
Abstract
Diabetic retinopathy (DR) is a devastating complication of diabetes with a prevalence rate of 35%, and no effective treatment options. Since the most visible clinical features of DR are microvascular irregularities, therapeutic interventions often attempt to reduce microvascular injury, but only after permanent retinal damage has ensued. However, recent data suggests that diabetes initially affects retinal neurons, leading to neurodegeneration as an early occurrence in DR, before onset of the more noticeable vascular abnormalities. In this review, we delineate the sequence of initiating events leading to retinal degeneration in DR, considering neuronal dysfunction as a primary event. Key molecular mechanisms and potential biomarkers associated with retinal neuronal degeneration in diabetes are discussed. In addition to glial reactivity and inflammation in the diabetic retina, the contribution of neurotrophic factors, cell adhesion molecules, apoptosis markers, and G protein signaling to neurodegenerative pathways warrants further investigation. These studies could complement recent developments in innovative treatment strategies for diabetic retinopathy, such as targeting retinal neuroprotection, promoting neuronal regeneration, and attempts to re-program other retinal cell types into functional neurons. Indeed, several ongoing clinical trials are currently attempting treatment of retinal neurodegeneration by means of such novel therapeutic avenues. The aim of this article is to highlight the crucial role of neurodegeneration in early retinopathy progression, and to review the molecular basis of neuronal dysfunction as a first step toward developing early therapeutic interventions that can prevent permanent retinal damage in diabetes. ClinicalTrials.gov: NCT02471651, NCT01492400
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes currently affects nearly half a billion people globally, and this number is projected to increase to 629 million by 2045, according to recent estimates by the International Diabetes Federation. The global healthcare cost of diabetes is now 727 billion US dollars per year, which is a tremendous economic burden on the society. USA spends the most on healthcare for people with diabetes, accounting for 48% of the global expenditure, followed by Europe which spends about 23% of the total expenditure (IDF diabetes atlas, eighth edition, 2017). While diabetes has already attained epidemic proportions worldwide with escalating economic costs, the prevalence of debilitating long-term complications associated with the disease such as retinopathy, nephropathy, neuropathy, and cardiovascular disease is also steadily increasing among diabetic patients. Retinopathy is the most common microvascular complication of diabetes and the chief cause of vision-impairment and blindness among working-age adults between 20 and 74 years of age (Yau et al. 2012). Risk factors associated with the development of diabetic retinopathy (DR) include hyperglycemia, dyslipidemia, and hypertension, as well as the duration of diabetes (Klein et al. 1984; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group et al. 2000; Kohner 2008; ACCORD Study Group et al. 2010; Frank 2014; Hammer and Busik 2017). Almost all type I diabetic patients and approximately 60% of type II patients develop retinopathy after 20 years of diabetes (Aiello et al. 1998; Fong et al. 2003). Despite such a high incidence of DR among diabetic patients, the condition tends to remain undiagnosed until permanent damage to the retina has already occurred; moreover, the focus of current therapy is on treating advanced stages of DR. Understanding the pathophysiology of DR is important for developing effective and early treatment options for this devastating complication of diabetes.
Pathophysiology of Diabetic Retinopathy
DR is usually characterized by microvascular abnormalities such as irregular blood vessels and microaneurysms, which lead to microhemorrhages (Antonetti et al. 2012). Such damaged blood vessels are prone to leak blood, lipids, and fluids in the retina, resulting in macular edema and blurry vision. These exudative changes further lead to ischemic changes, such as nerve fiber damage (observed as “cotton-wool spots”), formation of microvascular collaterals or shunt vessels, and dilatation of venules. The severity of DR is diagnosed by performing fundus photography and fluorescein angiography in patients and is clinically classified as mild, moderate, or severe based on the number of hemorrhages, microaneurysms, lipoprotein exudates, and microvascular abnormalities detected. In the later proliferative stage of DR, new and delicate capillaries emerge in the retina in an attempt to compensate for the obstruction of blood flow. These abnormal new vessels are very delicate and leak readily, causing edema. They may also extend into the vitreous body, leading to vitreous hemorrhage, and this bleeding is manifested as the presence of “floaters” or dark specks in the visual field (Ivanova et al. 2016). This leads to scar tissue formation on the retina, which along with the vitreous can contract, resulting in retinal tear or detachment, causing blindness if not repaired surgically (Williams et al. 2004). Impairment of vision can also occur through retinal vein occlusion, macular edema, or neovascular glaucoma; the discussion of which is beyond the scope of this review.
Neurodegeneration in the Diabetic Retina
Although vascular abnormalities are the most visible clinical features in DR, symptoms of neuropathy such as degeneration of the inner nuclear and ganglion cell layer, and pathological changes in the nerve fiber layer have been described in the early literature on DR (Wolter 1961; Bloodworth 1962). Over the years, structural as well as functional changes in neural elements have been documented in numerous studies in animal models of DR, diabetic patients, and post-mortem human retinas (Bresnick 1986; Falsini et al. 1989; Barber et al. 1998; Lieth et al. 2000; Abu-El-Asrar et al. 2004; Antonetti et al. 2006). We now know that diabetes impacts the complete neurovascular unit of the retina, leading to neuroinflammation, gliosis or activation of glial cells, and gradual neurodegeneration that occur before the more noticeable vascular pathologies (Abcouwer and Gardner 2014). In the normal retina, the retinal vascular endothelium interacts with neuronal glia and pericytes to control retinal blood flow in response to metabolic demand: a process termed autoregulation (Kur et al. 2012; Coughlin et al. 2017). Loss of neurovascular coupling in the diabetic retina leads to impaired autoregulation and may contribute to neuronal loss due to metabolic dysfunction. The retina is composed of distinct classes of neurons: ganglion cells, amacrine cells, horizontal cells, bipolar cells, and the light-sensitive photoreceptors (Fig. 1). These neurons are organized into specific layers within the retinal tissue and form a complex neural circuitry that transmits visual signals to the brain using graded electrical activity converted to action potentials. Assessment of neuroglial function by electroretinography (ERG) indicates that ganglion, bipolar, amacrine, and photoreceptor cell functions are altered by diabetes (Barber et al. 1998; Gastinger et al. 2006; Bui et al. 2009; Kern and Berkowitz 2015; Coughlin et al. 2017). Interestingly, a recent study of type 2 diabetic patients demonstrated thinning of the inner retina by high-resolution spectral-domain optical coherence tomography (SD-OCT) (Chhablani et al. 2015). Several groups have also demonstrated that diabetes leads to rapid and early apoptosis of retinal neurons, before the onset of vascular cell death (Martin et al. 2004; Barber et al. 2005; Kern et al. 2010). Further, ERG data accurately predicts the retinal location of future microvascular damage in diabetic patients. Together, these data support the theory that the onset of neurodegeneration in the retina is an early event and not the result of vascular damage (Ng et al. 2008; Harrison et al. 2011; Simó et al. 2012). The sequence of molecular changes believed to result in retinal degeneration in diabetes is outlined in Fig. 2.

Retinal section stained with hematoxylin and eosin, showing the structure of the mouse retina and localization of various types of neurons organized within specific layers in the retina. (GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; PL, photoreceptor layer)

Postulated sequence of molecular changes leading to retinal degeneration in diabetes: early on in diabetes, inflammatory markers such as TNF-α, IL-1β, IL-6, MCP-1, VEGF, ICAM-1, and VCAM-1 are increased partly due to activation of glial cells, and neuroprotective factors such as NGF, BDNF, and PEDF are reduced. This leads to neuronal dysfunction, apoptosis of retinal ganglion cells, and loss of retinal synapses. Neurodegenerative changes have been observed prior to vascular pathology, which is initially manifested as loss of pericytes and endothelial cells lining retinal capillaries, gradual breakdown of the blood-retinal barrier, and increased vascular permeability. Further increase in reactivity of retinal glial cells (Müller cells, astrocytes, microglia) is observed. Upregulation of cell adhesion molecules on retinal vasculature leads to increased leukostasis and infiltration of circulating monocytes, which contribute to the vicious cycle of releasing neurotoxins and inflammatory cytokines that damages the retinal tissue. Microaneurysms, hemorrhages, leakage of lipid exudates in the retinal tissue, and loss of functional capillaries occur, leading to blurry vision. If untreated, DR can progress to the proliferative stage, involving neovascularization, macular edema, vitreous hemorrhage, and scar tissue formation, eventually causing blindness. (TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; MCP-1, monocyte chemoattractant protein-1; VEGF, vascular endothelial growth factor; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cellular adhesion molecule-1; NGF, nerve growth factor; BDNF, brain-derived neurotrophic factor; PEDF, pigment epithelium-derived factor)
Factors Associated with Retinal Neuronal Degeneration
Inflammatory Cytokines and Neurotrophic Factors
In diabetes, the integrity of the blood-retinal barrier is compromised as a result of long-term hyperglycemia, leading to loss of autoregulation, changes in permeability of endothelial cells and pericytes lining retinal capillaries, and increased secretion of cytokines and growth factors. With breakdown of the normally tight blood-retinal barrier, chronic low-grade inflammation develops in the diabetic retina. Leukocytes migrate to the retina and adhere to retinal capillaries with the help of integrins CD11b/CD18 and CD11a. The integrins bind to cell adhesion molecules (CAMs) E-selectin, ICAM-1, and VCAM-1 on the capillary endothelial cells which subsequently leads to capillary obstruction and injury; further, death of pericytes and endothelial cells results in retinal capillary loss (McLeod et al. 1995; Miyamoto et al. 1999; Barouch et al. 2000; Joussen et al. 2001). Extensive studies have demonstrated the contribution of potent inflammatory cytokines and growth factors secreted by leukocytes and activated glial cells such as TNF-α, IL-1β, IL-6, IL-8, MCP-1, and VEGF toward propagating chronic inflammation in the early stages of DR (Aiello et al. 1994; Busik et al. 2008; Abcouwer 2013).
In contrast, some pro-survival neurotrophins such as brain-derived neurotrophic factor (BDNF) are believed to protect retinal ganglion and amacrine cells from degeneration (Johnson et al. 1986). Significantly, decreased levels of BDNF have been observed in diabetic patients as well as animal models (Ola et al. 2013; Guo et al. 2017). Nerve growth factor (NGF) has been shown to prevent apoptosis of Müller glia and neurons. By contrast, its precursor protein pro-NGF is believed to promote retinal neurodegeneration by activating pro-apoptotic pathways in retinal ganglion cells (Hammes et al. 1995; Mysona et al. 2014). The oxidative milieu of the diabetic retina impairs processing of pro-NGF to mature NGF which contributes to ganglion cell death, increased vascular permeability, and inflammation (Mysona et al. 2014). Pigment epithelium-derived factor (PEDF), a potent inhibitor of angiogenesis, is believed to protect neurons from light damage and oxidative stress. Some studies have reported a decrease in PEDF levels in the diabetic retina which may contribute to inflammation and vessel leakage (Zhang et al. 2006; Yoshida et al. 2009), while supplementation with PEDF reduces retinal vascular leakage in diabetic mice (Liu et al. 2012). Ciliary neurotrophic factor (CNTF) and fibroblast growth factor (FGF) have also been reported to promote survival of retinal neurons and protect ganglion cells from degeneration in animal models, possibly by preventing diabetes-induced apoptosis of neurons (Aizu et al. 2003; Unsicker 2013; Mathews et al. 2015; Guo and Liu 2017). Figure 3 summarizes the key factors believed to be associated with neuronal degeneration in the diabetic retina.

This review provides the readers an insight into novel perspectives on diabetic retinopathy, which consider neurodegeneration an early event before the onset of more visible microvascular abnormalities in the diabetic retina. Key molecular mechanisms underlying diabetic retinal neurodegeneration are discussed, such as imbalances in neurotrophins and cytokines, glial reactivity, changes in signaling pathways and cell adhesion molecules, activation of caspases, and mitochondrial dysfunction. Understanding the molecular basis of neuronal damage is the first step toward preventing permanent retinal damage in diabetes. (BDNF, brain-derived neurotrophic factor; NGF, nerve growth factor; PEDF, pigment epithelium-derived factor; CNTF, ciliary neurotrophic factor; EPO, erythropoietin; FGF, fibroblast growth factor; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; MCP-1, monocyte chemoattractant protein-1; VEGF, vascular endothelial growth factor; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cellular adhesion molecule-1; NCAM, neural cellular adhesion molecule; AIF, apoptosis inducing factor)
Glial Reactivity
In the healthy retina, glial cells help to maintain the integrity of the blood-retinal barrier and maintain tissue homeostasis. However, in diabetes, activation and proliferation of glia such as astrocytes, Müller cells, and microglia contributes to inflammation by secretion of pro-inflammatory mediators and neurotoxic factors (Rungger-Brändle et al. 2000). Müller cells normally help in maintaining neuronal health by recycling neurotransmitters, preventing neurotoxicity by removing excess glutamate, preventing accumulation of K+ ions by spatial buffering, participating in the retinoid cycle, and providing lactate as a fuel source for photoreceptors. Müller cell morphology is altered early on in diabetes, with enlargement of cell bodies and increased number of processes. They also demonstrate increased expression of a key marker for reactive gliosis: glial fibrillary acidic protein (GFAP) (Coughlin et al. 2017). Müller cells and astrocytes normally aid in autoregulation, provide neuronal support, and maintain ionic homeostasis in the retina. However, in early stages of diabetes, astrocyte cell numbers are decreased and their GFAP expression is markedly reduced (Rungger-Brändle et al. 2000). Although increased GFAP expression in Müller cells persists throughout DR while GFAP expression in astrocytes is seemingly downregulated, no studies have demonstrated any functional outcomes in connection with GFAP. However, these early changes in glial cells are coincident with reduced capillary density in the inner retina and functional deficits in ganglion cell responses (Ly et al. 2011).
Microglia play a critical role in initiating neural inflammatory responses. These normally dormant resident macrophages of the retina become activated in diabetes with retraction of their dendrites and contribute to neuronal damage by secreting neurotoxic factors (Colton and Wilcock 2010; Abcouwer 2013). Further, circulating monocytes are believed to contribute to inflammation by infiltrating the diabetic retina, transforming into microglia-like reactive cells, and leading to further proliferation of surrounding glial cells (Hinze and Stolzing 2011; Hu et al. 2013; Chakravarthy et al. 2016).
In the proliferative stage of retinopathy, activated glia, especially Müller cells contribute to the formation of fibrovascular scar tissue in the retina in an attempt to rebuild the injured tissue. Such gliotic scars prevent regrowth of neurons, compromise vision due to retinal detachment, and may ultimately lead to blindness (Roy et al. 2016). Major components of glial scar formation are extracellular matrix (ECM) glycoproteins such as collagen, fibronectin, chondroitin sulfate proteoglycans (CSPG), laminins, and tenascins (Ban and Twigg 2008; Barros et al. 2011). Increased synthesis and/or reduced degradation of specific ECM proteins have been shown to create an inhibitory environment that prevents axonal regeneration (Asher et al. 2001; Reinhard et al. 2017). Enzymatic digestion of CSPG reduces its inhibitory actions and promotes axonal regrowth and functional recovery in animal models of CNS injury (Bai et al. 2010; Pearson et al. 2018). These studies suggest that modulation of ECM proteins is likely to promote neuronal regeneration in the retina of diabetic individuals.
Markers of Neuronal Degeneration
Several studies have demonstrated significant reduction in the thickness of the inner plexiform layer of the retina in animal models of DR, indicating a progressive loss of neural dendrites and synapses in the inner retina (Barber et al. 1998, 2005). Structural remodeling of the ON-type ganglion cell dendrites, including changes in dendritic length and density, occurs in humans as well as animal models suggesting that a specific subset of retinal ganglia is altered by diabetes (Qin et al. 2006; Meyer-Rüsenberg et al. 2007; Gastinger et al. 2008). These pathological alterations presumably affect retinal function in diabetes, since differences in shape and size of the dendrites are believed to contribute to specificity of the visual response (Yang and Masland 1994; Brown et al. 2000). Recent studies in retinal neurons have demonstrated a critical role for CAMs in mediating cell-cell interactions, regulating dendritic morphogenesis, patterning of neuronal arbors, and maintaining synaptic specificity between the distinct types of neurons in the retina (Fuerst et al. 2008; Duan et al. 2014; Krishnaswamy et al. 2015). Focusing research attention toward CAMs may lead to better understanding of the immense diversity among retinal neuronal subtypes, which is crucial for developing neuronal regenerative therapies such as ganglion or photoreceptor cell replacement.
Neural cell adhesion molecule (NCAM), which is expressed on all neurons and glial cells of the retina, has a key role in maintaining neuronal plasticity and in preventing ganglion cell death due to retinal injury or aging (Murphy et al. 2007a). NCAM−/− mice demonstrate premature loss of visual acuity, impaired retinal function, and reduced survival of retinal neurons following injury (Luke et al. 2016a). In patients with neurodegenerative disorders such as Alzheimer’s disease, an increase in soluble NCAM is observed in serum and cerebrospinal fluid (Berezin 2010). NCAM is also believed to be involved in the modulation of pathological angiogenesis during proliferative retinopathy (Håkansson et al. 2011). These findings suggest that neuronal degeneration in the retina of diabetic individuals is likely associated with alterations in retinal and/or vitreal NCAM levels. The effect of diabetes on the function of CAMs, homophilic, and heterophilic interactions between different members of CAMs and their co-receptors on interneurons and retinal ganglia and downstream effect of these interactions on signal transduction within the neuronal cell interior are exciting new avenues that warrant further investigation.
Another set of molecular players are caspases which are known to function as central components in the apoptotic response of retinal ganglion cells. Caspases 1, 2, 6, 8, and 9 are activated as early as 8 weeks in diabetic mice (Mohr et al. 2002). Activities of caspase 1, 3, 4, 5, and 6 are increased at around 4 months after experimentally induced diabetes as well as in diabetic patients (Mohr et al. 2002; Thomas et al. 2017). Caspase 3 is regarded as a pivotal molecular player orchestrating apoptosis and bringing the cell to its demise. The pattern of caspase activation changes through the time course of diabetes and occurs as a gradual response to hyperglycemia. In general, levels of apoptotic markers such as caspase 1, 3, 6, 9, Fas, Bad, and Bax correlate highly with neuronal degeneration in DR models, as well as diabetic patients (Abu El-Asrar et al. 2007; Oshitari et al. 2010; Thomas et al. 2017).
Mitochondria play a crucial role in mediating caspase-independent apoptotic cell death, by the release of apoptogenic molecules such as apoptosis inducing factor (AIF) and cytochrome c (van Gurp et al. 2003; Schultz and Harrington 2003). Both photoreceptors and ganglion cells from diabetic patients express upregulated levels of AIF and increased immunoreactivity for cytochrome c, indicating mitochondrial dysfunction and caspase-independent apoptosis within the retina (Abu El-Asrar et al. 2007). Another molecule, a neural regeneration-associated membrane receptor expressed in retinal ganglia called Nogo receptor (NogoR), is also believed to mediate apoptosis of ganglion cells by activating the RhoA/Rock1 signaling pathway in diabetic animal models (Liu et al. 2014; Guo and Liu 2017).
Signaling Pathways in Neurodegeneration
Aberrant activation of a number of signaling cascades such as protein kinase C (PKC) and mitogen-activated protein kinase (MAPK) signaling has been implicated in structural and functional damage to retinal neurons in DR (Ahrén 2009). Increased activity of PKC-δ, an upstream regulator of the Akt cell survival pathway, was shown to contribute to retinal neuronal apoptosis in diabetic rats (Kim et al. 2008). Activation of p38 MAPK signaling has been reported in DR and is implicated in apoptosis of retinal ganglion cells, while its inhibition prevented the development of early stages of DR in a rat model (Kikuchi et al. 2000; Poulaki et al. 2002; Du et al. 2010).
Recent studies suggest a connection between G protein-coupled receptor (GPCR)-mediated signaling and diabetes (Du et al. 2015; Roy et al. 2017). Functional deterioration of G proteins, especially the inhibitory G protein (Gαi), has been observed in the diabetic rat retina (Kowluru et al. 1992). In another study, a receptor coupled to G protein, GPR91, was shown to accumulate in diabetic rat retinal ganglion cells and mediate retinal vascular damage via the MAPK signaling pathway (Li et al. 2014). The Gβγ heterodimer associates with the enzyme phosphoinositide 3-kinase (PI3K) which is an important mediator of insulin receptor signaling (Vadas et al. 2013). Furthermore, the activity of PI3K/Akt kinase pathway which is used by retinal neurons as a survival signal is reduced in diabetic rat retinas, leading to neuronal cell death (Reiter et al. 2006). GPCR-mediated regulation of neurodegenerative pathways in diabetes is a relatively new field and it is important to keep in mind the complexities of this system, such as post-translational regulation of G protein subunits which may be altered in diabetes (Kowluru 2010) or expression of additional modulatory factors that affect G protein signaling (Kowluru 2017). Some G protein isoforms may also compensate for the loss of another isoform; this was recently observed in a model of arterial thrombosis (Devanathan et al. 2015).
Current Treatment Strategies for Diabetic Retinopathy
Prevention and Control of DR
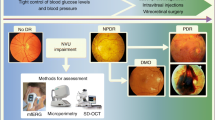
In the past, patients with diabetes had a very high risk of complete visual loss, due to a dearth of treatment options for DR, and poor control of blood glucose levels. Today, with the advent of intensive glycemic and blood pressure control, innovation in accurate monitoring of blood glucose levels, increased screening for DR and earlier diagnosis of diabetes, therapy for DR has improved dramatically. Numerous important clinical trials have demonstrated the advantages of tight control of blood glucose (with HbA1c < 7%) in the prevention of DR onset and decreasing DR progression (Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group et al. 2000; ACCORD Study Group et al. 2010). The results of the Diabetes Control and Complications Trial (DCCT) demonstrated that intensive insulin therapy significantly reduced the risk of development of DR by 75% and progression of DR by 50% after 6.5 years in type I diabetic patients, and the United Kingdom Prospective Diabetes Study (UKPDS) reported a 21% reduction in progression of DR in type II diabetic patients (Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group et al. 2000; Kohner 2008). However, highly intensive glycemic control (HbA1c of 6% or less) is not recommended since it may increase the risk of cardiovascular events and mortality due to hypoglycemia (Frank 2014).
Detection of Retinal Neurodegeneration
Clinically, DR is classified as a microvascular disease based on the observation and quantification of standard signs such as hemorrhages, microaneurysms, hard exudates, cotton-wool spots, retinal thickening, neovascularization, and other microvascular abnormalities by fundus photography, viewing only 20–70° of the retina (Wilkinson et al. 2003). However, functional, sub-clinical, and peripheral retinal changes are not incorporated in the conventional DR grading system. Novel methods such as ultra-widefield imaging allow up to 200° view of the retina in a single image, enabling correct estimation of DR severity (Kiss and Berenberg 2014). Multifocal ERG (mfERG) is another valuable technique which can be used by clinicians to predict the future location of clinical DR by mapping the precise location of visual dysfunction in the neuroretina (Ng et al. 2008; Harrison et al. 2011). This tool can help clinicians identify patients at risk of developing DR, as well as the nature and extent of retinal damage.
Typically, non-invasive techniques such as optical coherence tomography (OCT) are used for detecting total retinal thickness in cross-sectional images. Although commercial OCT devices have an axial resolution of 5–8 μm, smaller structural changes (0.5–1.0 μm) in the neuroretina can be measured using publicly available image analysis algorithms (www.iibi.uiowa.edu/content/shared-software-download) (van Dijk et al. 2011; Lynch and Abràmoff 2017). Recent developments such as the spectral-domain OCT (SD-OCT) technology allow clearer visualization of individual retinal layers with improved signal-to-noise ratio (de Boer et al. 2003). Studies using SD-OCT have demonstrated that substantial loss of neurons in patients, as indicated by reduced neuroretinal thickness, occurs very early in DR and in some cases, even in the absence of clinical DR (van Dijk et al. 2011; Chhablani et al. 2015; Sohn et al. 2016). The use of widefield technology, mfERG, and SD-OCT image analysis of the neuroretina by clinicians is essential for early detection, effective treatment, and monitoring of retinal neurodegeneration in diabetic patients.
Laser Photocoagulation
For several decades, treating patients having vision-threatening retinopathy with laser photocoagulation has been a standard therapeutic strategy for DR. The panretinal photocoagulation technique (PRP) burns the retinal tissue at several spots, sealing off leaky blood vessels and discouraging further vessel growth. This treatment has been demonstrated to reduce risk of severe impairment of sight due to abnormal vessel growth in macular degeneration and proliferative DR by approximately 50%. Although valuable, PRP may give rise to further problems like loss of peripheral vision and sub-retinal fibrosis, and it may worsen macula edema (Vander et al. 1991; Lövestam-Adrian and Agardh 2000).
Intravitreal Biologic Therapies
Recently, the management of proliferative DR has greatly benefited from a new class of biologics which directly inhibit pro-angiogenic vascular endothelial growth factor (VEGF) in the retina. Anti-VEGF antibodies, such as ranibizumab (Lucentis) and bevacizumab (Avastin), improve visual acuity in patients with macular edema and proliferative DR, as evidenced in randomized clinical trials (Nguyen et al. 2012; Brown et al. 2013; Osaadon et al. 2014; Schmidt-Erfurth et al. 2014). Despite improvements in visual acuity in the short term, the effectiveness of anti-VEGF injections for treatment of retinal diseases in the long term is still unclear. Approximately 30–40% of diabetic patients do not respond to anti-VEGF treatment, while many develop resistance over time after repeated anti-VEGF administration (Yang et al. 2016; Gonzalez et al. 2016). Follow-up studies have described complications such as increase in intraocular pressure, no significant reduction in central subfield thickness, persistence of macular edema, retinal atrophy, abnormal thinning of the retina, and suboptimal visual acuity in a substantial number of patients after 2–5 years of anti-VEGF treatment (Bressler et al. 2012; Comparison of Age-related Macular Degeneration Treatments Trials (CATT) Research Group et al. 2016; Gonzalez et al. 2016). In patients with severe proliferative DR, tractional retinal detachment may occur shortly following administration of intravitreal bevacizumab (Arevalo et al. 2008; Falavarjani and Nguyen 2013). Moreover, sustained inhibition of VEGF leads to neurodegeneration in animal models of DR, including loss of retinal ganglion, bipolar, and amacrine cells, likely due to loss of neurotrophic factors associated with VEGF depletion (Romano et al. 2012; Park et al. 2014; Hombrebueno et al. 2015). Additional data from clinical trials are needed to provide conclusive evidence regarding retinal neurodegeneration and other adverse outcomes of prolonged anti-VEGF treatment in DR.
Therapies such as intravitreal injection of corticosteroids or vitreoretinal surgery may also help reduce inflammation and prevent severe vision loss, but these approaches are associated with complications such as increased incidence of cataract and glaucoma (Diabetic Retinopathy Clinical Research Network et al. 2010; Stewart 2012).
Alternative Drug Delivery Systems
Currently, standardized long-term anti-VEGF therapy is not ideal since it requires patients to endure a high burden of healthcare visits with indefinite monthly rounds of invasive intravitreal injections (Blinder et al. 2017). It also poses a significant financial burden on patients and caregivers due to the requirement for special ophthalmic formulations, and a retina specialist to administer injections in the eye (Wallick et al. 2015). More effective methods of ophthalmic drug delivery, as well as innovative treatment strategies that target early neuroglial injury and help regenerate the retinal tissue, are clearly needed. In this context, the recent development of slow-release implants allows extended delivery of very low doses of a drug, thereby reducing many harmful side effects (Edelhauser et al. 2010). Such intravitreal implants that release dexamethasone are currently being evaluated in phase III and IV clinical trials for the treatment of diabetic macular edema, and preliminary data indicates favorable results in vision improvement (Boyer et al. 2014; Augustin et al. 2015), (ClinicalTrials.gov: NCT02471651, NCT01492400).
Topical administration of drugs such as somatostatin and glucagon-like peptide 1 (GLP-1) receptor agonists in the form of eye drops has been recently demonstrated to prevent retinal neurodegeneration in animal models of early DR (Hernández et al. 2013, 2016), and a phase II clinical trial (NCT01726075) in DR patients has shown promising initial outcomes in this direction (Simó 2017). Other routes of retinal drug delivery that are being tested in pre-clinical studies include therapeutic contact lenses containing hydrogels for molecular imprinting and mucoadhesive (poloxamer/chitosan) formulations (Gratieri et al. 2010; Hui et al. 2012).
Alternative Treatment Strategies
Apart from VEGF pathway, the angiopoietin-Tie receptor signaling pathway has been identified as an important regulator of blood-retinal barrier and vessel formation, thereby representing an alternative target for anti-angiogenic therapy in DR (Augustin et al. 2009). The concentrations of Ang-1 and Ang-2 are significantly high in animal models and DR patients, indicating an imbalance in this signaling pathway (Patel et al. 2005; Rangasamy et al. 2011; Khalaf et al. 2017). Several drugs such as MEDI3617 (AstraZeneca) and CVX-060 (Pfizer) that selectively inhibit Ang-2 (Leow et al. 2012; Wu et al. 2016), and AMG 386 (Amgen) targeting Ang1/2 are currently being evaluated in clinical trials for cancer therapy, while AMG 386 is also being tested in retinal diseases (Oliner et al. 2012; Marth et al. 2017). RG7716 (Roche) is a bispecific antibody targeting both VEGF and Ang-2, currently in phase 2 and 3 clinical trials for the treatment of diabetic macular edema (Regula et al. 2016). Sub-cutaneous injections of a Tie-2 activator, AKB-9778 (Aerpio) in combination with anti-VEGF therapy is in phase 2 clinical trials for DR and shows significantly improved visual acuity compared to anti-VEGF monotherapy (Campochiaro et al. 2016).
Retinal microvascular repair is currently being explored using stem/progenitor cell therapy. These vascular reparative cells reside in the adult bone marrow and can be harvested from peripheral blood after G-CSF administration, genetically modified ex vivo by adeno-associated virus transduction and infused intravenously to treat the same patient (Sekiguchi et al. 2009; Sen et al. 2010). These progenitor cells have the ability to home to areas of vascular injury in response to chemotactic gradients and aid in vascular repair (Vagima et al. 2011). This type of personalized treatment using stem/progenitor cells is an attractive therapeutic strategy, since these cells can potentially assist in repair of injured retinal microvasculature in DR as well as in other diabetic complications such as wound healing or limb ischemia. Additionally, intravenous delivery of progenitor cells does not involve invasive procedures or packaging into expensive ophthalmic preparations as is required for intravitreal injections, which makes this mode of therapy very attractive to patients. Currently, the safety of autologous transplantation of bone marrow stem cells for treatment is being evaluated in several phase I clinical trials (Table 1) (Park et al. 2015; Satarian et al. 2017; Cotrim et al. 2017).
Functional regeneration of retinal neurons is another promising strategy in the treatment of DR. Restoration of insulin signaling and strict glycemic control can help normalize inflammation in the diabetic retina, but they do not aid in regaining normal neuronal function (Masser et al. 2014). Although mammalian retinal cells lack the potential for spontaneous neuronal regeneration, modulation of specific proteins may help in repair of the injured retinal tissue and restoration of vision; these will be discussed in the next section.
Neuronal Regeneration in Diabetic Retinopathy
Promoters of Neuronal Growth
Cell adhesion molecules (CAMs) activate downstream signaling pathways leading to regulation of neurite outgrowth, and neuronal regeneration. Two kinds of CAMs are found on growing axons: the calcium-dependent cadherins and the immunoglobulin-superfamily CAMs (IgSF-CAMs). Interaction between these CAMs is crucial not only for forming physical attachments between cells, but also for activation of signaling cascades for growth cone navigation during neuronal development and regeneration. Cadherins have been shown to be important for regeneration of injured retinal ganglion cell axons in zebrafish and newt (Hirota et al. 2001; Liu et al. 2002). Polysialylated NCAM (PSA-NCAM) is also believed to support the survival of injured retinal ganglion cells and protect the retina from neurodegeneration (Murphy et al. 2007b, 2009; Lobanovskaya et al. 2015; Luke et al. 2016b).
Neurotrophins trigger neuroprotective signaling cascades in the neuron by interacting with receptors or integrins on the growth cone and can be used to potentially prevent neuronal degeneration. Brain-derived neurotrophic factor (BDNF) has been demonstrated to protect ganglion, glial, and amacrine cells in culture and in diabetic rats from cell death (Johnson et al. 1986; Hammes et al. 1995; Loeliger et al. 2008). Sigma 1 receptor in the retina is another promising therapeutic target for retinal degenerative diseases, and activation of this molecular chaperone has been shown to protect against ganglion cell loss and preserve cone photoreceptor function in vivo (Smith et al. 2008, 2018; Wang et al. 2016). Imbalance in levels of native neuroprotective factors in the retina such as somatostatin, insulin, PEDF, and CNTF can also be corrected by supplementation of the neurotrophin (Aizu et al. 2003; Reiter et al. 2006; Liu et al. 2012; Hernández et al. 2013). Alternatively, topical administration of a combination of neurotrophins may better protect the various types of retinal neurons and may prove more effective in preventing early retinal neurodegeneration, since intravitreal injections are not preferred in the early stages of DR when there are no visible symptoms.
Since apoptosis is the major pathway for neuronal death in DR, inhibition of apoptosis using caspase inhibitors, inhibition of microglia, and overexpressing Bcl-2 or pro-survival neurotrophic factors have been demonstrated to drastically reduce neuronal cell death in the degenerating mouse retina (Chierzi et al. 1998; Bode and Wolfrum 2003; Vigneswara and Ahmed 2016; Feng et al. 2016; Thomas et al. 2017). Interestingly, inhibition of caspases has also been demonstrated to promote limited axon regeneration in rat retinal ganglion cells (Vigneswara et al. 2014; Thomas et al. 2017).
Reprogramming Glia or RPE Cells
Reprograming other retinal cell types into functional neurons has been studied extensively in the last few years, in an attempt to recreate retinal regeneration from Müller or retinal pigmented epithelium (RPE) cells by the zebrafish and some amphibians. The RPE can potentially be reprogrammed to generate the neuroretina since RPE cells found in the chick embryo and some amphibians have a natural ability to differentiate and regenerate retinal neurons (Yoshii et al. 2007; Luz-Madrigal et al. 2014). Several studies have explored the possibility of gene-directed reprogramming of RPE cells in vitro and in vivo to differentiate toward retinal neurons (Ma et al. 2009; Li et al. 2010; Wang and Yan 2014).
Müller cells are the most abundant non-neural cell type in the retina and share a common progenitor cell lineage with photoreceptor cells. Recent studies have identified potential genes involved in reprogramming Müller cells into retinal neurons, such as Ascl1, Sox2, and Lin28 which can be modulated to stimulate functional neuronal regeneration (Pollak et al. 2013; Ueki et al. 2015; Gorsuch et al. 2017). More recently, Müller glia were successfully reprogrammed to express retinal neuronal markers and synapse with other neurons, as well as respond to light in adult mice (Jorstad et al. 2017).
Cell Replacement Strategies
The retinal ganglion cells are the most susceptible to cellular damage and neurotoxicity in response to hyperglycemia, among all neuronal types in the retina. Recent studies demonstrate that intravitreal injection of neural precursors or mature ganglion cells may help restore retinal function following neurodegeneration by integration of the transplanted neurons or progenitors in the adult retinas (Venugopalan et al. 2016; Divya et al. 2017). Once transplanted, ganglion cells must regenerate their axons in the host retina and form target-specific synapses within and beyond the eye. Supplementation with neurotrophins such as CNTF and PEDF has been demonstrated to aid axon regeneration and elongation after optic nerve injury (Zhang et al. 2016; Cen et al. 2017).
Data from ERG indicate that photoreceptors which are metabolically the most active neurons of the CNS become functionally impaired as early as 2 days after onset of diabetes in rats (Phipps et al. 2004; Kern and Berkowitz 2015). Replacement of degenerated photoreceptor cells with precursor or stem cells may improve visual function, as demonstrated by pre-clinical studies in animals (Pearson et al. 2012; Barber et al. 2013; Singh et al. 2013; Barnea-Cramer et al. 2016). Phase 1 and 2 clinical trials are ongoing for photoreceptor replacement by sub-retinal/intravitreal injection of retinal progenitor cells in patients with retinitis pigmentosa (ReNeuron, jCyte) (Table 1).
Although replacement of retinal neurons by transplantation is an attractive strategy to restore vision, the effectiveness and safety of these strategies remains to be established. Cell replacement may be hampered by the effect of the inflammatory microenvironment hosting the transplanted cells which impedes their survival and integration into the degenerating retinal tissue (Pearson et al. 2014; Singh et al. 2018). Long-term immunosuppression may be required following transplantation to prevent the immune system from destroying donor cells (Schwartz et al. 2015). Further, after in vitro expansion and transplantation of retinal progenitor cells, they must integrate into the host tissue and differentiate into the correct neuronal cell type. Although a small subset of transplanted cells derived from these cultures integrate into the host tissue and express characteristic neuronal markers, the majority of donor cells do not integrate or develop the full protein expression profile, morphology, and function of differentiated retinal neurons (Klassen et al. 2004; MacLaren and Pearson 2007; Barber et al. 2013). It is clear that major challenges lie ahead in the field of neuron replacement therapy before this technique can be applied in a clinical setting.
Other Approaches
Significant interest has been generated in developing multi-hit approaches that target neurovascular degeneration and retinal inflammation in early DR. In this context, PEDF secreted by RPE cells has multifunctional roles such as preventing neuronal dysfunction, retinal inflammation, and vascular hyperpermeability by reducing oxidative stress and glutamate excitotoxicity in the diabetic retina (Barnstable and Tombran-Tink 2004; Yoshida et al. 2009; Xie et al. 2012). Peptide derivatives of PEDF delivered in the form of eye drops have been demonstrated to reduce neurodegeneration, vascular permeability, and inflammation in retinas of diabetic mice (Liu et al. 2012). Erythropoietin (EPO) is another potent molecule that has shown promise in protecting against both neuroglial as well as vascular degeneration in DR animal models (Zhang et al. 2008; Wang et al. 2011; McVicar et al. 2011). Large, randomized clinical trials are required to confirm the potential benefits of PEDF or EPO treatment for DR in humans.
The clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system is an exciting new technique used for genome editing in animals as well as in cultured cells. The potential application of this revolutionary novel technology in the treatment of neurodegeneration in DR is worth exploring. Recently, this tool was used to prevent pathological angiogenesis in mouse models of retinal neovascularization by causing a 30% reduction in the expression of VEGFR2 gene in vascular endothelial cells in the mouse retina (Huang et al. 2017).
Other approaches include the use of surgically implanted electronic retinal prosthetics, transplantation of whole retinal sheets, viral expression of light-sensitive rhodopsin, and the use of synthetic photoswitches to restore a portion of the neuronal activity within the injured retina (Busskamp et al. 2010; Yue et al. 2016; Seiler et al. 2017; Tochitsky et al. 2017).
Future Directions
In order to accurately evaluate the feasibility of the above-mentioned treatment strategies for early retinal neurodegeneration in diabetes, the availability of robust animal models of DR is vital. Mice and rats are the most commonly used animal models, but dogs, pigs, rabbits, and zebrafish are also used to study the pathogenesis of DR (Robinson et al. 2012; Olivares et al. 2017). Most DR animal models whether generated via drug or diet-induced diabetes, galactosemia, or genetic models tend to display early vascular pathological changes such as pericyte loss, formation of microaneurysms, and basement membrane thickening (Kern and Engerman 1996; Agardh et al. 1997). However, none of these DR models completely mimics all the vascular and neuronal changes that occur in each stage of DR in humans (Lai and Lo 2013; Olivares et al. 2017). Type 1 DR rodent models such as streptozotocin-induced diabetes and some genetic models (NOD, Ins2Akita, db/db mice), show accelerated ganglion cell loss, distinct ONL, INL, and IPL thinning, and changes in glial responses indicating progressive neuronal damage in the diabetic retina (Rungger-Brändle et al. 2000; Martin et al. 2004; Cheung et al. 2005; Gastinger et al. 2008; Liu et al. 2012). Most of these studies have used type 1 diabetic animal models due to the lack of well-characterized type 2 diabetic animal models. A recent characterization of the ob/ob mouse model of type 2 diabetes shows promise as a model of early neurodegenerative changes in the diabetic retina (Lee et al. 2018). Since the majority of DR patients are type 2 diabetic, more future studies on early retinal neurodegeneration in type 2 DR models are warranted.
In summary, DR is a complication of diabetes that appears to involve neuropathy as well as microvasculopathy. Although historically, vision loss in diabetic patients has been attributed to microvascular damage to the retina, recent studies reveal that DR is a neurodegenerative disorder, with evidence strongly pointing to the occurrence of neuronal damage prior to overt vascular dysfunction (Sohn et al. 2016; Lynch and Abràmoff 2017). Several clinical trials have been initiated to test the safety and feasibility of using cell replacement therapies, stem cell transplantation, or the use of sub-retinal implants for the treatment of retinal neurodegeneration (Table 1).
Since the combination of retinal inflammation, neurodegeneration, and microvascular injury together contributes to the clinical signs of DR, treatment of DR should ideally aim to be three-fold: (1) addressing retinal inflammation, (2) functional regeneration of retinal neurons, and (3) repair of damaged retinal vasculature. Further studies are warranted to evaluate whether such combination therapies represent a viable choice as opposed to monotherapy and ultimately improve visual function in DR patients. Additionally, reliable biomarkers are needed to detect the extent of early retinal neurodegeneration and identify which patients would benefit the most from neuroprotective/regenerative treatment.
References
Abcouwer SF (2013) Angiogenic factors and cytokines in diabetic retinopathy. J Clin Cell Immunol Suppl 1. https://doi.org/10.4172/2155-9899
Abcouwer SF, Gardner TW (2014) Diabetic retinopathy: loss of neuroretinal adaptation to the diabetic metabolic environment. Ann N Y Acad Sci 1311:174–190. https://doi.org/10.1111/nyas.12412
Abu El-Asrar AM, Dralands L, Missotten L, Geboes K (2007) Expression of antiapoptotic and proapoptotic molecules in diabetic retinas. Eye 21:238–245. https://doi.org/10.1038/sj.eye.6702225
Abu-El-Asrar AM, Dralands L, Missotten L et al (2004) Expression of apoptosis markers in the retinas of human subjects with diabetes. Invest Ophthalmol Vis Sci 45:2760–2766. https://doi.org/10.1167/iovs.03-1392
ACCORD Study Group, ACCORD Eye Study Group, Chew EY et al (2010) Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med 363:233–244. https://doi.org/10.1056/NEJMoa1001288
Agardh CD, Agardh E, Zhang H, Ostenson CG (1997) Altered endothelial/pericyte ratio in Goto-Kakizaki rat retina. J Diabetes Complicat 11:158–162
Ahrén B (2009) Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov 8:369–385. https://doi.org/10.1038/nrd2782
Aiello LP, Avery RL, Arrigg PG et al (1994) Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 331:1480–1487. https://doi.org/10.1056/NEJM199412013312203
Aiello LP, Gardner TW, King GL et al (1998) Diabetic retinopathy. Diabetes Care 21:143–156
Aizu Y, Katayama H, Takahama S et al (2003) Topical instillation of ciliary neurotrophic factor inhibits retinal degeneration in streptozotocin-induced diabetic rats. Neuroreport 14:2067–2071. https://doi.org/10.1097/01.wnr.0000097044.56589.78
Antonetti DA, Barber AJ, Bronson SK et al (2006) Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes 55:2401–2411. https://doi.org/10.2337/db05-1635
Antonetti DA, Klein R, Gardner TW (2012) Diabetic retinopathy. N Engl J Med 366:1227–1239. https://doi.org/10.1056/NEJMra1005073
Arevalo JF, Maia M, Flynn HW et al (2008) Tractional retinal detachment following intravitreal bevacizumab (Avastin) in patients with severe proliferative diabetic retinopathy. Br J Ophthalmol 92:213–216. https://doi.org/10.1136/bjo.2007.127142
Asher RA, Morgenstern DA, Moon LD, Fawcett JW (2001) Chondroitin sulphate proteoglycans: inhibitory components of the glial scar. Prog Brain Res 132:611–619. https://doi.org/10.1016/S0079-6123(01)32106-4
Augustin AJ, Kuppermann BD, Lanzetta P et al (2015) Dexamethasone intravitreal implant in previously treated patients with diabetic macular edema: subgroup analysis of the MEAD study. BMC Ophthalmol 15:150. https://doi.org/10.1186/s12886-015-0148-2
Augustin HG, Koh GY, Thurston G, Alitalo K (2009) Control of vascular morphogenesis and homeostasis through the angiopoietin-tie system. Nat Rev Mol Cell Biol 10:165–177. https://doi.org/10.1038/nrm2639
Bai F, Peng H, Etlinger JD, Zeman RJ (2010) Partial functional recovery after complete spinal cord transection by combined chondroitinase and clenbuterol treatment. Pflugers Arch 460:657–666. https://doi.org/10.1007/s00424-010-0852-y
Ban CR, Twigg SM (2008) Fibrosis in diabetes complications: pathogenic mechanisms and circulating and urinary markers. Vasc Health Risk Manag 4:575–596
Barber AC, Hippert C, Duran Y et al (2013) Repair of the degenerate retina by photoreceptor transplantation. Proc Natl Acad Sci U S A 110:354–359. https://doi.org/10.1073/pnas.1212677110
Barber AJ, Antonetti DA, Kern TS et al (2005) The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci 46:2210–2218. https://doi.org/10.1167/iovs.04-1340
Barber AJ, Lieth E, Khin SA et al (1998) Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 102:783–791. https://doi.org/10.1172/JCI2425
Barnea-Cramer AO, Wang W, Lu S-J et al (2016) Function of human pluripotent stem cell-derived photoreceptor progenitors in blind mice. Sci Rep 6:29784. https://doi.org/10.1038/srep29784
Barnstable CJ, Tombran-Tink J (2004) Neuroprotective and antiangiogenic actions of PEDF in the eye: molecular targets and therapeutic potential. Prog Retin Eye Res 23:561–577. https://doi.org/10.1016/j.preteyeres.2004.05.002
Barouch FC, Miyamoto K, Allport JR et al (2000) Integrin-mediated neutrophil adhesion and retinal leukostasis in diabetes. Invest Ophthalmol Vis Sci 41:1153–1158
Barros CS, Franco SJ, Müller U (2011) Extracellular matrix: functions in the nervous system. Cold Spring Harb Perspect Biol 3:a005108. https://doi.org/10.1101/cshperspect.a005108
Berezin V (ed) (2010) Structure and function of the neural cell adhesion molecule NCAM. Springer New York, New York, NY
Blinder KJ, Dugel PU, Chen S et al (2017) Anti-VEGF treatment of diabetic macular edema in clinical practice: effectiveness and patterns of use (ECHO study report 1). Clin Ophthalmol Auckl NZ 11:393–401. https://doi.org/10.2147/OPTH.S128509
Bloodworth JM (1962) Diabetic retinopathy. Diabetes 11:1–22
Bode C, Wolfrum U (2003) Caspase-3 inhibitor reduces apototic photoreceptor cell death during inherited retinal degeneration in tubby mice. Mol Vis 9:144–150
Boyer DS, Yoon YH, Belfort R et al (2014) Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology 121:1904–1914. https://doi.org/10.1016/j.ophtha.2014.04.024
Bresnick GH (1986) Diabetic retinopathy viewed as a neurosensory disorder. Arch Ophthalmol Chic Ill 1960 104:989–990
Bressler SB, Qin H, Beck RW et al (2012) Factors associated with changes in visual acuity and central subfield thickness at 1 year after treatment for diabetic macular edema with ranibizumab. Arch Ophthalmol Chic Ill 1960 130:1153–1161. https://doi.org/10.1001/archophthalmol.2012.1107
Brown DM, Nguyen QD, Marcus DM et al (2013) Long-term outcomes of ranibizumab therapy for diabetic macular edema: the 36-month results from two phase III trials: RISE and RIDE. Ophthalmology 120:2013–2022. https://doi.org/10.1016/j.ophtha.2013.02.034
Brown SP, He S, Masland RH (2000) Receptive field microstructure and dendritic geometry of retinal ganglion cells. Neuron 27:371–383
Bui BV, Loeliger M, Thomas M et al (2009) Investigating structural and biochemical correlates of ganglion cell dysfunction in streptozotocin-induced diabetic rats. Exp Eye Res 88:1076–1083. https://doi.org/10.1016/j.exer.2009.01.009
Busik JV, Mohr S, Grant MB (2008) Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes 57:1952–1965. https://doi.org/10.2337/db07-1520
Busskamp V, Duebel J, Balya D et al (2010) Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science 329:413–417. https://doi.org/10.1126/science.1190897
Campochiaro PA, Khanani A, Singer M et al (2016) Enhanced benefit in diabetic macular edema from AKB-9778 Tie2 activation combined with vascular endothelial growth factor suppression. Ophthalmology 123:1722–1730. https://doi.org/10.1016/j.ophtha.2016.04.025
Cen L-P, Liang J-J, Chen J-H et al (2017) AAV-mediated transfer of RhoA shRNA and CNTF promotes retinal ganglion cell survival and axon regeneration. Neuroscience 343:472–482. https://doi.org/10.1016/j.neuroscience.2016.12.027
Chakravarthy H, Beli E, Navitskaya S et al (2016) Imbalances in mobilization and activation of pro-inflammatory and vascular reparative bone marrow-derived cells in diabetic retinopathy. PLoS One 11:e0146829. https://doi.org/10.1371/journal.pone.0146829
Cheung AKH, Fung MKL, Lo ACY et al (2005) Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes 54:3119–3125
Chhablani J, Sharma A, Goud A et al (2015) Neurodegeneration in type 2 diabetes: evidence from spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci 56:6333–6338. https://doi.org/10.1167/iovs.15-17334
Chierzi S, Cenni MC, Maffei L et al (1998) Protection of retinal ganglion cells and preservation of function after optic nerve lesion in bcl-2 transgenic mice. Vis Res 38:1537–1543. https://doi.org/10.1016/S0042-6989(97)00332-5
Colton CA, Wilcock DM (2010) Assessing activation states in microglia. CNS Neurol Disord Drug Targets 9:174–191
Comparison of Age-related Macular Degeneration Treatments Trials (CATT) Research Group, Maguire MG, Martin DF et al (2016) Five-year outcomes with anti-vascular endothelial growth factor treatment of neovascular age-related macular degeneration: the comparison of age-related macular degeneration treatments trials. Ophthalmology 123:1751–1761. https://doi.org/10.1016/j.ophtha.2016.03.045
Cotrim C, Toscano L, Messias A, et al (2017) Intravitreal use of bone marrow mononuclear fraction containing CD34+ stem cells in patients with atrophic age-related macular degeneration. Clin Ophthalmol Volume 11:931–938. doi: https://doi.org/10.2147/OPTH.S133502
Coughlin BA, Feenstra DJ, Mohr S (2017) Müller cells and diabetic retinopathy. Vis Res 139:93–100. https://doi.org/10.1016/j.visres.2017.03.013
de Boer JF, Cense B, Park BH, et al (2003) Improved signal-to-noise ratio in spectral-domain compared with time-domain optical coherence tomography. Opt Lett 28:2067–2069
Devanathan V, Hagedorn I, Köhler D et al (2015) Platelet G i protein Gα i2 is an essential mediator of thrombo-inflammatory organ damage in mice. Proc Natl Acad Sci 112:6491–6496. https://doi.org/10.1073/pnas.1505887112
Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group, Lachin JM, Genuth S et al (2000) Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med 342:381–389. https://doi.org/10.1056/NEJM200002103420603
Diabetic Retinopathy Clinical Research Network, Elman MJ, Aiello LP et al (2010) Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology 117:1064–1077.e35. https://doi.org/10.1016/j.ophtha.2010.02.031
Divya MS, Rasheed VA, Schmidt T et al (2017) Intraocular injection of ES cell-derived neural progenitors improve visual function in retinal ganglion cell-depleted mouse models. Front Cell Neurosci 11:295. https://doi.org/10.3389/fncel.2017.00295
Du Y, Cramer M, Lee CA et al (2015) Adrenergic and serotonin receptors affect retinal superoxide generation in diabetic mice: relationship to capillary degeneration and permeability. FASEB J Off Publ Fed Am Soc Exp Biol 29:2194–2204. https://doi.org/10.1096/fj.14-269431
Du Y, Tang J, Li G et al (2010) Effects of p38 MAPK inhibition on early stages of diabetic retinopathy and sensory nerve function. Invest Ophthalmol Vis Sci 51:2158–2164. https://doi.org/10.1167/iovs.09-3674
Duan X, Krishnaswamy A, De la Huerta I, Sanes JR (2014) Type II cadherins guide assembly of a direction-selective retinal circuit. Cell 158:793–807. https://doi.org/10.1016/j.cell.2014.06.047
Edelhauser HF, Rowe-Rendleman CL, Robinson MR et al (2010) Ophthalmic drug delivery systems for the treatment of retinal diseases: basic research to clinical applications. Invest Ophthalmol Vis Sci 51:5403–5420. https://doi.org/10.1167/iovs.10-5392
Falavarjani KG, Nguyen QD (2013) Adverse events and complications associated with intravitreal injection of anti-VEGF agents: a review of literature. Eye Lond Engl 27:787–794. https://doi.org/10.1038/eye.2013.107
Falsini B, Porciatti V, Scalia G et al (1989) Steady-state pattern electroretinogram in insulin-dependent diabetics with no or minimal retinopathy. Doc Ophthalmol Adv Ophthalmol 73:193–200
Feng L, Chen H, Yi J et al (2016) Long-term protection of retinal ganglion cells and visual function by brain-derived neurotrophic factor in mice with ocular hypertension. Invest Ophthalmol Vis Sci 57:3793–3802. https://doi.org/10.1167/iovs.16-19825
Fong DS, Aiello L, Gardner TW et al (2003) Diabetic retinopathy. Diabetes Care 26(Suppl 1):S99–S102
Frank RN (2014) Systemic therapies for diabetic retinopathy: the accord eye study. Ophthalmology 121:2295–2296. https://doi.org/10.1016/j.ophtha.2014.08.019
Fuerst PG, Koizumi A, Masland RH, Burgess RW (2008) Neurite arborization and mosaic spacing in the mouse retina require DSCAM. Nature 451:470–474. https://doi.org/10.1038/nature06514
Gastinger MJ, Kunselman AR, Conboy EE et al (2008) Dendrite remodeling and other abnormalities in the retinal ganglion cells of Ins2 Akita diabetic mice. Invest Ophthalmol Vis Sci 49:2635–2642. https://doi.org/10.1167/iovs.07-0683
Gastinger MJ, Singh RSJ, Barber AJ (2006) Loss of cholinergic and dopaminergic amacrine cells in streptozotocin-diabetic rat and Ins2Akita-diabetic mouse retinas. Invest Ophthalmol Vis Sci 47:3143–3150. https://doi.org/10.1167/iovs.05-1376
Gonzalez VH, Campbell J, Holekamp NM et al (2016) Early and long-term responses to anti-vascular endothelial growth factor therapy in diabetic macular edema: analysis of protocol I data. Am J Ophthalmol 172:72–79. https://doi.org/10.1016/j.ajo.2016.09.012
Gorsuch RA, Lahne M, Yarka CE et al (2017) Sox2 regulates Müller glia reprogramming and proliferation in the regenerating zebrafish retina via Lin28 and Ascl1a. Exp Eye Res 161:174–192. https://doi.org/10.1016/j.exer.2017.05.012
Gratieri T, Gelfuso GM, Rocha EM et al (2010) A poloxamer/chitosan in situ forming gel with prolonged retention time for ocular delivery. Eur J Pharm Biopharm Off J Arbeitsgemeinschaft Pharm Verfahrenstechnik EV 75:186–193. https://doi.org/10.1016/j.ejpb.2010.02.011
Guo M, Liu H, Li S-S et al (2017) Low serum brain-derived neurotrophic factor but not brain-derived neurotrophic factor gene VAL66MET polymorphism is associated with diabetic retinopathy in Chinese type 2 diabetic patients. Retina Phila Pa 37:350–358. https://doi.org/10.1097/IAE.0000000000001132
Guo X, Liu X (2017) Nogo receptor knockdown and ciliary neurotrophic factor attenuate diabetic retinopathy in streptozotocin-induced diabetic rats. Mol Med Rep 16:2030–2036. https://doi.org/10.3892/mmr.2017.6850
Håkansson J, Ståhlberg A, Wolfhagen Sand F et al (2011) N-CAM exhibits a regulatory function in pathological angiogenesis in oxygen induced retinopathy. PLoS One 6:e26026. https://doi.org/10.1371/journal.pone.0026026
Hammer SS, Busik JV (2017) The role of dyslipidemia in diabetic retinopathy. Vis Res 139:228–236. https://doi.org/10.1016/j.visres.2017.04.010
Hammes HP, Federoff HJ, Brownlee M (1995) Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol Med Camb Mass 1:527–534
Harrison WW, Bearse MA, Ng JS et al (2011) Multifocal electroretinograms predict onset of diabetic retinopathy in adult patients with diabetes. Invest Ophthalmol Vis Sci 52:772–777. https://doi.org/10.1167/iovs.10-5931
Hernández C, Bogdanov P, Corraliza L, et al (2016) Topical administration of GLP-1 receptor agonists prevents retinal neurodegeneration in experimental diabetes. Diabetes 65:172–187. doi: https://doi.org/10.2337/db15-0443
Hernández C, García-Ramírez M, Corraliza L et al (2013) Topical administration of somatostatin prevents retinal neurodegeneration in experimental diabetes. Diabetes 62:2569–2578. https://doi.org/10.2337/db12-0926
Hinze A, Stolzing A (2011) Differentiation of mouse bone marrow derived stem cells toward microglia-like cells. BMC Cell Biol 12:35. https://doi.org/10.1186/1471-2121-12-35
Hirota K, Kaneko Y, Matsumoto G, Hanyu Y (2001) Cadherin expression during retinal regeneration in the adult newt. Zool Sci 18:145–149. https://doi.org/10.2108/zsj.18.145
Hombrebueno JR, Ali IH, Xu H, Chen M (2015) Sustained intraocular VEGF neutralization results in retinal neurodegeneration in the Ins2Akita diabetic mouse. Sci Rep 5. https://doi.org/10.1038/srep18316
Hu P, Thinschmidt JS, Yan Y et al (2013) CNS inflammation and bone marrow neuropathy in type 1 diabetes. Am J Pathol 183:1608–1620. https://doi.org/10.1016/j.ajpath.2013.07.009
Huang X, Zhou G, Wu W et al (2017) Genome editing abrogates angiogenesis in vivo. Nat Commun 8:112. https://doi.org/10.1038/s41467-017-00140-3
Hui A, Sheardown H, Jones L (2012) Acetic and acrylic acid molecular imprinted model silicone hydrogel materials for ciprofloxacin-HCl delivery. Mater Basel Switz 5:85–107. https://doi.org/10.3390/ma5010085
Ivanova T, Jalil A, Antoniou Y et al (2016) Vitrectomy for primary symptomatic vitreous opacities: an evidence-based review. Eye Lond Engl 30:645–655. https://doi.org/10.1038/eye.2016.30
Johnson JE, Barde YA, Schwab M, Thoenen H (1986) Brain-derived neurotrophic factor supports the survival of cultured rat retinal ganglion cells. J Neurosci 6:3031–3038
Jorstad NL, Wilken MS, Grimes WN et al (2017) Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 548:103–107. https://doi.org/10.1038/nature23283
Joussen AM, Murata T, Tsujikawa A et al (2001) Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 158:147–152
Kern TS, Berkowitz BA (2015) Photoreceptors in diabetic retinopathy. J Diabetes Investig 6:371–380. https://doi.org/10.1111/jdi.12312
Kern TS, Engerman RL (1996) A mouse model of diabetic retinopathy. Arch Ophthalmol Chic Ill 1960 114:986–990
Kern TS, Tang J, Berkowitz BA (2010) Validation of structural and functional lesions of diabetic retinopathy in mice. Mol Vis 16:2121–2131
Khalaf N, Helmy H, Labib H et al (2017) Role of angiopoietins and Tie-2 in diabetic retinopathy. Electron Physician 9:5031–5035. https://doi.org/10.19082/5031
Kikuchi M, Tenneti L, Lipton SA (2000) Role of p38 mitogen-activated protein kinase in axotomy-induced apoptosis of rat retinal ganglion cells. J Neurosci 20:5037–5044
Kim Y-H, Kim Y-S, Park C-H et al (2008) Protein kinase C-δ mediates neuronal apoptosis in the retinas of diabetic rats via the Akt signaling pathway. Diabetes 57:2181–2190. https://doi.org/10.2337/db07-1431
Kiss S, Berenberg TL (2014) Ultra widefield fundus imaging for diabetic retinopathy. Curr Diab Rep 14:514. https://doi.org/10.1007/s11892-014-0514-0
Klassen HJ, Ng TF, Kurimoto Y et al (2004) Multipotent retinal progenitors express developmental markers, differentiate into retinal neurons, and preserve light-mediated behavior. Invest Ophthalmol Vis Sci 45:4167–4173. https://doi.org/10.1167/iovs.04-0511
Klein R, Klein BE, Moss SE et al (1984) The Wisconsin epidemiologic study of diabetic retinopathy. III. Prevalence and risk of diabetic retinopathy when age at diagnosis is 30 or more years. Arch Ophthalmol Chic Ill 1960 102:527–532
Kohner EM (2008) Microvascular disease: what does the UKPDS tell us about diabetic retinopathy? Diabet Med J Br Diabet Assoc 25(Suppl 2):20–24. https://doi.org/10.1111/j.1464-5491.2008.02505.x
Kowluru A (2010) Small G proteins in islet beta-cell function. Endocr Rev 31:52–78. https://doi.org/10.1210/er.2009-0022
Kowluru A (2017) Role of G-proteins in islet function in health and diabetes. Diabetes Obes Metab 19(Suppl 1):63–75. https://doi.org/10.1111/dom.13011
Kowluru A, Kowluru RA, Yamazaki A (1992) Functional alterations of G-proteins in diabetic rat retina: a possible explanation for the early visual abnormalities in diabetes mellitus. Diabetologia 35:624–631
Krishnaswamy A, Yamagata M, Duan X et al (2015) Sidekick 2 directs formation of a retinal circuit that detects differential motion. Nature 524:466–470. https://doi.org/10.1038/nature14682
Kur J, Newman EA, Chan-Ling T (2012) Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog Retin Eye Res 31:377–406. https://doi.org/10.1016/j.preteyeres.2012.04.004
Lai AKW, Lo ACY (2013) Animal models of diabetic retinopathy: summary and comparison. J Diabetes Res 2013:106594. https://doi.org/10.1155/2013/106594
Lee VK, Hosking BM, Holeniewska J et al (2018) BTBR ob/ob mouse model of type 2 diabetes exhibits early loss of retinal function and retinal inflammation followed by late vascular changes. Diabetologia. https://doi.org/10.1007/s00125-018-4696-x
Leow CC, Coffman K, Inigo I et al (2012) MEDI3617, a human anti-angiopoietin 2 monoclonal antibody, inhibits angiogenesis and tumor growth in human tumor xenograft models. Int J Oncol 40:1321–1330. https://doi.org/10.3892/ijo.2012.1366
Li T, Hu J, Du S, et al (2014) ERK1/2/COX-2/PGE2 signaling pathway mediates GPR91- dependent VEGF release in streptozotocin-induced diabetes. Mol Vis 13
Li X, Ma W, Zhuo Y et al (2010) Using neurogenin to reprogram chick RPE to produce photoreceptor-like neurons. Invest Ophthalmol Vis Sci 51:516–525. https://doi.org/10.1167/iovs.09-3822
Lieth E, Gardner TW, Barber AJ et al (2000) Retinal neurodegeneration: early pathology in diabetes. Clin Exp Ophthalmol 28:3–8
Liu Q, Londraville RL, Azodi E et al (2002) Up-regulation of cadherin-2 and cadherin-4 in regenerating visual structures of adult zebrafish. Exp Neurol 177:396–406
Liu X, Zuo Z, Liu W et al (2014) Upregulation of Nogo receptor expression induces apoptosis of retinal ganglion cells in diabetic rats. Neural Regen Res 9:815–820. https://doi.org/10.4103/1673-5374.131597
Liu Y, Leo LF, McGregor C et al (2012) Pigment epithelium-derived factor (PEDF) peptide eye drops reduce inflammation, cell death and vascular leakage in diabetic retinopathy in Ins2(Akita) mice. Mol Med Camb Mass 18:1387–1401. https://doi.org/10.2119/molmed.2012.00008
Lobanovskaya N, Zharkovsky T, Jaako K et al (2015) PSA modification of NCAM supports the survival of injured retinal ganglion cells in adulthood. Brain Res 1625:9–17. https://doi.org/10.1016/j.brainres.2015.08.008
Loeliger MM, Briscoe T, Rees SM (2008) BDNF increases survival of retinal dopaminergic neurons after prenatal compromise. Invest Ophthalmol Vis Sci 49:1282–1289. https://doi.org/10.1167/iovs.07-0521
Lövestam-Adrian M, Agardh E (2000) Photocoagulation of diabetic macular oedema—complications and visual outcome. Acta Ophthalmol Scand 78:667–671
Luke MP, LeVatte TL, O’Reilly AM et al (2016a) Effect of NCAM on aged-related deterioration in vision. Neurobiol Aging 41:93–106. https://doi.org/10.1016/j.neurobiolaging.2016.02.003
Luke MP-S, LeVatte TL, Rutishauser U et al (2016b) Polysialylated neural cell adhesion molecule protects against light-induced retinal degeneration. Invest Ophthalmol Vis Sci 57:5066–5075. https://doi.org/10.1167/iovs.16-19499
Luz-Madrigal A, Grajales-Esquivel E, McCorkle A et al (2014) Reprogramming of the chick retinal pigmented epithelium after retinal injury. BMC Biol 12:28. https://doi.org/10.1186/1741-7007-12-28
Ly A, Yee P, Vessey KA et al (2011) Early inner retinal astrocyte dysfunction during diabetes and development of hypoxia, retinal stress, and neuronal functional loss. Invest Ophthalmol Vis Sci 52:9316–9326. https://doi.org/10.1167/iovs.11-7879
Lynch SK, Abràmoff MD (2017) Diabetic retinopathy is a neurodegenerative disorder. Vis Res 139:101–107. https://doi.org/10.1016/j.visres.2017.03.003
Ma W, Yan R-T, Li X, Wang S-Z (2009) Reprogramming retinal pigment epithelium to differentiate toward retinal neurons with Sox2. Stem Cells Dayt Ohio 27:1376–1387. https://doi.org/10.1002/stem.48
MacLaren RE, Pearson RA (2007) Stem cell therapy and the retina. Eye Lond Engl 21:1352–1359. https://doi.org/10.1038/sj.eye.6702842
Marth C, Vergote I, Scambia G et al (2017) ENGOT-ov-6/TRINOVA-2: randomised, double-blind, phase 3 study of pegylated liposomal doxorubicin plus trebananib or placebo in women with recurrent partially platinum-sensitive or resistant ovarian cancer. Eur J Cancer Oxf Engl 1990 70:111–121. https://doi.org/10.1016/j.ejca.2016.09.004
Martin PM, Roon P, Van Ells TK et al (2004) Death of retinal neurons in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci 45:3330–3336. https://doi.org/10.1167/iovs.04-0247
Masser DR, VanGuilder Starkey HD, Bixler GV et al (2014) Insulin treatment normalizes retinal neuroinflammation but not markers of synapse loss in diabetic rats. Exp Eye Res 125:95–106. https://doi.org/10.1016/j.exer.2014.06.005
Mathews MK, Guo Y, Langenberg P, Bernstein SL (2015) Ciliary neurotrophic factor (CNTF)-mediated ganglion cell survival in a rodent model of non-arteritic anterior ischaemic optic neuropathy (NAION). Br J Ophthalmol 99:133–137. https://doi.org/10.1136/bjophthalmol-2014-305969
McLeod DS, Lefer DJ, Merges C, Lutty GA (1995) Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol 147:642–653
McVicar CM, Hamilton R, Colhoun LM et al (2011) Intervention with an erythropoietin-derived peptide protects against neuroglial and vascular degeneration during diabetic retinopathy. Diabetes 60:2995–3005. https://doi.org/10.2337/db11-0026
Meyer-Rüsenberg B, Pavlidis M, Stupp T, Thanos S (2007) Pathological changes in human retinal ganglion cells associated with diabetic and hypertensive retinopathy. Graefes Arch Clin Exp Ophthalmol Albrecht Von Graefes Arch Klin Exp Ophthalmol 245:1009–1018. https://doi.org/10.1007/s00417-006-0489-x
Miyamoto K, Khosrof S, Bursell SE et al (1999) Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A 96:10836–10841
Mohr S, Xi X, Tang J, Kern TS (2002) Caspase activation in retinas of diabetic and galactosemic mice and diabetic patients. Diabetes 51:1172–1179
Murphy JA, Franklin TB, Rafuse VF, Clarke DB (2007a) The neural cell adhesion molecule is necessary for normal adult retinal ganglion cell number and survival. Mol Cell Neurosci 36:280–292. https://doi.org/10.1016/j.mcn.2007.07.006
Murphy JA, Hartwick ATE, Rutishauser U, Clarke DB (2009) Endogenous polysialylated neural cell adhesion molecule enhances the survival of retinal ganglion cells. Invest Ophthalmol Vis Sci 50:861–869. https://doi.org/10.1167/iovs.08-2334
Murphy JA, Nickerson PEB, Clarke DB (2007b) Injury to retinal ganglion cell axons increases polysialylated neural cell adhesion molecule (PSA-NCAM) in the adult rodent superior colliculus. Brain Res 1163:21–32. https://doi.org/10.1016/j.brainres.2007.05.069
Mysona BA, Shanab AY, Elshaer SL, El-Remessy AB (2014) Nerve growth factor in diabetic retinopathy: beyond neurons. Expert Rev Ophthalmol 9:99–107. https://doi.org/10.1586/17469899.2014.903157
Ng JS, Bearse MA, Schneck ME et al (2008) Local diabetic retinopathy prediction by multifocal ERG delays over 3 years. Invest Ophthalmol Vis Sci 49:1622–1628. https://doi.org/10.1167/iovs.07-1157
Nguyen QD, Brown DM, Marcus DM et al (2012) Ranibizumab for diabetic macular edema: results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology 119:789–801. https://doi.org/10.1016/j.ophtha.2011.12.039
Ola MS, Nawaz MI, El-Asrar AA et al (2013) Reduced levels of brain derived neurotrophic factor (BDNF) in the serum of diabetic retinopathy patients and in the retina of diabetic rats. Cell Mol Neurobiol 33:359–367. https://doi.org/10.1007/s10571-012-9901-8
Oliner JD, Bready J, Nguyen L et al (2012) AMG 386, a selective angiopoietin 1/2-neutralizing peptibody, inhibits angiogenesis in models of ocular neovascular diseases. Invest Ophthalmol Vis Sci 53:2170–2180. https://doi.org/10.1167/iovs.11-7381
Olivares AM, Althoff K, Chen GF et al (2017) Animal models of diabetic retinopathy. Curr Diab Rep 17:93. https://doi.org/10.1007/s11892-017-0913-0
Osaadon P, Fagan XJ, Lifshitz T, Levy J (2014) A review of anti-VEGF agents for proliferative diabetic retinopathy. Eye Lond Engl 28:510–520. https://doi.org/10.1038/eye.2014.13
Oshitari T, Yoshida-Hata N, Yamamoto S (2010) Effect of neurotrophic factors on neuronal apoptosis and neurite regeneration in cultured rat retinas exposed to high glucose. Brain Res 1346:43–51. https://doi.org/10.1016/j.brainres.2010.05.073
Park H-YL, Kim JH, Park CK (2014) Neuronal cell death in the inner retina and the influence of vascular endothelial growth factor inhibition in a diabetic rat model. Am J Pathol 184:1752–1762. https://doi.org/10.1016/j.ajpath.2014.02.016
Park SS, Bauer G, Abedi M et al (2015) Intravitreal autologous bone marrow CD34+ cell therapy for ischemic and degenerative retinal disorders: preliminary phase 1 clinical trial findings. Invest Ophthalmol Vis Sci 56:81–89. https://doi.org/10.1167/iovs.14-15415
Patel JI, Hykin PG, Gregor ZJ et al (2005) Angiopoietin concentrations in diabetic retinopathy. Br J Ophthalmol 89:480–483. https://doi.org/10.1136/bjo.2004.049940
Pearson CS, Mencio CP, Barber AC, et al (2018) Identification of a critical sulfation in chondroitin that inhibits axonal regeneration. eLife 7:. doi: https://doi.org/10.7554/eLife.37139
Pearson RA, Barber AC, Rizzi M et al (2012) Restoration of vision after transplantation of photoreceptors. Nature 485:99–103. https://doi.org/10.1038/nature10997
Pearson RA, Hippert C, Graca AB, Barber AC (2014) Photoreceptor replacement therapy: challenges presented by the diseased recipient retinal environment. Vis Neurosci 31:333–344. https://doi.org/10.1017/S0952523814000200
Phipps JA, Fletcher EL, Vingrys AJ (2004) Paired-flash identification of rod and cone dysfunction in the diabetic rat. Invest Ophthalmol Vis Sci 45:4592–4600. https://doi.org/10.1167/iovs.04-0842
Pollak J, Wilken MS, Ueki Y et al (2013) ASCL1 reprograms mouse Muller glia into neurogenic retinal progenitors. Dev Camb Engl 140:2619–2631. https://doi.org/10.1242/dev.091355
Poulaki V, Qin W, Joussen AM et al (2002) Acute intensive insulin therapy exacerbates diabetic blood-retinal barrier breakdown via hypoxia-inducible factor-1alpha and VEGF. J Clin Invest 109:805–815. https://doi.org/10.1172/JCI13776
Qin Y, Xu G, Wang W (2006) Dendritic abnormalities in retinal ganglion cells of three-month diabetic rats. Curr Eye Res 31:967–974. https://doi.org/10.1080/02713680600987674
Rangasamy S, Srinivasan R, Maestas J et al (2011) A potential role for angiopoietin 2 in the regulation of the blood-retinal barrier in diabetic retinopathy. Invest Ophthalmol Vis Sci 52:3784–3791. https://doi.org/10.1167/iovs.10-6386
Regula JT, Lundh von Leithner P, Foxton R et al (2016) Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol Med 8:1265–1288. https://doi.org/10.15252/emmm.201505889
Reinhard J, Roll L, Faissner A (2017) Tenascins in retinal and optic nerve neurodegeneration. Front Integr Neurosci 11. https://doi.org/10.3389/fnint.2017.00030
Reiter CEN, Wu X, Sandirasegarane L et al (2006) Diabetes reduces basal retinal insulin receptor signaling: reversal with systemic and local insulin. Diabetes 55:1148–1156
Robinson R, Barathi VA, Chaurasia SS et al (2012) Update on animal models of diabetic retinopathy: from molecular approaches to mice and higher mammals. Dis Model Mech 5:444–456. https://doi.org/10.1242/dmm.009597
Romano MR, Biagioni F, Besozzi G et al (2012) Effects of bevacizumab on neuronal viability of retinal ganglion cells in rats. Brain Res 1478:55–63. https://doi.org/10.1016/j.brainres.2012.08.014
Roy S, Amin S, Roy S (2016) Retinal fibrosis in diabetic retinopathy. Exp Eye Res 142:71–75. https://doi.org/10.1016/j.exer.2015.04.004
Roy S, Kern TS, Song B, Stuebe C (2017) Mechanistic insights into pathological changes in the diabetic retina: Implications for Targeting Diabetic Retinopathy. Am J Pathol 187:9–19. https://doi.org/10.1016/j.ajpath.2016.08.022
Rungger-Brändle E, Dosso AA, Leuenberger PM (2000) Glial reactivity, an early feature of diabetic retinopathy. Invest Ophthalmol Vis Sci 41:1971–1980
Satarian L, Nourinia R, Safi S et al (2017) Intravitreal injection of bone marrow mesenchymal stem cells in patients with advanced retinitis pigmentosa; a safety study. J Ophthalmic Vis Res 12:58–64. https://doi.org/10.4103/2008-322X.200164
Schmidt-Erfurth U, Lang GE, Holz FG et al (2014) Three-year outcomes of individualized ranibizumab treatment in patients with diabetic macular edema: the RESTORE extension study. Ophthalmology 121:1045–1053. https://doi.org/10.1016/j.ophtha.2013.11.041
Schultz DR, Harrington WJ (2003) Apoptosis: programmed cell death at a molecular level. Semin Arthritis Rheum 32:345–369. https://doi.org/10.1053/sarh.2003.50005
Schwartz SD, Regillo CD, Lam BL et al (2015) Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet Lond Engl 385:509–516. https://doi.org/10.1016/S0140-6736(14)61376-3
Seiler MJ, Lin RE, McLelland BT et al (2017) Vision recovery and connectivity by fetal retinal sheet transplantation in an immunodeficient retinal degenerate rat model. Invest Ophthalmol Vis Sci 58:614–630. https://doi.org/10.1167/iovs.15-19028
Sekiguchi H, Ii M, Losordo DW (2009) The relative potency and safety of endothelial progenitor cells and unselected mononuclear cells for recovery from myocardial infarction and ischemia. J Cell Physiol 219:235–242. https://doi.org/10.1002/jcp.21672
Sen S, Merchan J, Dean J et al (2010) Autologous transplantation of endothelial progenitor cells genetically modified by adeno-associated viral vector delivering insulin-like growth factor-1 gene after myocardial infarction. Hum Gene Ther 21:1327–1334. https://doi.org/10.1089/hum.2010.006
Simó R (2017) Topical administration of somatostatin and brimonidine in the early stages of diabetic retinopathy
Simó R, Hernández C, European Consortium for the Early Treatment of Diabetic Retinopathy (EUROCONDOR) (2012) Neurodegeneration is an early event in diabetic retinopathy: therapeutic implications. Br J Ophthalmol 96:1285–1290. https://doi.org/10.1136/bjophthalmol-2012-302005
Singh MS, Charbel Issa P, Butler R et al (2013) Reversal of end-stage retinal degeneration and restoration of visual function by photoreceptor transplantation. Proc Natl Acad Sci U S A 110:1101–1106. https://doi.org/10.1073/pnas.1119416110
Singh R, Cuzzani O, Binette F et al (2018) Pluripotent stem cells for retinal tissue engineering: current status and future prospects. Stem Cell Rev 14:463–483. https://doi.org/10.1007/s12015-018-9802-4
Smith SB, Duplantier J, Dun Y et al (2008) In vivo protection against retinal neurodegeneration by sigma receptor 1 ligand (+)-pentazocine. Invest Ophthalmol Vis Sci 49:4154–4161. https://doi.org/10.1167/iovs.08-1824
Smith SB, Wang J, Cui X et al (2018) Sigma 1 receptor: a novel therapeutic target in retinal disease. Prog Retin Eye Res. https://doi.org/10.1016/j.preteyeres.2018.07.003
Sohn EH, van Dijk HW, Jiao C et al (2016) Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc Natl Acad Sci U S A 113:E2655–E2664. https://doi.org/10.1073/pnas.1522014113
Stewart MW (2012) Corticosteroid use for diabetic macular edema: old fad or new trend? Curr Diab Rep 12:364–375. https://doi.org/10.1007/s11892-012-0281-8
Thomas CN, Berry M, Logan A et al (2017) Caspases in retinal ganglion cell death and axon regeneration. Cell Death Discov 3:17032. https://doi.org/10.1038/cddiscovery.2017.32
Tochitsky I, Trautman J, Gallerani N et al (2017) Restoring visual function to the blind retina with a potent, safe and long-lasting photoswitch. Sci Rep 7:45487. https://doi.org/10.1038/srep45487
Ueki Y, Wilken MS, Cox KE et al (2015) Transgenic expression of the proneural transcription factor Ascl1 in Müller glia stimulates retinal regeneration in young mice. Proc Natl Acad Sci U S A 112:13717–13722. https://doi.org/10.1073/pnas.1510595112
Unsicker K (2013) Neurotrophic molecules in the treatment of neurodegenerative disease with focus on the retina: status and perspectives. Cell Tissue Res 353:205–218. https://doi.org/10.1007/s00441-013-1585-y
Vadas O, Dbouk HA, Shymanets A et al (2013) Molecular determinants of PI3Kγ-mediated activation downstream of G-protein-coupled receptors (GPCRs). Proc Natl Acad Sci U S A 110:18862–18867. https://doi.org/10.1073/pnas.1304801110
Vagima Y, Lapid K, Kollet O et al (2011) Pathways implicated in stem cell migration: the SDF-1/CXCR4 axis. Methods Mol Biol Clifton NJ 750:277–289. https://doi.org/10.1007/978-1-61779-145-1_19
van Dijk HW, Verbraak FD, Stehouwer M et al (2011) Association of visual function and ganglion cell layer thickness in patients with diabetes mellitus type 1 and no or minimal diabetic retinopathy. Vis Res 51:224–228. https://doi.org/10.1016/j.visres.2010.08.024
van Gurp M, Festjens N, van Loo G, et al (2003) Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Commun 304:487–497
Vander JF, Duker JS, Benson WE et al (1991) Long-term stability and visual outcome after favorable initial response of proliferative diabetic retinopathy to panretinal photocoagulation. Ophthalmology 98:1575–1579
Venugopalan P, Wang Y, Nguyen T et al (2016) Transplanted neurons integrate into adult retinas and respond to light. Nat Commun 7:10472. https://doi.org/10.1038/ncomms10472
Vigneswara V, Ahmed Z (2016) Long-term neuroprotection of retinal ganglion cells by inhibiting caspase-2. Cell Death Discov 2:16044. https://doi.org/10.1038/cddiscovery.2016.44
Vigneswara V, Akpan N, Berry M et al (2014) Combined suppression of CASP2 and CASP6 protects retinal ganglion cells from apoptosis and promotes axon regeneration through CNTF-mediated JAK/STAT signalling. Brain J Neurol 137:1656–1675. https://doi.org/10.1093/brain/awu037
Wallick CJ, Hansen RN, Campbell J et al (2015) Comorbidity and health care resource use among commercially insured non-elderly patients with diabetic macular edema. Ophthalmic Surg Lasers Imaging Retina 46:744–751. https://doi.org/10.3928/23258160-20150730-09
Wang J, Saul A, Roon P, Smith SB (2016) Activation of the molecular chaperone, sigma 1 receptor, preserves cone function in a murine model of inherited retinal degeneration. Proc Natl Acad Sci U S A 113:E3764–E3772. https://doi.org/10.1073/pnas.1521749113
Wang Q, Gorbey S, Pfister F et al (2011) Long-term treatment with suberythropoietic Epo is vaso- and neuroprotective in experimental diabetic retinopathy. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol 27:769–782. https://doi.org/10.1159/000330085
Wang S-Z, Yan R-T (2014) The retinal pigment epithelium: a convenient source of new photoreceptor cells? J Ophthalmic Vis Res 9:83–93
Wilkinson CP, Ferris FL, Klein RE et al (2003) Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology 110:1677–1682. https://doi.org/10.1016/S0161-6420(03)00475-5
Williams R, Airey M, Baxter H et al (2004) Epidemiology of diabetic retinopathy and macular oedema: a systematic review. Eye Lond Engl 18:963–983. https://doi.org/10.1038/sj.eye.6701476
Wolter JR (1961) Diabetic retinopathy. Am J Ophthalmol 51:1123–1141
Wu FTH, Man S, Xu P et al (2016) Efficacy of cotargeting angiopoietin-2 and the VEGF pathway in the adjuvant postsurgical setting for early breast, colorectal, and renal cancers. Cancer Res 76:6988–7000. https://doi.org/10.1158/0008-5472.CAN-16-0888
Xie B, Jiao Q, Cheng Y et al (2012) Effect of pigment epithelium-derived factor on glutamate uptake in retinal Muller cells under high-glucose conditions. Invest Ophthalmol Vis Sci 53:1023–1032. https://doi.org/10.1167/iovs.11-8695
Yang G, Masland RH (1994) Receptive fields and dendritic structure of directionally selective retinal ganglion cells. J Neurosci 14:5267–5280
Yang S, Zhao J, Sun X (2016) Resistance to anti-VEGF therapy in neovascular age-related macular degeneration: a comprehensive review. Drug Des Devel Ther 10:1857–1867. https://doi.org/10.2147/DDDT.S97653
Yau JWY, Rogers SL, Kawasaki R et al (2012) Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 35:556–564. https://doi.org/10.2337/dc11-1909
Yoshida Y, Yamagishi S-I, Matsui T et al (2009) Protective role of pigment epithelium-derived factor (PEDF) in early phase of experimental diabetic retinopathy. Diabetes Metab Res Rev 25:678–686. https://doi.org/10.1002/dmrr.1007
Yoshii C, Ueda Y, Okamoto M, Araki M (2007) Neural retinal regeneration in the anuran amphibian Xenopus laevis post-metamorphosis: transdifferentiation of retinal pigmented epithelium regenerates the neural retina. Dev Biol 303:45–56. https://doi.org/10.1016/j.ydbio.2006.11.024
Yue L, Weiland JD, Roska B, Humayun MS (2016) Retinal stimulation strategies to restore vision: fundamentals and systems. Prog Retin Eye Res 53:21–47. https://doi.org/10.1016/j.preteyeres.2016.05.002
Zhang J, Wu Y, Jin Y et al (2008) Intravitreal injection of erythropoietin protects both retinal vascular and neuronal cells in early diabetes. Invest Ophthalmol Vis Sci 49:732–742. https://doi.org/10.1167/iovs.07-0721
Zhang SX, Wang JJ, Gao G et al (2006) Pigment epithelium-derived factor (PEDF) is an endogenous antiinflammatory factor. FASEB J Off Publ Fed Am Soc Exp Biol 20:323–325. https://doi.org/10.1096/fj.05-4313fje
Zhang W-M, Zhang Z-R, Zhang Y-G, Gao Y-S (2016) Neural stem cell-based intraocular administration of pigment epithelium-derived factor promotes retinal ganglion cell survival and axon regeneration after optic nerve crush injury in rat: an experimental study. Iran J Med Sci 41:382–390
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Chakravarthy, H., Devanathan, V. Molecular Mechanisms Mediating Diabetic Retinal Neurodegeneration: Potential Research Avenues and Therapeutic Targets. J Mol Neurosci 66, 445–461 (2018). https://doi.org/10.1007/s12031-018-1188-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-018-1188-x