi J, Mortazavi-Tabatabaei SA, Sharif S, Verdi H, Shoae-Hassani A (2014a) Citalopram increases the differentiation efficacy of bone marrow mesenchymal stem cells into neuronal-like cells. Neural Regen Res 9(8):845–850
Verdi J, Sharif S, Banafshe HR, Shoae-Hassani A (2014b) Sertraline increases the survival of retinoic acid induced neuronal cells but not glial cells from human mesenchymal stem cells. Cell Biol Int 38(8):901–909
Wirth B, Brichta L, Schrank B, Lochmüller H, Blick S, Baasner A, Heller R (2006) Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet 119(4):422–428
Wu CY et al (2012) Efficient differentiation of mouse embryonic stem cells into motor neurons. J Vis Exp 9(64):e3813
Yang YM, Gupta SK, Kim KJ, Powers BE, Cerqueira A, Wainger BJ, Ngo HD, Rosowski KA, Schein PA, Ackeifi CA, Arvanites AC, Davidow LS, Woolf CJ, Rubin LL (2013) A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell 12(6):713–726
Zerres K, Rudnik-Schöneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I (1997) A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci 146(1):67–72
Zhang Z, Cai L, Zhou X, Su C, Xiao F, Gao Q, Luo H (2015) Methyl 3,4-dihydroxybenzoate promote rat cortical neurons survival and neurite outgrowth through the adenosine A2a receptor/PI3K/Akt signaling pathway. Neuroreport 26:367–373
Zheleznyakova GY, Kiselev AV, Vakharlovsky VG, Rask-Andersen M, Chavan R, Egorova AA, Schiöth HB, Baranov VS (2011) Genetic and expression studies of SMN2 gene in Russian patients with spinal muscular atrophy type II and III. BMC Med Genet 12:96
Zhou J, Du T, Li B, Rong Y, Verkhratsky A, Peng L (2015) Crosstalk between MAPK/ERK and PI3K/AKT signal pathways during brain ischemia/reperfusion. ASN Neuro 6;7(5). https://doi.org/10.1177/1759091415602463
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The fat tissue was obtained after proper consenting process and approvals from Tehran University of Medical Sciences ethics committee.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mohseni, R., Ashrafi, M.R., Ai, J. et al. Overexpression of SMN2 Gene in Motoneuron-Like Cells Differentiated from Adipose-Derived Mesenchymal Stem Cells by Ponasterone A. J Mol Neurosci 67, 247–257 (2019). https://doi.org/10.1007/s12031-018-1232-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-018-1232-x