
Abstract
Epigenetics is the array of the chromatin modifications that customize in cell-, stage-, or condition-specific manner the information encloses in plain DNA molecules. Increasing evidences suggest the importance of epigenetic mechanisms for development and maintenance of central nervous system. In fact, a large number of newly discovered genetic causes of neurodevelopmental disorders such as intellectual disability, autism spectrum disorders, and many other syndromes are mutations within genes encoding for chromatin remodeling enzymes. Here, we review recent findings on the epigenetic origin of human diseases, with emphasis on disorders that affect development of the nervous system, and discuss novel therapeutic avenues that target epigenetic mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurodevelopmental disorders (NDDs) represent a wide and variegated spectrum of diseases occurring in the childhood characterized by altered brain functionality leading to mild to severe psychiatric impairment. NDDs are relatively common mental illnesses including but not limited to intellectual disability (ID), autism spectrum disorders (ASD), and schizophrenia. These disorders occur often as syndromes with complex clinical manifestations that show a high grade of symptomatic overlap: cognitive deficits, poor learning, limitations in adaptive behavior, and seizures often found in affected children (Geschwind 2009; Maski et al. 2011).
Unfortunately, such a broad spectrum of phenotypic manifestations mirrors the extreme heterogeneity in the etiopathology of the disorders posing many limitations to both diagnosis and treatment. In fact, environmental factors, malnutrition, alcohol abuse, and prenatal disease of the mother are proven causes or cofactors of NDD together with a high genetic component. The introduction and widespread application of high-throughput sequencing platforms has marked a “new era” in understanding the genetics of NDD, with the addition of a number of new rare alterations, either heritable or de novo (e.g., microscopic duplication/deletions, copy number variations, single nucleotide mutations), to the list of the historically known chromosomal aberrations or gene mutations (e.g., trisomy 21 and expansion of CGG repeat in FMR1 gene for Down syndrome and fragile X syndrome, respectively, two common causes of ID) (Lejeune et al. 1959; Vissers et al. 2015). Altogether, these novel datasets are proving extremely useful to infer the biological processes and the molecular mechanisms at the basis of the altered brain functions and thus new potential target for therapies. An unexpected result of these genome sequencing efforts is the increasing evidence that a large array of new, rare NDD-linked mutations are found in chromatin remodeling factors, thus pointing to epigenetics as a converging pathogenetic mechanism (Pinto et al. 2014; De Rubeis et al. 2014; Iossifov et al. 2014). In 1942, Conrad Waddington was the first to use the term epigenetics to describe environmental factors able to induce differences in equal cells; today, it is employed to group many and different phenomena that, acting on chromatin, can induce changes in gene expression, often in heritable way, without any modifications in underlying DNA sequence (Goldberg et al. 2007; Pujadas and Feinberg 2012). Examples of these mechanisms are DNA methylation, covalent modification of histone proteins, modifications in nucleosome number/composition, and the action of noncoding RNAs that are responsible to make the chromatin, and thus the genes, more or less tightly packed, influencing the accessibility of the transcriptional machinery, as well as regulators of DNA replication, recombination, and repair. The importance of epigenetic control is further witnessed by the fact that it is established throughout the entire life of an organism—and beyond, since the transgenerational inheritance of epigenetic traits has been recently demonstrated (Siklenka et al. 2015). Thus, it is not a surprise that mutations in genes that encode for enzymes involved in the imposition, removal, or recognition of epigenetic marks often result in pathological conditions.
Herein we summarize some classical and recent findings on the epigenetic origin of NDDs (Table 1), their implication in the molecular mechanisms at the basis of the defective neurological functions, and useful experimental models to mimic and study the pathological conditions.
DNA Methylation
Mutations in Methylation-Associated Machinery
DNA methylation on cytosine (5-methylcytosine—5mC) residues in CpG sequences is a stable covalent modification that persist in post-mitotic cells throughout their lifetime. This modification is one of the most well-known example of epigenetic regulation that generally correlates with close chromatin conformation and thus transcriptional silencing. The general content of DNA methylation in the central nervous system (CNS) increases in adulthood and its role is of cardinal importance (Chen et al. 2013; Lister et al. 2013). De novo DNA methyltransferases (DNMTs) of DNMT3 family catalyze the chemical transformation that is preserved by DNMT1 (Kinney and Pradhan 2011). Genetic engineering in mouse has informed that Dnmt1 and 3b are essential for embryonic development, while Dnmt3a KO mice survive for few weeks after birth (Li et al. 1992; Okano et al. 1999). Strikingly, defects in the regulation or detection of 5mC cause severe disorders in humans (Klein et al. 2011; Xu et al. 1999; Jin et al. 2008).
An example of the importance of these regulatory mechanisms is provided by Rett syndrome, a devastating NDD characterized by reduced brain growth, gait abnormalities, distinctive hand movements, seizures, and ID. Rett syndrome affects predominantly girls in their early childhood (usually after 6 months of age), primarily due to mutations within X chromosome gene MECP2 that codify for a reader of methyl-CpG in the DNA (Amir et al. 1999). Occasionally boys are affected, very often in a more severe way (most of them die prenatally or in early infancy), rarely in less aggressive form. More than 20 years of research have revealed multiple functionality in MECP2 spanning between negative regulator of transcription, silencer of repetitive elements and retrotransposons, transcriptional activator through the recruitment of CREB1, and other cell type-specific cofactors (Lyst et al. 2013; Muotri et al. 2010; Chahrour et al. 2008; Guy et al. 2011; Lyst and Bird 2015). The importance of its ability to bind CpG islands is underlined by the fact that most the missense mutations causing Rett syndrome are located within the methyl-CpG-binding domain (MBD) (Ballestar et al. 2000). Importantly, it has been demonstrated that re-expression of Mecp2 can reverse neurological defects in mouse model indicating that the altered brain functions are not due to irreversible developmental malformations opening new expectations for novel therapies (Guy et al. 2007).
It has been recently reported that heterozygous mutations in DNMT3A cause a rare overgrowth condition called Tatton-Brown-Rahman syndrome (tall stature, large head, distinctive facial appearance, mild to moderate ID) (Tatton-Brown et al. 2014). Several de novo mutations (ten nonsynonymous, two small frameshift insertions, and one in-frame deletion) are found within the three functional domains of the protein: a proline–tryptophan–tryptophan–proline (PWWP) domain (three mutations), an ATRX-DNMT3A-DNMT3L-type zinc finger domain (three), and DNA methyltransferase (MTase) domain (seven) suggesting a pathological mechanism due to protein with an aberrant function rather than simple haploinsufficiency (Tatton-Brown et al. 2014). In addition, Xin et al. have recently reported six cases of inherited Tatton-Brown–Rahman overgrowth syndrome (TBRS) due to novel DNMT3A germline mutations in C-terminal DNA MTase domain (Xin et al. 2016). Studies in mouse models have evidenced defects in memory formation due to altered synaptic functions (Feng et al. 2010; Swiech et al. 2015). Regarding the overgrowth mechanisms, it has been reported that somatic mutations in DNMT3A are recognized causes of hematological malignancies and thus linked with uncontrolled cell proliferation (Hou et al. 2012; Scourzic et al. 2016). Moreover, a recent study has indicated DNA methylation, mediated by elevated expression of DNMT3A, as the main player in the silencing of CDK inhibitor p18INK4C that induces overproliferation in gastric carcinogenesis (Cui et al. 2015). This cancer-NDD comparison would suggest the presence of dominant negative alleles leading to a gain of DNMT3A function of the pathological variants causing overgrowth phenotype both in tumors and in Tatton-Brown-Rahman syndrome. However, this fascinating hypothesis is challenged by a recently reported new TBRS patient with 2p23 microdeletion including DNMT3A, which would support the idea of DNMT3A haploinsufficiency as cause of the overgrowth syndrome (Okamoto et al. 2016).
Immunodeficiency, centromere instability, and facial anomalies syndrome 1 (ICF1) (ID, facial dysmorphism, recurrent and prolonged infections) is largely due to homozygous or compound heterozygous mutations in DNMT3B (Hansen et al. 1999; Jin et al. 2008). These aberrations result in hypomorphic proteins since the complete loss of the enzyme is probably lethal as demonstrated in mice (Okano et al. 1999). The reduced DNMT3B activity in patients and cell lines leads to significant changes in transcription in subset of genes important for immune functions, development, and neurogenesis which, at least in part, bear subtle but significant hypomethylation in their promoters associated with alteration in the histone modifications in the surrounded area (loss of H3K27me3, gain of H3K9ac, and K4me3) that together sustain genic upregulation (Jin et al. 2008).
Recent genome-wide studies of schizophrenic patients have revealed an enrichment in DNA methylation quantitative trait loci in regions associated with the disease (Jaffe et al. 2016; Hannon et al. 2016). Interestingly the majority of the methylation-sensitive sites associated with schizophrenia are implicated in the transition from fetal to neonatal life, strengthening the hypothesis that genetic or environmental factors acting at this critical developmental stage may influence susceptibility to schizophrenia through alteration in DNA methylation. This concept is supported by several reports of increase expression of DNMT1 and DNMT3A in schizophrenia and bipolar patients that results in hypermethylation nearby the controlling regions of specific GABAergic genes (e.g., RLN and GAD67) causing their strong downregulation (for review Guidotti et al. 2011).
Aberrant Methylation Pattern
Another important role of DNA methylation is occurring in genomic imprinting; examples linked with NDDs are given by mutations within chromosomal region 15q11-13 leading to Angelman or Prader-Willi syndrome depending on which parental chromosome is affected (Knol et al. 1989; Buiting et al. 1995). In neurons, differently to other cell types, 15q11-13 region, that control the expression of the ubiquitin ligase E3A (UBE3A), is strongly imprinted. In particular, in the Prader-Willi/Angelman syndrome (PWS-AS) locus, the heavy DNA methylation in the PWS/AS imprinting centers found in the maternal chromosome suppresses the downstream genes that, on the contrary, in the paternal chromosome leads to the production of long non coding RNAs (lncRNAs) including the UBE3A-antisense transcript (ATS) to suppress in cis UBE3A gene expression (Nicholls and Knepper 2001). When the genetic defects (mutations or deletions) are carried by the maternal allele, they result in UBE3A KO in neuronal cells of patients with Angelman syndrome (ID, absent speech, ataxia, seizures, episodes of inappropriate happy demeanor). Conversely, when the paternal allele is affected, the loss of paternal-derived genes causes the Prader-Willi syndrome (hyperphagia, short stature, cognitive dysfunctions, hypotonia, behavioral problems) (Nicholls and Knepper 2001; Sell and Margolis 2015). This complex local regulation is maintained in human-induced pluripotent stem cells (hIPSC), deriving from AS and PWS patients, that retain the appropriate methylation imprinting in 15q11-13 following the reprogramming process resulting in a robust model to study these disorders (Chamberlain et al. 2010). Moreover, the neural differentiation of AS-hIPSC is effectual into generating mature neurons in which the paternal UBE3A is “correctly” repressed through the action of UBE3A-ATS (Chamberlain et al. 2010). The strong epigenetic architecture of the locus is a complication for our understanding of the diseases but offers interesting intervention options to try to de-repress imprinted alleles that remain intact in patients, to restore functional gene products. For instance, Philpot and co-workers have achieved the pharmacological reactivation of the paternal Ube3a in maternal Ube3a null neurons (Huang et al. 2011). Surprisingly, this was not due to drugs known to influence the epigenetic marks (e.g., DNA methyltransferase inhibitors or chromatin remodeling compounds) but thanks to topoisomerase inhibitors (e.g., the FDA-approved drug Topotecan) that silence Ube3a-ATS in epigenetic independent manner (Huang et al. 2011). Comparable results have been obtained by silencing Ube3a-ATS genetically or with antisense oligonucleotides that can ameliorate the cognitive deficits of AS mouse model (Meng et al. 2013, 2015).
In early 90s, it has been identified the abnormal expansion of CGG repeats in the 5′ UTR of the FMRI gene on the X chromosome as the molecular cause of the most common form of inherited mental retardation, the fragile X syndrome (FXS) (moderate to severe mental retardation, facial dysmorphisms) (Pieretti et al. 1991; Verkerk et al. 1991; Kremer et al. 1991). The CGG repeat region is located immediately downstream of a CpG island that become hyper-methylated upon the strong methylation associated to the CGG repeats when their number exceeds 200; this entails histone modifications nearby (H3K9, K27, and H4K20 methylation and H3K4 demethylation, histone deacetylation) resulting in deficiency of the fragile X mental retardation protein (FMRP), a neuronal mRNA-binding protein encoded by FMRI. Treatment with a potent agent against DNA methylation (5′-azadeoxycytidine) leads to the re-expression of FMRI strongly indicating DNA methylation as the pioneering mechanism in the etiology of the pathology (Coffee et al. 1999). However, the findings of (i) cases of CGG expansion with un-methylated FMRI and (ii) the phenotype of pre-mutation carriers (55–200 repeats) than can display autistic traits or develop fragile X associated tremor/ataxia syndrome (FXTAS) suggest that alternative pathogenetic processes involving aberrant functions of RNA molecules might also be involved (Tassone et al. 2000; Greco et al. 2006; Hagerman et al. 2001).
Histone Tail Modifications
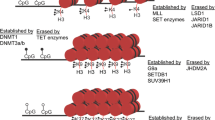
Histone proteins can be covalently modified on specific residues, especially in their N- or C- terminus, or tails, with functional consequences on chromatin biology and ultimately in gene expression (Strahl and Allis 2000). These modifications are an array of biochemical transformations of a great variety of amino acids in the histone tails including lysine acetylation and multiple methylation, arginine mono- and di-methylation, serine/threonine phosphorylation, and others that compose a proper histone code (Strahl and Allis 2000; Sweatt 2013). As for the DNA methylation, dedicated enzymes, working as writers, erasers, or readers, grant the dynamical regulation and interpretation of this code in cell- and stage-specific manner. Novel evidences started to strongly link this epigenetic level of regulation with CNS and its development. An example is given by the dynamic state of gene promoters during differentiation from stem cells to neural precursors and, finally, to differentiated neurons. Lineage-specific genes, that in stem/progenitor level are kept silenced but prompted to be activated in the following stages, present their promoters/regulative regions with both activating (e.g., H3K4me3, H3K9ac) and repressive marks (H3K27me3). During fate transitions, specific enzymes (e.g., KDM6B, an H3K27-specific demethylase) can resolve these “bivalent” or “poised” state removing the first or the latter, to keep the genes silenced (e.g., neuronal genes in astrocytes) or to switch on them (Mikkelsen et al. 2007; Mohn et al. 2008; Hirabayashi and Gotoh 2010; Broccoli et al. 2015). Accordingly, to their importance in neural progenitors and post-mitotic neural cells, dysregulation in histone posttranslational modifications can result in major human NDDs. Moreover, recent large studies have identified new disease variants in confirmed or putative chromatin-modifying enzymes or epigenetic readers, such as KDM6B, KDM5B MLL5, PHF2, CHD2, CHD8, and SETD5, causing autistic phenotype and/or intellectual disabilities (Iossifov et al. 2012; Rauch et al. 2012; Neale et al. 2012; O’Roak et al. 2012; Gilissen et al. 2014; De Rubeis et al. 2014; Iossifov et al. 2014).
Repressive Histone Methylation Marks
Mutations in one allele of EHMT1 gene decrease the level of functional euchromatic histone-lysine N-methyltransferase 1, an H3K9-specific methyltransferase, and are the genetic basis of Kleefstra syndrome (developmental delay, ID, limited or absent speech, and hypotonia) (Kleefstra et al. 2009). H3K9me3 is typically found in highly compacted heterochromatin or anyway silenced genomic regions. Mouse model that mimics the genetic alteration exhibits clinical manifestations of the disease. In particular, this deficiency causes learning and memory defects associated with significant reduction in dendritic arborization and in the number of functional spines (Balemans et al. 2013). The molecular mechanisms underlying these defects are poorly understood; however, it has been reported how EHMT1 and even its homologue EHMT2 interact with core members of polycomb repressive complex, namely enhancer of zeste homologue 2 (EZH2) that catalyzes the deposition of H3K27me3 negative mark (Mozzetta et al. 2014). Thus, an important functional relationship between H3K9me3 and K27me3 takes place at the promoter of neuronal genes, since EZH2 recruitment is dependent on the specific H3K9 methyltransferase activity, suggesting they both contribute to gene silencing (Mozzetta et al. 2014).
Mutations in EZH2 lead to an overgrowth condition called Weaver syndrome (WVS) (skeletal abnormalities, macrocephaly, mild ID, poor coordination, and balance) (Tatton-Brown et al. 2011; Gibson et al. 2012; Seal et al. 2013). EZH2 haploinsufficency is unlikely the sole cause since the vast majority of the mutations are missense and the few leading to premature STOP codons are located at the end of the gene and therefore maintain an intact SET domain (Tatton-Brown et al. 2013). Moreover, heterozygous and homozygous activating and inactivating mutations in EZH2 cause hematological cancers (Chase and Cross 2011). The molecular basis of the disorders seems linked to defects in the content of H3K27me3 due to the reduced function as HTM of WVS variants of EZH2 protein (Cohen et al. 2016), although other mechanisms, as the lacking of some important protein-protein interactions, cannot be excluded.
Activating Histone Methylation Marks
Kabuki syndrome (KS) (ID, microcephaly, postnatal dwarfism, long palpebral fissures) clearly illustrates the importance of histone tail modifications for human health (Kuroki et al. 1981; Niikawa et al. 1981; Bögershausen and Wollnik 2013). KS is an autosomal dominant disorder caused by the heterozygous loss-of-function mutations in either KMT2D or KDM6A. These codify for a lysine-specific methyltransferase (KMT2D) and a lysine-specific demethylase (KDM6A), which are involved in the apposition of H3K4me3 and the removal of H3K27me3, respectively. The two histone tail modifications generally have opposite effects: K4me3 marks open and permissive chromatin while H3K27me3 labels silenced inactive genes. Therefore, the two KS-linked genes promote chromatin laxity and gene expression (Ng et al. 2010; Hannibal et al. 2011; Kokitsu-Nakata et al. 2012; Lederer et al. 2012).
The majority of the mutations are loss of function, thus both in patients with mutation KMT2D, the more frequent genetic cause of KS, and in those with aberrant KDM6A, the final outcome is chromatin in closed conformation. A mouse model for KS, in which the part of the sequence of one Kmt2d allele codifying for the SET (Suvar, enhancer of zeste, Thritorax) domain, responsible for the enzymatic activity has been replaced by β-Geo cassette, showed a striking defect in hippocampal memory with a decrease in dentate gyrus volume and altered neurogenesis (Bjornsson et al. 2014). Interestingly, it has been demonstrated that usage of histone deacetylase (HDAC) inhibitor AR-42 is able to rescue the H3K4 hypomethylation and importantly the defects of the mouse model opening new possibilities for therapeutic intervention not only for KS but possibly for other disorders involving histone modifications (Bjornsson et al. 2014). In fact, the involvement of H3K4 methylation with NDD results deep and intricate since other enzymes, homologous of Drosophila Trithorax genes, are associated with congenital disorders of the developing CNS. Rare loss of function variants found in SETD1A codifying for an H3K4 HMT have been recently associated with schizophrenia and developmental disorders (Singh et al. 2016). Wiedemann-Steiner syndrome (WSS) (facial features, short stature, ID) is a rare genetic disease, initially known as atypical Kabuki syndrome, caused by the loss of one allele of KMT2A gene responsible to produce another HMT-specific for H3K4 residue (Jones et al. 2012; Miyake et al. 2016). Thus, understanding the role and the broad impact of K4 hypomethylation on gene enhancers and promoters in pathological contexts could help to better comprehend and possibly cure a high number of NDD.
Haploinsufficiency of another HMT, NSD1, leads to an overgrowth syndrome called Sotos syndrome (SS) (narrow face, high forehead, behavioral problems including attention deficit hyperactivity disorder (ADHD), ID) (Kurotaki et al. 2002). However, NSD1 specifically controls a different set of epigenetic modifications, namely the methylation of H3K36 (up to the di-methyl form) and H4K20 (Kudithipudi et al. 2014). H3K36 methylation is commonly associated with active transcription (gene body) but even other processes including alternative splicing, dosage compensation, DNA repair, and recombination while H4K20 is usually linked to gene silencing, chromatin condensation, and S-phase progression (Wagner and Carpenter 2012). It has been demonstrated that loss of NSD1 causes hypomethylation at these two marks. Interestingly, this is not only observed in Sotos patients but also in neuroblastomas and glioblastoma cell lines, where NSD1 expression is inhibited by DNA hypermethylation (Berdasco et al. 2009), suggesting that NSD1 may function as a tumor suppressor-like gene, inhibiting key factors responsible for either overgrowth syndrome or tumorigenesis. The ability of NSD1 to bind H3K4me and K9me through its plant homeodomains (PHDs) might also play a role. In fact, when SS single point mutations hit these domains, they produce the same phenotype of the deletion of the entire gene or the SET domain (Cecconi et al. 2005; Pasillas et al. 2011).
Mutations in NSD2, a paralog of NSD1, cause Wolf-Hirschhorn syndrome (WHS) (delayed growth, heart defects, ID, seizures) (Nimura et al. 2009). As for NSD1, NSD2 deficiency causes defects in H3K36 methylation, although WHS is not an overgrowth syndrome and Nsd2 knockout mice do not exhibit gigantism but rather smaller size (Nimura et al. 2009). Moreover, the translocation t(4;14), implicated in multiple myeloma, results in NSD2 overexpression causing changes in H3K36 and K27 methylation and alterations in EZH2 binding at genome-wide level (Kuo et al. 2011; Popovic et al. 2014). Thus, in this case, it appears that the increase rather than lowering of H3K36 methyltransferase activity results in cell cycle progression. Consistent with these observations, mutations in SETD2, the only known enzyme able to directly catalyze H3K36 tri-methylation, produce a variegated phenotype ranging from cancer (deletions/missense mutations) to Sotos-like syndrome and ASD (likely through its haploinsufficiency) (Wagner and Carpenter 2012; Luscan et al. 2014; O’Roak et al. 2012; Iossifov et al. 2014; Lumish et al. 2015). Therefore, the changes in the levels of the H3K36 epigenetic mark do not have a unique clinical manifestation but are instead influenced by other factors, such as the complex domain structure of the enzymes implicated in H3K36 regulation, association with other chromatin marks, cell type-specific requirements, etc. Further comparison of the mechanisms of action of the different H3K36 HMTs will clarify their role as promoter or suppressor of cell proliferation as well as of other pathological processes.
A striking example of the effects of either deficiency or overactivation of an epigenetic regulator is provided by two genetic syndromes due to copy number variation of 7q11.23 (26–28 genes) which exhibit both some similar and some symmetrical, opposite features (Merla et al. 2010). Both Williams-Beuren syndrome (WBS), due to the loss of the region in one allele, and Williams-Beuren region duplication syndrome (7dupASD), due to its heterozygous microduplication, are characterized by anxiety and ADHD; conversely, hypersociability, difficulties in visual tasks, cardiovascular symptoms, and facial dimorphisms are WBS-specific while speech impairment and autistic behavior were found only in 7dupASD. A recent study using human iPSCs derived from healthy donors, as well as WBS and 7dupASD patients, has helped to clarify the molecular mechanisms at the basis of the diseases. The gene GTF2I has been identified as particularly relevant for the establishment of the opposite phenotypes since its dosage is important for transcriptional dysregulation (Adamo et al. 2015). This seems due to the ability of the relative protein, a transcription factor also known as TFII-I, to form repressive complexes with two repressive chromatin modifiers, the H3K4- and K9-specific demethylase LSD1 and histone deacetylase HDAC2, that accordingly are hypo- or hyper-activated in WBS and 7dupASD, respectively (Adamo et al. 2015).
Histone Acetylation
The acetylation of residues in histone tails removes the positive charges thereby decreasing the interaction with the negatively charged DNA resulting in a more relaxed chromatin. The relative activities of histone acetyltransferases (HATs) and HDACs help to define the DNA regions competent for gene transcription, and their balance is required for the correct development and maintenance of the brain. Mutations in CREBBP and EP300 genes codifying for CBP and P300, two known HATs, are responsible for Rubenstein-Taybi syndrome (RTS) (ID, short stature, broad thumbs and first toes) (Lopez-Atalaya et al. 2014). Cbp +/− mice display behavioral impairment and inhibition of neuronal and glial differentiation due to a decrease in histone acetylation of crucial lineage-specific genes (Wang et al. 2010). Most of RTS cases are due to emideletions that decrease protein levels; however, CBP overexpression due to duplications of the region 16p13.3 is also associated with the disease (Stef et al. 2007). Likewise, loss of function mutations in HDAC4 gene are responsible for the another mental retardation syndrome (brachydactyly–mental retardation syndrome) (Williams et al. 2010). Mutations or deletion of one allele of ANKRD11 gene cause KGB syndrome (severe cognitive dysfunctions, hyperactivity, anxiety) (Lo-Castro et al. 2013). The related protein has been revealed as an important chromatin regulator through its binding to HDAC3 which is crucial for the deacetylase activity. In vivo and in vitro studies have revealed a general upregulation of gene expression upon loss of ANKRD11 function, which affects neural progenitor proliferation. Notably, forced expression of HDAC3 or HDAC inhibitors is, once again, able to rescue these defects (Gallagher et al. 2015).
Although it is not classified as a NDD, Friedreich’s ataxia (FRDA) provides an interesting example of a severe degenerative disorder in which GAA repeats within intron 1 of the Frataxin (FXN) gene cause heterochromatin-mediated gene silencing leading to decreased levels of the mitochondrial protein Frataxin (Campuzano et al. 1996) (Saveliev et al. 2003). Therefore, epigenetic-based therapies have been proposed to cure FRDA. An approach tested in clinical trial is based on the use of nicotinamide (vitamin B3), a histone deacetylase inhibitor (Libri et al. 2014). Patients treated daily with nicotinamide showed higher levels of frataxin concentration and reduced heterochromatin modification at the FXN locus, although there were no significant clinical improvements.
ATP-Dependent Chromatin Remodeling
In addition to the aforementioned covalent histone modifications, non-covalent energy-dependent chromatin modifications are important epigenetic mechanisms for nucleosome mobility and transcriptional control. In fact, chromatin or nucleosome remodeling complexes can drift, remove, or reshuffle nucleosome structure in an ATP-dependent fashion (López and Wood 2015). The enzymatic complexes that act as remodelers are subdivided in four groups: SWI/SNF, INO80/SWR1, ISWI, and CHD (Hargreaves and Crabtree 2011). Their remodeling activity, well known in other systems, is only moderately studied in neuronal cells. Nevertheless, a number of mutations in genes related to this process are found associated with NDDs, including ASD, ID, schizophrenia, and related syndromes (López and Wood 2015).
Vertebrate SWI/SNF (switching defective/sucrose nonfermentable), a.k.a. BAF (Brg1/Brm associated factor) complex, is formed by several subunits inclusive of core ATPases (Brg1 or Brm), invariant core subunits (SMARCB1/BAF47, SMARCC1/BAF155, SMARCC2/BAF170), and a number of cell-specific components (Narayanan and Tuoc 2014). In fact, BAF complex exists in different flavors even within neural cell types, e.g., in neural progenitors (npBAF), neurons (nBAF), oligodendrocytes (olBAF), etc. (Lessard et al. 2007). Several members of this complex are linked to Coffin-Siris syndrome (CSS) (mild to severe ID or delayed development of speech, hypoplasia of the tips of the fingers) (Tsurusaki et al. 2012; Kleefstra et al. 2014). The most common causes are mutations in ARID1B gene, while mutations in ARID1A, SMARCA4, SMARCB1, and SMARCE1 are rare (Tsurusaki et al. 2012; Kosho et al. 2014). Mutations in another SWI/SNF component, namely SMARCA2, cause a very similar condition called Nicolaides-Baraitser syndrome, while alterations in the ADNP gene are responsible for ASD (Van Houdt et al. 2012). These genes encode for subunits of neural chromatin remodeling complexes and, although the mutational spectrum and genotype/phenotype correlation are not completely resolved, disease-linked mutations seem to lead to hypofunctional complexes. Mouse transgenic models have recently demonstrated the dramatic effect of BAF loss-of-function on brain development, inducing rearrangement of global chromatin modifications (H3K27me2/3) by abolishing the activity of the specific demethylases KDM6B/UTX on H3 histones (Tuoc et al. 2013; Narayanan et al. 2015). Interestingly, npBAF complex directly interacts with PAX6, a key transcription factor expressed in cortical progenitors and adult NSCs that collaborate with SOX11 to promote neurogenic programs (Ninkovic et al. 2013). Recently, heterozygous mutations in SOX11 have been identified as additional causes of CSS (Tsurusaki et al. 2014).
Within the chromodomain helicase DNA binding (CHD) protein subfamily, three proteins have been found related to NDDs. Mutations in CHD7 are the most common cause of coloboma, hearth anomaly, choanal atresia, retardation, and genital and ear anomalies (CHARGE) syndrome (Vissers et al. 2004). Pathogenic CHD7 variants range from inactivating (nonsense and frameshift) to missense mutations with a broad spectrum of clinical manifestation; however, the major and common pathogenic mechanism seems to be related to a loss or decrease in CHD activity (Hale et al. 2016). Studies in mouse model of CHD7 haploinsufficiency have showed severe malformation in the ear and reduced proliferation in neural stem cells, while in vitro approaches have underlined the role of CHD7 in binding H3K4me3-enriched chromatin to directly enhance gene transcription (Layman et al. 2009; Hurd et al. 2010; Schnetz et al. 2009). Proteomic and genomic approaches in NSCs have identified CHD7 as transcriptional cofactor of SOX2, an HMG-box transcription factor critical for neurogenesis (Engelen et al. 2011). The interaction between these two factors regulates Notch and Sonic Hedgehog pathways, which explains why CHARGE syndrome patients display penetrant alterations in semicircular canals of the inner ear and the accompanying vestibular defects. CHD2 haploinsufficiency, either caused by deletion of the entire gene (Chénier et al. 2014) or by missense mutations (Hamdan et al. 2014), is associated to ID, epilepsy, and ASD. Moreover, studies have shown that de novo CHD2 mutations are correlated with epileptic encephalopathy (Carvill et al. 2013; Lund et al. 2014). The precise molecular mechanism is still unclear, although a mouse mutant for Chd2 (gene trap) has revealed the tumor suppressor activity of CHD2 that can modulate DNA damage responses and in turn genome stability (Nagarajan et al. 2009). Finally, large studies on ASD cohorts have identified recurrent de novo inactivating mutations in CHD8 associated with ASD and macrocephaly (Neale et al. 2012; O’Roak et al. 2012; Iossifov et al. 2014, Wang et al. 2016). Inactivation of cdh8 in zebrafish resembles the human phenotype including the increased head size (Bernier et al. 2014; Sugathan et al. 2014). In line with these findings, moderate gene expression changes and physical interaction with neuronal silencing factor REST cause macrocephaly in Chd8 heterozygous mice (Katayama et al. 2016). Conversely, a developmental study in the mouse brain has illustrated the important role of Chd8 in neural progenitor proliferation through WNT-dependent activation of cell cycle genes; moreover, the authors showed the repression of PRC2 complex genes (e.g., Ezh2 and Suz12) causing reduced dendritic complexity and behavioral abnormalities in adult mice (Durak et al. 2016). Whether the contradiction on cell cycle dynamics maybe due to different experimental setup (chronic vs. acute downregulation of Chd8), this latter aspect could explain the autistic behavior in Chd8 heterozygous mice and in patients. As reported in a recent study in human NSCs and fetal brain, CHD8 genomic targets include a number of ASD risk gens (e.g., ARID1B, NCOR1, POGZ, PTEN, TBL1XR1) that are dysregulated upon CHD8 knockdown (Cotney et al. 2015). CHD8 binding to promoter regions enriched in active marks H3K4me3 and K27ac is positively correlated with gene expression (Cotney et al. 2015). An interesting study mapped the genome-wide interactions with nucleosomes for a number of ATP-remodeling complexes including CHD2 and CHD8 in mouse ESCs revealing that they are able to bind one or both the nucleosomes surrounding the nucleosome free promoter regions (NFRs) (De Dieuleveult et al. 2016). From this work emerged that the remodelers function either in positive or negative manner altering nucleosome dynamics at active gene level or at bivalent promoters. Specifically, CHD2 has high correlation with H3K36me3 in active genes while CHD8, at least in mESCs, is mainly involved in transcriptional repression through association with H3K4me3 in active or bivalent promoters (De Dieuleveult et al. 2016). Expanding this analysis to other cell type/organism will help to unravel the role of these enzymatic complexes in the pathogenic processes of human disorders.
Conclusions
All the observations above indicate an intricate landscape in which the epigenetic modifications are involved, a wide array of consequences upon their defects, and the need of a strong crosstalk among the different modifications to ensure the biological processes. Questions as “is there a common theme underlying in neurodevelopmental disorders?” are difficult to answer for many reasons, starting from the fact that we are still far from a complete understanding of the pathological processes at the basis of the different “chromatinopathies.” We could try to identify general rules from the phenotypes associated to certain epigenetic traits, but again the situation is nonlinear and complex; for example, a situation in which an increase of cell proliferation resulting in overgrowth phenotype could be due to a general relaxation of the chromatin as in polycomb defective Weaver syndrome or as in the cases of Sotos syndrome in which mutations in NSD1 should cause less permissivity to gene transcription. Moreover, there are debated cases, as for the mutations in DNMT3A in which overproliferation could be due by either closed or opened conformation. Besides these considerations, another important aspect to consider is that chromatin enzyme can act also on non-chromatin targets, and therefore a disease could be the results of multifactorial events; an example is provided by SETD2, known as methyltransferase specific for H3K36me3 that has been recently described to be able to methylate also lysine 40 in alpha-tubulin, and thus controlling cytoskeleton dynamics (Park et al. 2016).
Very informative are those cases in which the very same disease is caused by different genetic factors, as in Kabuki syndrome or different kind of mutations on the same genetic traits are causative for different diseases, as for Williams-Beuren syndrome and 7dupASD. In the first example, we can easily relate a closed chromatinic landscape with developmental delay and microcephaly resulting in intellectual impairment. In the cases of deletion or duplication of GTF2I, a state in which the genes are prone to be activated is linked to hypersociability while chromatin marks leading to gene silencing are associated with an opposite, autistic behavior.
The challenge of the near future for physicians and researchers is to identify more of these dualisms. To do so, it is necessary to establish a strong correlation between mutational spectrum and clinical manifestations and consider the possibly divergent effects of either changes at the gene level (true loss-of-functions, duplications, etc.) or missense mutations. Whether the former are useful to gain information on the complete lack or the overrepresentation of the main enzymatic product, the latter represent extremely valuable tool to possibly comprehend the functions of different modules of the protein that can shed new light on their role and on the different symptoms, in the affected people. In this direction, the use of cellular (e.g., hIPSCs) and animal models coupled with new ways to generate site-specific editing (e.g., CRISPR/Cas9 system) will help addressing these unsolved questions in the near future.
References
Adamo A, Atashpaz S, Germain PL, Zanella M, D’Agostino G, Albertin V, Chenoweth J, Micale L, Fusco C, Unger C, Augello B, Palumbo O, Hamilton B, Carella M, Donti E, Pruneri G, Selicorni A, Biamino E, Prontera P, McKay R, Merla G, Testa G (2015) 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat Genet 47(2):132–141. doi:10.1038/ng.3169
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genet 23:185–188
Balemans MC, Kasri NN, Kopanitsa MV, Afinowi NO, Ramakers G, Peters TA, Beynon AJ, Janssen SM, van Summeren RC, Eeftens JM, Eikelenboom N, Benevento M, Tachibana M, Shinkai Y, Kleefstra T, van Bokhoven H, Van der Zee CE (2013) Hippocampal dysfunction in the Euchromatin histone methyltransferase 1 heterozygous knockout mouse model for Kleefstra syndrome. Hum Mol Genet 22(5):852–866. doi:10.1093/hmg/dds490
Ballestar E, Yusufzai TM, Wolffe AP (2000) Effects of Rett syndrome mutations of the methyl-CpG binding domain of the transcriptional repressor MeCP2 on selectivity for association with methylated DNA. Biochemistry 39:7100–7106
Berdasco M, Ropero S, Setien F, Fraga MF, Lapunzina P, Losson R, Alaminos M, Cheung NK, Rahman N, Esteller M (2009) Epigenetic inactivation of the Soto’s overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc Natl Acad Sci U S A 106(51):21830–21835. doi:10.1073/pnas.0906831106
Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C et al (2014) Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158(2):263–276. doi:10.1016/j.cell.2014.06.017
Bjornsson HT, Benjamin JS, Zhang L, Weissman J, Gerber EE, Chen YC, Vaurio RG, Potter MC, Hansen KD, Dietz HC (2014) Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci Transl Med 6(256):256ra135. doi:10.1126/scitranslmed.3009278
Bögershausen N, Wollnik B (2013) Unmasking Kabuki syndrome. Clin Genet 83(3):201–211. doi:10.1111/cge.12051
Broccoli V, Colasante G, Sessa A, Rubio A (2015) Histone modifications controlling native and induced neural stem cell identity. Curr Opin Genet Dev 34:95–101. doi:10.1016/j.gde.2015.08.003
Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S, Nicholls RD, Horsthemke B (1995 Apr) Inherited microdeletions in the Angelman and Prader–Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet 9(4):395–400
Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F et al (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271(5254):1423–1427
Carvill GL, Heavin SB, Yendle SC, McMahon JM, O’Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S et al (2013) Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45(7):825–830. doi:10.1038/ng.2646
Cecconi M, Forzano F, Milani D, Cavani S, Baldo C, Selicorni A, Pantaleoni C, Silengo M, Ferrero GB et al (2005) Mutation analysis of the NSD1 gene in a group of 59 patients with congenital overgrowth. Am J Med Genet A.134:247–253
Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320:1224–1229
Chamberlain SJ, Chen PF, Ng KY et al (2010) Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc Natl Acad Sci U S A 107:17668–17673
Chase A, Cross NC (2011) Aberrations of EZH2 in cancer. Clin Cancer Res 17(9):2613–2618. doi:10.1158/1078-0432.CCR-10-2156
Chen Y, Ozturk NC, Zhou FC (2013) DNA methylation program in developing hippocampus and its alteration by alcohol. PLoS One 8(3):e60503. doi:10.1371/journal.pone.0060503
Chénier S, Yoon G, Argiropoulos B, Lauzon J, Laframboise R, Ahn JW et al (2014) CHD2 haploinsufficiency is associated with developmental delay, intellectual disability, epilepsy and neurobehavioural problems. J Neurodev Disord 6(1):9. doi:10.1186/1866-1955-6-9
Coffee B, Zhang F, Warren ST, Reines D (1999) Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat Genet 22(2):209–209. doi:10.1038/9738
Cohen AS, Yap DB, Lewis ME, Chijiwa C, Ramos-Arroyo MA, Tkachenko N, Milano V, Fradin M et al (2016) Weaver syndrome-associated EZH2 protein variants show impaired histone methyltransferase function in vitro. Hum Mutat 37(3):301–307. doi:10.1002/humu.22946
Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, Liu W, Klei L, Lei J et al (2015) The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun 6:6404. doi:10.1038/ncomms7404
Cui H, Zhao C, Gong P, Wang L, Wu H, Zhang K, Zhou R, Wang L, Zhang T, Zhong S, Fan H (2015) DNA methyltransferase 3A promotes cell proliferation by silencing CDK inhibitor p18INK4C in gastric carcinogenesis. Sci Rep 5:13781. doi:10.1038/srep13781
De Dieuleveult M, Yen K, Hmitou I, Depaux A, Boussouar F, Dargham DB, Jounier S, Humbertclaude H, Ribierre F, Baulard C, Farrell NP, Park B, Keime C, Carrière L, Berlivet S, Gut M, Gut I, Werner M, Deleuze JF, Olaso R, Aude JC, Chantalat S, Pugh BF, Gérard M (2016) Genome-wide nucleosome specificity and function of chromatin remodellers in ES cells. Nature 530(7588):113–116. doi:10.1038/nature16505
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y et al (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515(7526):209–215. doi:10.1038/nature13772
Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, Liu CY, Watson LA, Tsai LH (2016) Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci 19(11):1477–1488. doi:10.1038/nn.4400
Engelen E, Akinci U, Bryne JC, Hou J, Gontan C, Moen M, Szumska D, Kockx C, van Ijcken W, Dekkers DH, Demmers J, Rijkers EJ, Bhattacharya S, Philipsen S, Pevny LH, Grosveld FG, Rottier RJ, Lenhard B, Poot RA (2011) Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet 43(6):607–611. doi:10.1038/ng.825
Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G (2010) Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13(4):423–430. doi:10.1038/nn.2514
Gallagher D, Voronova A, Zander MA, Cancino GI, Bramall A, Krause MP, Abad C, Tekin M, Neilsen PM, Callen DF, Scherer SW, Keller GM, Kaplan DR, Walz K, Miller FD (2015) Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev Cell 32(1):31–42. doi:10.1016/j.devcel.2014.11.031
Geschwind DH (2009) Advances in autism. Annu Rev Med 60:367–380. doi:10.1146/annurev.med.60.053107.121225
Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, Marra MA, FORGE Canada Consortium, Chitayat D, Boycott KM, Weaver DD, Jones SJ (2012) Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet 90(1):110–118. doi:10.1016/j.ajhg.2011.11.018
Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH et al (2014) Genome sequencing identifies major causes of severe intellectual disability. Nature 511(7509):344–347. doi:10.1038/nature13394
Goldberg AD, Allis CD, Bernstein E (2007) Epigenetics: a landscape takes shape. Cell 128(4):635–638 Review
Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, Trapp BD, Iwahashi C, Brunberg J, Grigsby J, Hessl D, Becker EJ, Papazian J, Leehey MA, Hagerman RJ, Hagerman PJ (2006) Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 129(Pt 1):243–255
Guidotti A, Auta J, Chen Y, Davis JM, Dong E, Gavin DP, Grayson DR, Matrisciano F, Pinna G, Satta R, Sharma RP, Tremolizzo L, Tueting P (2011) Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 60(7–8):1007–1016. doi:10.1016/j.neuropharm.2010.10.021
Guy J, Gan J, Selfridge J, Cobb S, Bird A (2007) Reversal of neurological defects in a mouse model of Rett syndrome. Science 315(5815):1143–1147
Guy J, Cheval H, Selfridge J, Bird A (2011) The role of MeCP2 in the brain. Annu Rev Cell Dev Biol 27:631–652. doi:10.1146/annurev-cellbio-092910-154121
Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, Grigsby J, Gage B, Hagerman PJ (2001) Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57(1):127–130
Hale CL, Niederriter AN, Green GE, Martin DM (2016) Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A 170(2):344–354. doi:10.1002/ajmg.a.37435
Hamdan FF, Srour M, Capo-Chichi JM, Daoud H, Nassif C, Patry L, Massicotte C et al (2014) De novo mutations in moderate or severe intellectual disability. PLoS Genet 10(10):e1004772. doi:10.1371/journal.pgen.1004772
Hannibal MC, Buckingham KJ, Ng SB et al (2011) Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet A 155A:1511–1516
Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM, Troakes C, Turecki G, O’Donovan MC, Schalkwyk LC, Bray NJ, Mill J (2016) Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci 19(1):48–54. doi:10.1038/nn.4182
Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, Gartler SM (1999) The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci U S A 96(25):14412–14417
Hargreaves DC, Crabtree GR (2011) ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21:396e420
Hirabayashi Y, Gotoh Y (2010) Epigenetic control of neural precursor cell fate during development. Nat Rev Neurosci 11(6):377–388. doi:10.1038/nrn2810 Review
Hou HA, Kuo YY, Liu CY, Chou WC, Lee MC, Chen CY, Lin LI, Tseng MH, Huang CF, Chiang YC, Lee FY, Liu MC, Liu CW, Tang JL, Yao M, Huang SY, Ko BS, Hsu SC, Wu SJ, Tsay W, Chen YC, Tien HF (2012) DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood 119(2):559–568. doi:10.1182/blood-2011-07-369934
Huang HS, Allen JA, Mabb AM, King IF, Miriyala J, Taylor-Blake B, Sciaky N, Dutton JW Jr, Lee HM, Chen X, Jin J, Bridges AS, Zylka MJ, Roth BL, Philpot BD (2011) Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 481:185–189
Hurd EA, Poucher HK, Cheng K, Raphael Y, Martin DM (2010) The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development 137(18):3139–3150. doi:10.1242/dev.047894
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A et al (2012) De novo gene disruptions in children on the autistic spectrum. Neuron 74(2):285–299. doi:10.1016/j.neuron.2012.04.009
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L et al (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515(7526):216–221. doi:10.1038/nature13908
Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, Kleinman JE (2016) Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci 19(1):40–47. doi:10.1038/nn.4181
Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, Fields CR, Delmas AL, Liu X, Qiu J, Robertson KD (2008) DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet 17:690–709
Jones WD, Dafou D, McEntagart M, Woollard WJ, Elmslie FV, Holder-Espinasse M, Irving M, Saggar AK, Smithson S, Trembath RC, Deshpande C, Simpson MA (2012) De novo mutations in MLL cause Wiedemann-Steiner syndrome. Am J Hum Genet 91(2):358–364. doi:10.1016/j.ajhg.2012.06.008
Katayama Y, Nishiyama M, Shoji H, Ohkawa Y, Kawamura A, Sato T, Suyama M, Takumi T, Miyakawa T, Nakayama KI (2016) CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature 537(7622):675–679. doi:10.1038/nature19357
Kinney SR, Pradhan S (2011) Regulation of expression and activity of DNA (cytosine-5) methyltransferases in mammalian cells. Prog Mol Biol Transl Sci 101:311–333
Kleefstra T, van Zelst-Stams WA, Nillesen WM, Cormier-Daire V, Houge G, Foulds N, van Dooren M, Willemsen MH, Pfundt R, Turner A, Wilson M, McGaughran J, Rauch A, Zenker M, Adam MP, Innes M, Davies C, López AG, Casalone R, Weber A, Brueton LA, Navarro AD, Bralo MP, Venselaar H, Stegmann SP, Yntema HG, van Bokhoven H, Brunner HG (2009) Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J Med Genet 46(9):598–606. doi:10.1136/jmg.2008.062950
Kleefstra T, Schenck A, Kramer JM, van Bokhoven H (2014) The genetics of cognitive epigenetics. Neuropharmacology 80:83–94. doi:10.1016/j.neuropharm.2013.12.025
Klein CJ, Botuyan MV, Wu Y, Ward CJ, Nicholson GA, Hammans S, Hojo K, Yamanishi H et al (2011) Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 43(6):595–600
Knol JH, Nicholls RD, Magenis RE, Graham JM Jr, Lalande M, Latt SA (1989) Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. Am J Med Genet 32(2):285–290
Kokitsu-Nakata NM, Petrin AL, Heard JP et al (2012) Analysis of MLL2 gene in the first Brazilian family with Kabuki syndrome. Am J Med Genet A 158A(8):2003–2008
Kosho T, Miyake N, Carey JC (2014) Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am J Med Genet C Semin Med Genet 166C(3):241–251. doi:10.1002/ajmg.c.31415
Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, Warren ST, Schlessinger D, Sutherland GR, Richards RI (1991) Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252(5013):1711–1714
Kudithipudi S, Lungu C, Rathert P, Happel N, Jeltsch A (2014) Substrate specificity analysis and novel substrates of the protein lysine methyltransferase NSD1. Chem Biol 21(2):226–237. doi:10.1016/j.chembiol.2013.10.016
Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, Li W, Gozani O (2011) NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell 44(4):609–620. doi:10.1016/j.molcel.2011.08.042
Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I (1981) A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr 99(4):570–573. doi:10.1016/S0022-3476(81)80256-9
Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, Ohashi H, Naritomi K, Tsukahara M, Makita Y, Sugimoto T, Sonoda T, Hasegawa T, Chinen Y, Tomita Ha HA, Kinoshita A, Mizuguchi T, Yoshiura Ki K, Ohta T, Kishino T, Fukushima Y, Niikawa N, Matsumoto N (2002) Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 30:365–366
Layman WS, McEwen DP, Beyer LA, Lalani SR, Fernbach SD, Oh E, Swaroop A, Hegg CC, Raphael Y, Martens JR, Martin DM (2009) Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum Mol Genet 18(11):1909–1923. doi:10.1093/hmg/ddp112
Lederer D, Grisart B, Digilio MC et al (2012) Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet 90(1):119–124
Lejeune J, Gautier M, Turpin R (1959) Etudes des chromosomes somatique de neuf enfants mongoliens. CR Hebd Seances Acad Sci 248:1721–1722
Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR (2007) An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55:201–215
Li E, Bestor TH, Jaenisch R (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69:915–926
Libri V, Yandim C, Athanasopoulos S, Loyse N, Natisvili T, Law PP, Chan PK, Mohammad T, Mauri M, Tam KT, Leiper J, Piper S, Ramesh A, Parkinson MH, Huson L, Giunti P, Festenstein R (2014) Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich’s ataxia: an exploratory, open-label, dose-escalation study. Lancet 384(9942):504–513. doi:10.1016/S0140-6736(14)60382-2
Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, Yu M, Tonti-Filippini J, Heyn H, Hu S, Wu JC, Rao A, Esteller M, He C, Haghighi FG, Sejnowski TJ, Behrens MM, Ecker JR (2013) Global epigenomic reconfiguration during mammalian brain development. Science 341(6146):1237905. doi:10.1126/science.1237905
Lo-Castro A, Brancati F, Digilio MC, Garaci FG, Bollero P, Alfieri P, Curatolo P (2013) Neurobehavioral phenotype observed in KBG syndrome caused by ANKRD11 mutations. Am J Med Genet B Neuropsychiatr Genet 162B(1):17–23. doi:10.1002/ajmg.b.32113
López AJ, Wood MA (2015) Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front Behav Neurosci 9:100. doi:10.3389/fnbeh.2015.00100 eCollection 2015. Review
Lopez-Atalaya JP, Valor LM, Barco A (2014) Epigenetic factors in intellectual disability: the Rubinstein-Taybi syndrome as a paradigm of neurodevelopmental disorder with epigenetic origin. Prog Mol Biol Transl Sci 128:139–176. doi:10.1016/B978-0-12-800977-2.00006-1 Review
Lumish HS, Wynn J, Devinsky O, Chung WK (2015) Brief report: SETD2 mutation in a child with autism, intellectual disabilities and epilepsy. J Autism Dev Disord 45(11):3764–3770. doi:10.1007/s10803-015-2484-8
Lund C, Brodtkorb E, Øye AM, Røsby O, Selmer KK (2014) CHD2 mutations in Lennox-Gastaut syndrome. Epilepsy Behav 33:18–21. doi:10.1016/j.yebeh.2014.02.005
Luscan A, Laurendeau I, Malan V, Francannet C, Odent S, Giuliano F, Lacombe D, Touraine R, Vidaud M, Pasmant E, Cormier-Daire V (2014) Mutations in SETD2 cause a novel overgrowth condition. J Med Genet 51(8):512–517. doi:10.1136/jmedgenet-2014-102402
Lyst MJ, Bird A (2015) Rett syndrome: a complex disorder with simple roots. Nat Rev Genet 16(5):261–275. doi:10.1038/nrg3897
Lyst M, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima AF, Rappsilber J, Greenberg ME, Bird A (2013) Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nature Neurosci 16:898–902
Maski KP, Jeste SS, Spence SJ (2011) Common neurological co-morbidities in autism spectrum disorders. Curr Opin Pediatr 23:609–615
Meng L, Person RE, Huang W, Zhu PJ, Costa-Mattioli M, Beaudet AL (2013) Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet 9(12):e1004039. doi:10.1371/journal.pgen.1004039
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F (2015) Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518(7539):409–412. doi:10.1038/nature13975
Merla G, Brunetti-Pierri N, Micale L, Fusco C (2010) Copy number variants at Williams-Beuren syndrome 7q11.23 region. Hum Genet 128(1):3–26. doi:10.1007/s00439-010-0827-2
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK et al (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448(7153):553–560
Miyake N, Tsurusaki Y, Koshimizu E, Okamoto N, Kosho T, Brown NJ, Tan TY, Yap PJ, Suzumura H, Tanaka T, Nagai T, Nakashima M, Saitsu H, Niikawa N, Matsumoto N (2016) Delineation of clinical features in Wiedemann-Steiner syndrome caused by KMT2A mutations. Clin Genet 89(1):115–119. doi:10.1111/cge.12586
Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schübeler D (2008) Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30(6):755–766. doi:10.1016/j.molcel.2008.05.007
Mozzetta C, Pontis J, Fritsch L, Robin P, Portoso M, Proux C, Margueron R, Ait-Si-Ali S (2014) The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol Cell 53(2):277–289. doi:10.1016/j.molcel.2013.12.005
Muotri AR, Marchetto MC, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH (2010) L1 retrotransposition in neurons is modulated by MeCP2. Nature 468:443–446
Nagarajan P, Onami TM, Rajagopalan S, Kania S, Donnell R, Venkatachalam S (2009) Role of chromodomain helicase DNA-binding protein 2 in DNA damage response signaling and tumorigenesis. Oncogene 28(8):1053–1062
Narayanan R, Tuoc TC (2014) Roles of chromatin remodeling BAF complex in neural differentiation and reprogramming. Cell Tissue Res 356(3):575–584. doi:10.1007/s00441-013-1791-7
Narayanan R, Pirouz M, Kerimoglu C, Pham L, Wagener RJ, Kiszka KA, Rosenbusch J, Seong RH, Kessel M, Fischer A, Stoykova A, Staiger JF, Tuoc T (2015) Loss of BAF (mSWI/SNF) complexes causes global transcriptional and chromatin state changes in forebrain development. Cell Rep 13(9):1842–1854. doi:10.1016/j.celrep.2015.10.046
Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A et al (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485(7397):242–245. doi:10.1038/nature11011
Ng SB, Bigham AW, Buckingham KJ et al (2010) Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42:790–793
Nicholls RD, Knepper JL (2001) Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet 2:153–175
Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T (1981) Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr 99(4):565–569
Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, Kaneda Y (2009) A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 460(7252):287–291. doi:10.1038/nature08086
Ninkovic J, Steiner-Mezzadri A, Jawerka M, Akinci U, Masserdotti G, Petricca S, Fischer J, von Holst A, Beckers J, Lie CD, Petrik D, Miller E, Tang J, Wu J, Lefebvre V, Demmers J, Eisch A, Metzger D, Crabtree G, Irmler M, Poot R, Gotz M (2013) The BAF complex interacts with Pax6 in adult neural progenitors to establish a neurogenic cross-regulatory transcriptional network. Cell Stem Cell 13:403–418
Okamoto N, Toribe Y, Shimojima K, Yamamoto T (2016) Tatton-Brown-Rahman syndrome due to 2p23 microdeletion. Am J Med Genet A 170A(5):1339–1342. doi:10.1002/ajmg.a.37588
Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257
O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O’Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J (2012) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338(6114):1619–1622. doi:10.1126/science.1227764
Park IY, Powell RT, Tripathi DN, Dere R, Ho TH, Blasius TL, Chiang YC, Davis IJ et al (2016) Dual chromatin and cytoskeletal remodeling by SETD2. Cell 166(4):950–962. doi:10.1016/j.cell.2016.07.005
Pasillas MP, Shah M, Kamps MP (2011) NSD1 PHD domains bind methylated H3K4 and H3K9 using interactions disrupted by point mutations in human sotos syndrome. Hum Mutat 32(3):292–298. doi:10.1002/humu.21424
Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL (1991) Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66:817–822
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B et al (2014) Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 94(5):677–694. doi:10.1016/j.ajhg.2014.03.018
Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, Shah MY, Zheng Y, Will CM et al (2014) Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet 10(9):e1004566. doi:10.1371/journal.pgen.1004566 eCollection 2014 Sep
Pujadas E, Feinberg AP (2012) Regulated noise in the epigenetic landscape of development and disease. Cell 148(6):1123–1131. doi:10.1016/j.cell.2012.02.045
Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T et al (2012) Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380(9854):1674–1682. doi:10.1016/S0140-6736(12)61480-9
Saveliev A, Everett C, Sharpe T, Webster Z, Festenstein R (2003) DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature 422(6934):909–913
Schnetz MP, Bartels CF, Shastri K, Balasubramanian D, Zentner GE, Balaji R, Zhang X, Song L, Wang Z, Laframboise T, Crawford GE, Scacheri PC (2009) Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res 19(4):590–601. doi:10.1101/gr.086983.108
Scourzic L, Couronné L, Pedersen MT, Della Valle V, Diop M, Mylonas E, Calvo J, Mouly E, Lopez CK, Martin N, Fontenay M, Bender A, Guibert S, Dubreuil P, Dessen P, Droin N, Pflumio F, Weber M, Gaulard P, Helin K, Mercher T, Bernard OA (2016) DNMT3A<sup>R882H</sup> mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia. doi:10.1038/leu.2016.29
Seal S, Childhood Overgrowth Consortium, Rahman N (2013) Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet A 161A(12):2972–2980. doi:10.1002/ajmg.a.36229
Sell GL, Margolis SS (2015) From UBE3A to Angelman syndrome: a substrate perspective. Front Neurosci 9:322. doi:10.3389/fnins.2015.00322 eCollection 2015. Review
Siklenka K, Erkek S, Godmann M, Lambrot R, McGraw S, Lafleur C, Cohen T, Xia J, Suderman M, Hallett M, Trasler J, Peters AH, Kimmins S (2015) Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science 350(6261):aab2006. doi:10.1126/science.aab2006
Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, Suvisaari J et al (2016) Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci 19(4):571–577. doi:10.1038/nn.4267
Stef M, Simon D, Mardirossian B, Delrue MA, Burgelin I, Hubert C, Marche M, Bonnet F, Gorry P, Longy M, Lacombe D, Coupry I, Arveiler B (2007) Spectrum of CREBBP gene dosage anomalies in Rubinstein-Taybi syndrome patients. Eur J Hum Genet 15:843–847
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403(6765):41–45
Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, Ragavendran A, Brand H, Lucente D, Miles J, Sheridan SD, Stortchevoi A, Kellis M, Haggarty SJ, Katsanis N, Gusella JF, Talkowski ME (2014) CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci 111(42):E4468–E4477
Sweatt JD (2013) The emerging field of neuroepigenetics. Neuron 80(3):624–632. doi:10.1016/j.neuron.2013.10.023 Review
Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, Zhang F (2015) In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol 33(1):102–106. doi:10.1038/nbt.3055
Tassone F, Hagerman RJ, Taylor AK, Mills JB, Harris SW, Gane LW, Hagerman PJ (2000) Clinical involvement and protein expression in individuals with the FMR1 premutation. Am J Med Genet 91(2):144–152
Tatton-Brown K, Hanks S, Ruark E, Zachariou A, Duarte Sdel V, Ramsay E, Snape K, Murray A, Perdeaux ER et al (2011) Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2(12):1127–1133
Tatton-Brown K, Murray A, Hanks S, Douglas J, Armstrong R, Banka S, Bird LM, Clericuzio CL, Cormier-Daire V et al (2013) Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet A 161A(12):2972–2980. doi:10.1002/ajmg.a.36229
Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, del Vecchio DS, Zachariou A, Hanks S, O’Brien E et al (2014) Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nature Genet 46:385–388 2014
Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H et al (2012) Mutations affecting components of the SWI/SNF complex cause Coffin–Siris syndrome. Nat Genet 44:376–378
Tsurusaki Y, Koshimizu E, Ohashi H, Phadke S, Kou I, Shiina M, Suzuki T, Okamoto N, Imamura S et al (2014) De novo SOX11 mutations cause Coffin-Siris syndrome. Nat Commun 5:4011
Tuoc TC, Boretius S, Sansom SN, Pitulescu ME, Frahm J, Livesey FJ, Stoykova A (2013) Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev Cell 25(3):256–269. doi:10.1016/j.devcel.2013.04.005
Van Houdt JK, Nowakowska BA, Sousa SB, van Schaik BD, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, van den Boogaard MJ et al (2012) Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet 44(4):445–449 . doi:10.1038/ng.1105S1
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP et al (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65:905–914
Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG (2004) Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 36(9):955–957
Vissers LELM, Gilissen C, Veltman JA (2015) Genetic studies in intellectual disability and related disorders. Nat Rev Genet 17(1):9–18
Wagner EJ, Carpenter PB (2012) Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 13(2):115–126. doi:10.1038/nrm3274 Review
Wang J, Weaver IC, Gauthier-Fisher A, Wang H, He L, Yeomans J, Wondisford F, Kaplan DR, Miller FD (2010) CBP histone acetyltransferase activity regulates embryonic neural differentiation in the normal and Rubinstein-Taybi syndrome brain. Dev Cell 18(1):114–125. doi:10.1016/j.devcel.2009.10.023
Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, Turner TN, Liu Y et al (2016) De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun 7:13316. doi:10.1038/ncomms13316
Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, Zondag S, Toriello HV, Magenis RE, Elsea SH (2010) Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet 87(2):219–228. doi:10.1016/j.ajhg.2010.07.011
Xin B, Cruz Marino T, Szekely J, Leblanc J, Cechner K, Sency V, Wensel C, Barabas M, Therriault V, Wang H (2016) Novel DNMT3A germline mutations are associated with inherited Tatton-Brown-Rahman syndrome. Clin Genet. doi:10.1111/cge.12878
Xu GL, Bestor TH, Bourc’his D et al (1999) Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402:187–191
Acknowledgements
We thank people from our laboratory for helpful discussions. We are deeply indebted to D. Bonanomi for critical reading of the manuscript. Work in our laboratory is supported by the Telethon grant (GGP15096) and the Italian Ministry of Health (Young investigator grant no. GR-2013-02355540) to A.S.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mastrototaro, G., Zaghi, M. & Sessa, A. Epigenetic Mistakes in Neurodevelopmental Disorders. J Mol Neurosci 61, 590–602 (2017). https://doi.org/10.1007/s12031-017-0900-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-017-0900-6