
Abstract
Spinal and bulbar muscular atrophy (SBMA) is a late-onset neuromuscular disease caused by a polyglutamine expansion in the androgen receptor gene which results in progressive spinal and bulbar motor neuron degeneration, and muscle atrophy. Although the causative genetic defect is known, until recently, the molecular pathogenesis of the disease was unclear, resulting in few, if any, targets for therapy development. However, over the past decade, our understanding of the pathomechanisms that play a role in SBMA has increased dramatically, and several of these pathways and mechanisms have now been investigated as possible therapeutic targets. In this review, we discuss some of the key pathomechanisms implicated in SBMA and describe some of the therapeutic strategies that have been tested in SBMA to date, which fall into four main categories: (i) gene silencing; (ii) protein quality control and/or increased protein degradation; (iii) androgen deprivation; and (iv) modulation of AR function. Finally, it is also now clear that in addition to a greater understanding of the molecular mechanisms that underlie disease, the development of an effective disease modifying therapy for SBMA will require the coordinated, collaborative effort of research teams with diverse areas of expertise, clinicians, pharmaceutical companies as well as patient groups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spinal and bulbar muscular atrophy (SBMA) is a late-onset neuromuscular disease caused by a polyglutamine expansion in the androgen receptor (AR) gene. The disease is clinically characterized by slowly progressive muscle weakness and atrophy. During the last two decades, basic and clinical research has provided important insights into the disease phenotype and pathophysiology. Nevertheless, a treatment for this disease is currently unavailable. Among other polyglutamine diseases, SBMA is an exception in that the main functions of the mutant protein are well characterized, which is key to the identification of a successful therapeutic target. Other crucial factors for the development of an effective therapy are a good understanding of the underlying molecular mechanisms of a disease, the availability of predictive models and the application of relevant target validation technologies (Gashaw et al. 2012). A druggable target is a protein, peptide or nucleic acid with activity that can be modulated by a drug (Imming et al. 2006). Successful identification of a novel target is followed by detailed molecular target assessments consisting of experimental studies on pharmacodynamic properties according to disease hypothesis. These targets should play critical and preferably un-substitutable roles in the disease processes, have a certain level of functional and structural characteristics to allow for drug specificity, should not be significantly involved in other important processes in humans to limit potential side effects, and their expression should be selective for tissues that are relevant for the disease pathogenesis (Zheng et al. 2006). However, a major drawback in target validation arises from the limited ability of preclinical disease models to predict benefit in patients (McGonigle and Ruggeri 2014): analyses have shown that most failures are in phase II trials, and at least 50 % of these are due to lack of efficacy and 25 % due to toxicity (Wehling 2009). Therefore, the validity of a given target in a specific disease can only finally be judged after a clinical proof of concept trial. It follows that a thoroughly performed target validation is essential to reduce attrition rate in the later stages of drug development.
In this review, we discuss the molecular pathogenesis and the therapeutic strategies currently being developed for SBMA.
Molecular Pathogenesis
The AR is a member of the nuclear hormone receptor superfamily, residing in the cytoplasm when inactive, bound to a complex of heat shock proteins (HSP), including HSP90, which stabilize its protein conformation, prevent misfolding and enable binding to its main ligand, dihydrotestosterone (DHT). Upon DHT binding, the AR dimerizes, releases the chaperone complex and translocates to the nucleus where it binds to its site-specific DNA sequence termed the androgen response element (ARE), leading to the transcriptional regulation of target genes. All these are requisite steps in disease pathogenesis (Nedelsky et al. 2010). Animal models and human studies have shown that gender specificity in SBMA is due to higher levels of circulating androgens in males (Katsuno et al. 2012; Takeyama et al. 2002).
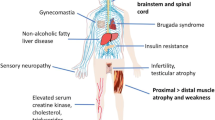
The possibility that a toxic gain of function mechanism explains disease pathogenesis in SBMA is mostly supported by evidence which shows that mutant AR accumulates into intra-nuclear and intra-cytoplasmic inclusions in motor neurons in the brainstem and spinal cord, as well as in non-neuronal tissues including the prostate, testes and skin (Li et al. 1998). Such cytoplasmic inclusions are the pathological hallmark of SBMA and other polyglutamine diseases. Nevertheless, patients often display signs of mild AR insensitivity, such as gynecomastia, testicular atrophy and reduced fertility, suggesting a more complicated scenario, where mechanisms of both gain of function and loss of function are not mutually exclusive. Recent studies in spinocerebellar ataxia type 1 (SCA1) and other polyglutamine diseases, including SBMA, have provided evidence in favour of a model in which the normal function of the disease protein is tied to the mechanism of pathogenesis (Emamian et al. 2003; Friedman et al. 2007; Lim et al. 2008; McMahon et al. 2005; Palhan et al. 2005; Monks et al. 2007). Loss of lower motor neurons in the anterior horn of the spinal cord as well as in the brainstem motor nuclei, the fundamental histopathological finding in SBMA (Sobue et al. 1989), ultimately result from the accumulation of a number of hits, such as transcriptional dysregulation (McCampbell et al. 2000; Minamiyama et al. 2004), mitochondrial impairment and oxidative stress (Ranganathan et al. 2009), and possibly, disruption of axonal transport (Morfini et al. 2006; Katsuno et al. 2006; Malik et al. 2011).
Therapeutic Targets in SBMA
During the last few years, a number of potential therapeutic strategies for SBMA have emerged from work using in vitro and in vivo models of the disease, some of which are starting to be tested in clinical trials. These strategies mostly fall into the following categories: (i) gene silencing; (ii) protein quality control and/or increased protein degradation; (iii) androgen deprivation; and (iv) modulation of AR function.
Gene Silencing
Therapies designed to target and suppress production of the mutant protein, upstream of its deleterious effects, represent a promising therapeutic strategy for SBMA and other polyglutamine diseases. In particular, RNA-based approaches, such as RNA interference (RNAi) and antisense oligonucleotides (AONs), hold great potential, although they are currently hampered by technical obstacles associated with design, discrimination and delivery, which must be overcome before they can be effectively applied in the clinical setting (Watson and Wood 2012).
A single intra-cerebroventricular administration of AONs targeting the AR has been shown to result in efficient suppression of the mutant gene expression in the CNS, and delayed onset and progression of motor dysfunction in a SBMA mouse model (Sahashi et al. 2015). Nevertheless, AONs recently developed to suppress AR gene expression in the periphery, but not the CNS, were shown to improve the disease phenotype in mutant mice after subcutaneous administration (Lieberman et al. 2014). This work, together with the evidence that muscle-specific excision of a human AR121Q transgene in a mouse model of SBMA prevents the development of both systemic and neuromuscular disease phenotypes (Cortes et al. 2014a), suggests that expression of mutant AR in the muscle is an important contributor to disease pathogenesis and that peripheral administration of therapeutics should be explored in SBMA patients (Lieberman et al. 2014).
Over the last decade, micro-RNAs (miRNAs) have become another viable option for target suppression in vivo, and several miRNA-based therapies are currently in pre-clinical or clinical development for a variety of conditions, including polyglutamine diseases (Elmén et al. 2008; Rodríguez-Lebrón et al. 2013; Keiser et al. 2014). Compared to AONs, miRNAs are likely to be less toxic and less immunogenic, given that they make use of naturally occurring RNAi mechanisms and can be more easily delivered into cells due to their smaller size (Huang et al. 2014). PolyQ-AR suppression was recently achieved in a mouse model of SBMA using viral delivery of miR-196a, which indirectly enhanced the decay of the AR mRNA by silencing CELF2, a protein involved in regulation of AR mRNA stability (Miyazaki et al. 2012).
Protein Quality Control and Increased Protein Degradation
Clearance of mutant polyQ-expanded AR by increased protein degradation and/or manipulation of protein quality control systems, such as protein chaperones and the heat shock response (HSR), represents an attractive therapeutic strategy in disorders such as SBMA, where protein aggregation is a key pathological feature. In SBMA, the characteristic nuclear and cytoplasmic inclusions which contain the polyQ-AR protein (Adachi et al. 2005) also harbour components of the ubiquitin–proteasome system (UPS) as well as protein chaperones, including HSPs (Stenoien et al. 1999; Adachi et al. 2001). This finding suggests that these cellular defence mechanisms may be involved in SBMA pathology.
Cells usually respond to the presence of misfolded proteins by either activating protein quality control systems, through the action of chaperones and their co-chaperones, or by encouraging the degradation of misfolded proteins via the UPS and autophagy pathway. Chaperones function to stabilize unfolded proteins and support their correct refolding and assembly into their native conformation. The AR is normally stabilized in the cytoplasm by interacting with a complex that includes HSP90 and HSP70. However, HSP levels have been reported to be reduced in mouse models of SBMA (Adachi et al. 2003; Katsuno et al. 2005). Therefore, the effects of (i) decreasing protein misfolding and/or (ii) enhancing clearance of the expanded polyQ-AR on SBMA pathology have been widely studied.
Several approaches have been taken to reduce protein misfolding in SBMA, by targeting of various components of the HSR as a means of upregulating inducing overexpression of HSPs (Bailey et al. 2002; Waza et al. 2005; Adachi et al. 2007), and many of these approaches have proved successful, at least in models of SBMA. Treatment with geranylgeranylacetone (GGA), which increases levels of Hsp70, Hsp90 and Hsp105 via activation of the HSF-1 transcription factor, ameliorates the disease phenotype in SBMA mice (Katsuno et al. 2005). Similarly, treatment with the Hsp90 inhibitor, 17-allylamino-17-demethoxygeldanamycin (17-AAG), has been shown to increase degradation of the polyQ-AR, via the proteasomal pathway (Waza et al. 2005), as does a more potent version of this compound, 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) (Tokui et al. 2009). Furthermore, overexpression of the C terminus of Hsc70 (heat shock cognate protein 70-interacting protein, CHIP), which interacts with both Hsp90 and Hsp70, has been shown to be beneficial in AR-97Q SBMA mice, by promoting degradation of mutant protein via the UPS (Adachi et al. 2007). Overexpression of Hip, a co-chaperone that enhances binding of Hsp70 to its substrates, promotes protein ubiquitination and increases polyQ-AR clearance (Wang et al. 2013). These authors also identified a small molecule that acts similarly to Hip, promoting Hsp70 binding to unfolded proteins (Wang et al. 2013). Most recently, a compound called paeoniflorin has also been reported to increase the expression of chaperones and enhance both the UPS and autophagy pathway in an SBMA model (Tohnai et al. 2014).
Although several of these compounds are effective in increasing the expression of HSPs, non-specific upregulation of HSP levels can have significant consequences. For example, intracellular heat shock proteins are highly expressed in cancerous cells and are essential to their survival. It is therefore not surprising that small molecule inhibitors of HSP expression have been investigated as potential anticancer agents.
However, one agent that can upregulate HSP expression yet avoid the potential problems of non-specific elevation in HSPs is Arimoclomol, a co-inducer of the HSR which has been shown to be effective in an SBMA mouse model (Malik et al. 2013). Arimoclomol upregulates HSP expression via prolonged activation of HSF-1 but has the significant advantage of enhancing the HSR only in stressed cells in which the HSR is already activated (Vigh et al. 1997; Kieran et al. 2004; Kalmar et al. 2008). Arimoclomol therefore acts like a smart drug, reducing the likelihood of non-specific side effects which can occur by using direct inducers of the HSR. Arimoclomol has been shown to ameliorate the disease phenotype and improve motor neuron survival in the SOD1G93A mouse model of ALS (Kieran et al. 2004; Kalmar et al. 2008). Similarly, arimoclomol was found to delay disease progression in the AR100 mouse model of SBMA, by both preventing motor neuron degeneration and alleviating muscle atrophy. Significantly, arimoclomol is currently undergoing a phase 2/3 trial in the USA in SOD1-ALS patients (www.ClinicalTrials.gov) (Cudkowicz et al. 2008), and a phase II trial in patients with the muscle disorder inclusion body myositis (IBM) has just been completed (Machado et al. 2013).
Although inclusion formation is a characteristic, pathological feature of several neurodegenerative diseases including SBMA, the role they play in disease pathology remains unclear. Indeed, it has been suggested that inclusion formation may initially be protective to cells, but as disease progresses, the inclusions may become detrimental to normal cellular function and viability (Simeoni et al. 2000; Montie et al. 2009; Rusmini et al. 2013), at which stage, clearance of the expanded polyQ-AR may become essential. However, failure of autophagic flux and the autophagic protein degradation pathway has been shown to contribute to cytoplasmic accumulation of polyQ-AR within motor neurons (Montie et al. 2009; Rusmini et al. 2010). Furthermore, polyQ-AR can directly affect and compromise autophagy, resulting in development of a disease phenotype in models of SBMA (Rusmini et al. 2010; Cortes et al. 2014b). Therefore, enhancing the autophagy pathway may be advantageous in alleviating disease progression in SBMA. Bicalutamide, the anti-androgen, has been reported to enhance cytoplasmic degradation via autophagy of polyQ-AR (Orr et al. 2010), and the autophagy activator, rapamycin, was found to be neuroprotective in a drosophila model of SBMA (Pandey et al. 2007). Additionally, trehalose has been reported to augment the degradation of polyQ-AR via autophagy (Montie et al. 2009; Rusmini et al. 2013), and the combined treatment regime of bicalutamide and trehalose has a potent inhibitory effect on polyQ-AR accumulation in the mouse model of SBMA (Giorgetti et al. 2015). Finally, since activation of the UPR by polyQ-AR and the resultant ER stress has been shown to occur in several models of SBMA (Yu et al. 2011; Montague et al. 2014), targeting of ER stress with salubrinal, a modulator of the PERK arm of the UPR, was also shown to be effective in reducing ER stress-induced apoptosis in the AR100 SBMA mouse model (Montague et al. 2014).
Androgen Deprivation
Several studies in animal models have demonstrated that the neurological symptoms and histopathological findings in SBMA depend on the circulating level of testosterone (Katsuno et al. 2002; Takeyama et al. 2002). In support of this view, surgical castration (Katsuno et al. 2002; Chevalier-Larsen et al. 2004) and androgen deprivation using leuprorelin (Katsuno et al. 2003) in male mice reverses the SBMA phenotype. Leuprorelin, a potent luteinizing hormone-releasing hormone (LHRH) analogue which suppresses the release of gonadotropins, luteinizing hormone and follicle-stimulating hormone, has been extensively employed for medical castration in the therapy of prostate cancer (Attard et al. 2015). Its safety and tolerability have been widely approved, although it can have significant side effects, including decreased libido, impotence, osteoporosis and fatigue. The effects of leuprorelin treatment in mice (prolonged life span, reversal of the behavioural and histopathological phenotypes) were ascribed to the almost complete androgen blockade, as testosterone exacerbated phenotypic expression in leuprorelin-treated male transgenic mice (Katsuno et al. 2003).
In order to determine whether androgen deprivation could be translated into the clinic, a two-arm, randomized, placebo-controlled, phase 2 clinical trial was conducted. Fifty SBMA patients underwent subcutaneous injections of leuprorelin acetate or placebo for 48 weeks, followed by an open-label trial for an additional 96 weeks. The treatment significantly extended the duration of cricopharyngeal opening in videofluorography and decreased mutant AR accumulation in scrotal skin biopsy. Autopsy of one patient who received leuprorelin suggested that androgen deprivation inhibits the nuclear accumulation and/or stabilization of mutant AR in the motor neurons of the spinal cord and brainstem (Banno et al. 2009).
These promising results led to the establishment of a larger, multicentre, randomized, placebo-controlled clinical trial of leuprorelin in SBMA, where the primary endpoint was pharyngeal barium residue, measured by videofluorography. A total of 199 SBMA male patients were assigned to receive either leuprorelin or placebo subcutaneous injections every 3 months for 12 months. The treatment did not show significant effects on swallowing function in SBMA patients. Interestingly, a significantly greater reduction in barium residue was observed in patients with disease duration of less than 10 years, strongly suggesting that early treatment is critical (Katsuno et al. 2010). More recently, a randomized, double-blind, placebo-controlled, single-site clinical trial was also conducted in 50 SBMA patients, randomly assigned to receive oral daily doses of either dutasteride or placebo for 24 months. Dutasteride is a potent inhibitor of the enzyme 5-α-reductase, which mediates the conversion of testosterone to DHT. The primary outcome measure was the quantitative muscle assessment (QMA), a scale for muscle strength and fatigue. This clinical trial showed that dutasteride was safe and well-tolerated and had a slight effect on the progression of muscle weakness, although non-significant (Fernández-Rhodes et al. 2011).
Based on the evidence shown in studies in animals and in patients, the possibility that androgen depleting therapies slow the progression of SBMA needs to be further verified in clinical trials with a rigorous and efficient design (Banno et al. 2011).
Modulation of AR Function
In recent years, studies on SBMA and other polyglutamine diseases have showed that alterations in the normal properties of the mutant protein, rather than acquisition of novel toxic functions, are determinant for the disease pathogenesis (Zoghbi and Orr 2009). Therapeutic strategies aimed at restoring the normal function of the mutant protein are particularly relevant for SBMA since only one copy of the gene is present and complete knockdown of the AR may result in worsening of the signs and symptoms linked to the loss of function.
Ligand binding induces conformational changes that lead to intra- and inter-molecular N/C interactions; mutations that prevent the N/C interaction counteract polyQ-mediated toxicity in SBMA, suggesting that this ligand-induced conformational change is a potential therapeutic target (Nedelsky et al. 2010; Orr et al. 2010). Selective androgen receptor modulators, which prevent the N/C interaction, ameliorated AR aggregation and toxicity while maintaining AR function, further supporting the relevance of this conformational change in SBMA pathogenesis and highlighting a novel therapeutic strategy to prevent the SBMA phenotype while retaining AR transcriptional function (Orr et al. 2010). Furthermore, curcumin-related compound 5- hydroxy-1,7-bis[3,4-dimethoxyphenyl]-1,4,6-heptatrien-3-one (ASC-J9), found to ameliorate the disease phenotype in SBMA mice, is believed to disrupt the interaction between the AR and its co-regulator ARA70, although the mechanism of action has not yet been completely unravelled (Yang et al. 2007).
Post-translational protein modifications, such as phosphorylation, SUMOylation and acetylation, modulate AR toxicity and therefore represent another possible target for therapeutic intervention.
Studies in vitro and in vivo have shown that phosphorylation of mutant AR by the kinase Akt is followed by reduced ligand binding, ligand-dependent nuclear translocation, transcriptional activation and toxicity and increased AR clearance via the ubiquitin–proteasome system (Palazzolo et al. 2007; Palazzolo et al. 2009). Genetic overexpression of a muscle-specific isoform of IGF-1 selectively in skeletal muscle in SBMA mice results in increased Akt activation and AR phosphorylation in muscle and rescue of the behavioural and histopathological abnormalities in SBMA mice (Palazzolo et al. 2009). Moreover, pharmacologic manipulation of IGF-1/Akt signalling in SBMA mice attenuated disease manifestations, even when treatment was started after disease onset, suggesting that compounds acting on the IGF-1/Akt pathway may be of therapeutic value for SBMA (Rinaldi et al. 2012).
Among other post-translational modifications that hold potential therapeutic value are acetylation and SUMOylation. Nuclear sirtuin 1 (SIRT1) deacetylates hyperacetylated polyQ-expanded AR and reduces DHT-dependent aggregation and toxicity in cell models of SBMA (Montie et al. 2011). Relieving the inhibitory effect of AR SUMOylation (SUMO: small ubiquitin-like modifier) resulted in mitigation of the transcriptional deficits of the mutant AR, prolonged survival and rescued exercise endurance and type I muscle fibre atrophy in a SBMA mouse model genetically modified to be SUMOylation resistant (Chua et al. 2015).
Concluding Remarks
Efforts in trying to unravel the pathophysiology of SBMA, and other polyglutamine diseases, have clearly shown that it is likely that more than one mechanism contributes to the pathogenesis. These include the following: (1) formation of toxic oligomeric complexes; (2) misfolding of the disease protein resulting in altered protein homeostasis; (3) deleterious protein interactions; (4) transcriptional dysregulation; (5) mitochondrial dysfunction; and (6) possibly, impaired axonal transport (Katsuno et al. 2012). Toxicity ultimately results when the ability of the cell to cope with the cumulative damage is worn out. The tissue-selectivity can be at least in part explained by the observation that these mechanisms of defence are not equivalent between cells. Although each of these molecular mechanisms could be targeted for therapeutic interventions, upstream events are more likely to result in effective strategies.
The evidence of a toxic gain of function as the main mechanism of disease pathogenesis suggests that reducing the levels of the mutant protein represent the ideal target for an SBMA therapy and possibly for other polyglutamine diseases. Nevertheless, this strategy may be potentially hampered by an exacerbation of the symptoms of loss of function, which in the case of SBMA undoubtedly affect the patients’ quality of life. Targeting highly selective functions of the mutant protein relevant for the disease pathogenesis may be particularly appealing because, whilst attempting to model the complexity of the disease, it may also provide a specific target for therapy.
Although SBMA is believed to be primarily a motor neuron disease, increasing evidence suggests that myogenic defects contribute significantly to the disease pathogenesis. Serum creatine kinase levels are higher than normal in SBMA patients (Lee et al. 2005), and this elevation can be detected prior to the onset of SBMA clinical symptoms (Chahin and Sorenson 2009). Moreover, muscle biopsies from SBMA patients show myogenic changes as well as neurogenic atrophy (Sorarù et al. 2008), muscle pathology precedes spinal cord pathology in a knock-in mouse model of SBMA (Yu et al. 2006) and muscle-specific overexpression of wild-type AR leads to an SBMA-like phenotype (Monks et al. 2007). Recent studies in mouse models of SBMA have demonstrated a crucial role for muscle expression of polyQ-AR in the disease and suggested muscle-directed therapies as effective treatments (Cortes et al. 2014a; Lieberman et al. 2014). These findings can have a significant impact on designing a therapeutic strategy for SBMA because muscle is readily accessible to systemic treatment. Further work is needed to answer the question whether this option can be translated into an effective treatment in patients (Rinaldi et al. 2014).
Once a molecular target is chosen and optimized to reach the required beneficial effect and with an acceptable safety profile, a key question which remains is how to run conclusive therapeutic trials in slowly progressive diseases like SBMA. The recent clinical trials testing androgen-deprivation therapies in SBMA patients have certainly provided clues for the design of future studies involving human subjects in this disease. Quantitative analysis of natural history, including genetic, biological, and clinical data, and suitable surrogate endpoints, which follow the disease progression and response to therapy are both necessary for long-term evaluation of therapeutic agents for SBMA.
Finally, given the rarity of the disease, patient groups are small, and the risk of a trial being underpowered to test the efficacy has to be taken into account.
Research into the pathomechanisms of SBMA has progressed dramatically over the last few years, shedding new insights into the causation of this disease. In order to harness these discoveries into a more complete understanding of the molecular events leading to neurodegeneration and to be able to ultimately translate this knowledge into disease modifying treatments—or even a cure—for SBMA, collaboration between research teams, clinicians, pharmaceutical companies and patient groups is an urgent requirement.
References
Adachi H, Katsuno M, Minamiyama M, et al. (2003) Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 23:2203–2211
Adachi H, Katsuno M, Minamiyama M, et al. (2005) Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain 128:659–670
Adachi H, Kume A, Li M, et al. (2001) Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum Mol Genet 10:1039–1048
Adachi H, Waza M, Tokui K, et al. (2007) CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J Neurosci. 27:5115–5126
Attard G, Parker C, Eeles RA, et al. (2015) Prostate cancer. Lancet pii: S0140-6736(14):61947–61944
Bailey CK, Andriola IF, Kampinga HH, Merry DE (2002) Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet 11:515–523
Banno H, Katsuno M, Suzuki K (2009) Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann Neurol 65:140–150
Banno H, Katsuno M, Suzuki K, Sobue G (2011) Dutasteride for spinal and bulbar muscular atrophy. Lancet Neurol 10:113–115
Chahin N, Sorenson EJ (2009) Serum creatine kinase levels in spinobulbar muscular atrophy and amyotrophic lateral sclerosis. Muscle Nerve 40:126–129
Chevalier-Larsen ES, O’Brien CJ, Wang H, et al. (2004) Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. 24:4778–4786
Chua JP, Reddy SL, Yu Z, et al. (2015) Disrupting SUMOylation enhances transcriptional function and ameliorates polyglutamine androgen receptor-mediated disease. J Clin Invest 125:831–845
Cortes CJ, Ling SC, Guo LT, et al. (2014a) Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 82:295–307
Cortes CJ, Miranda HC, Frankowski H, et al. (2014b) Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat Neurosci 17:1180–1189
Cudkowicz ME, Shefner JM, Simpson E, et al. (2008) Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve 38:837–844
Elmén J, Lindow M, Schütz S, et al. (2008) LNA-mediated microRNA silencing in non-human primates. Nature 452:896–899
Emamian ES, Kaytor MD, Duvick LA, et al. (2003) Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38:375–387
Fernández-Rhodes LE, Kokkinis AD, White MJ, et al. (2011) Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: a randomised placebo-controlled trial. Lancet Neurol 10:140–147
Friedman MJ, Shah AG, Fang ZH, et al. (2007) Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci 10:1519–1528
Gashaw I, Ellinghaus P, Sommer A, Asadullah K (2012) What makes a good drug target? Drug Discov Today 16:1037–1043
Giorgetti E, Rusmini P, Crippa V, et al. (2015) Synergic prodegradative activity of bicalutamide and trehalose on the mutant androgen receptor responsible for spinal and bulbar muscular atrophy. Hum Mol Genet 24:64–75
Huang J, Lyu H, Wang J, Liu B (2014) MicroRNA regulation and therapeutic targeting of survivin in cancer. Am J Cancer Res 5:20–31
Imming P, Sinning C, Meyer A (2006) Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 5:821–834
Kalmar B, Novoselov S, Gray A, Cheetham ME, Margulis B, Greensmith L (2008) Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J Neurochem 107:339–350
Katsuno M, Adachi H, Doyu M, et al. (2003) Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med 9:768–773
Katsuno M, Adachi H, Kume A, et al. (2002) Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35:843–854
Katsuno M, Adachi H, Minamiyama M, et al. (2006) Reversible disruption of dynactin 1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration. J Neurosci. 26:12106–12117
Katsuno M, Banno H, Suzuki K, et al. (2010) Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 9:875–884
Katsuno M, Sang C, Adachi H, et al. (2005) Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A 102:16801–16806
Katsuno M, Tanaka F, Adachi H, et al. (2012) Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA). Prog Neurobiol 99:246–256
Keiser MS, Boudreau RL, Davidson BL (2014) Broad therapeutic benefit after RNAi expression vector delivery to deep cerebellar nuclei: implications for spinocerebellar ataxia type 1 therapy. Mol Ther 22:588–595
Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L (2004) Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med 10:402–405
Lee JH, Shin JH, Park KP, et al. (2005) Phenotypic variability in Kennedy’s disease: implication of the early diagnostic features. Acta Neurol Scand 112:57–63
Lieberman AP, Yu Z, Murray S, et al. (2014) Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep 7:774–784
Lim J, Crespo-Barreto J, Jafar-Nejad P, et al. (2008) Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 452:713–718
Li M, Miwa S, Kobayashi Y, et al. (1998) Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol 44:249–254
Machado P, Brady S, Hanna MG (2013) Update in inclusion body myositis. Curr Opin Rheumatol 25:763–771
Malik B, Nirmalananthan N, Bilsland LG, et al. (2011) Absence of disturbed axonal transport in spinal and bulbar muscular atrophy. Hum Mol Genet 20:1776–1786
Malik B, Nirmalananthan N, Gray AL, La Spada AR, Hanna MG, Greensmith L (2013) Co-induction of the heat shock response ameliorates disease progression in a mouse model of human spinal and bulbar muscular atrophy: implications for therapy. Brain 136:926–943
McCampbell A, Taylor JP, Taye AA, et al. (2000) CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet 9:2197–2202
McGonigle P, Ruggeri B (2014) Animal models of human disease: challenges in enabling translation. Biochem Pharmacol 87:162–171
McMahon SJ, Pray-Grant MG, Schieltz D, Yates 3rd JR, Grant PA (2005) Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SLIK histone acetyltransferase activity. Proc Natl Acad Sci U S A 102:8478–8482
Minamiyama M, Katsuno M, Adachi H, et al. (2004) Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 13:1183–1192
Miyazaki Y, Adachi H, Katsuno M, et al. (2012) Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat Med 18:1136–1141
Monks DA, Johansen JA, Mo K, et al. (2007) Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc Natl Acad Sci U S A 104:18259–18264
Montague K, Malik B, Gray AL, et al. (2014) Endoplasmic reticulum stress in spinal and bulbar muscular atrophy: a potential target for therapy. Brain 137:1894–1906
Montie HL, Cho MS, Holder L, et al. (2009) Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 18:1937–1950
Montie HL, Pestell RG, Merry DE (2011) SIRT1 modulates aggregation and toxicity through deacetylation of the androgen receptor in cell models of SBMA. J Neurosci. 31:17425–17436
Morfini G, Pigino G, Szebenyi G, You Y, Pollema S, Brady ST (2006) JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat Neurosci 9:907–916
Nedelsky NB, Pennuto M, Smith RB, et al. (2010) Native functions of the androgen receptor are essential to pathogenesis in a drosophila model of spinobulbar muscular atrophy. Neuron 67:936–952
Orr CR, Montie HL, Liu Y, et al. (2010) An interdomain interaction of the androgen receptor is required for its aggregation and toxicity in spinal and bulbar muscular atrophy. J Biol Chem. 285:35567–33577
Palazzolo I, Burnett BG, Young JE, et al. (2007) Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum Mol Genet 16:1593–1603
Palazzolo I, Stack C, Kong L, et al. (2009) Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 63:316–328
Palhan VB, Chen S, Peng GH, et al. (2005) Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci U S A 102:8472–8477
Pandey UB, Nie Z, Batlevi Y, et al. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447:859–863
Ranganathan S, Harmison GG, Meyertholen K, Pennuto M, Burnett BG, Fischbeck KH (2009) Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum Mol Genet 18:27–42
Rinaldi C, Bott LC, Chen KL, et al. (2012) Insulinlike growth factor (IGF)-1 administration ameliorates disease manifestations in a mouse model of spinal and bulbar muscular atrophy. Mol Med 18:1261–1268
Rinaldi C, Bott LC, Fischbeck KH (2014) Muscle matters in Kennedy’s disease. Neuron 82:251–253
Rodríguez-Lebrón E, Costa Mdo C, Luna-Cancalon K, et al. (2013) Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol Ther 21:1909–1918
Rusmini P, Bolzoni E, Crippa V, et al. (2010) Proteasomal and autophagic degradative activities in spinal and bulbar muscular atrophy. Neurobiol Dis 40:361–369
Rusmini P, Crippa V, Giorgetti E, et al. (2013) Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol Aging 34:2585–2603
Sahashi K, Katsuno M, Hung G, et al. (2015) Silencing neuronal mutant androgen receptor in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. pii: ddv300.
Simeoni S, Mancini MA, Stenoien DL, et al. (2000) Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum Mol Genet 9:133–144
Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A (1989) X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain 112:209–232
Sorarù G, D’Ascenzo C, Nicolao P, et al. (2008) Muscle histopathology in upper motor neuron-dominant amyotrophic lateral sclerosis. Amyotroph Lateral Scler 9:287–293
Stenoien DL, Cummings CJ, Adams HP, et al. (1999) Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet 8:731–741
Takeyama K, Ito S, Yamamoto A, et al. (2002) Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in drosophila. Neuron 35:855–864
Tohnai G, Adachi H, Katsuno M, et al. (2014) Paeoniflorin eliminates a mutant AR via NF-YA-dependent proteolysis in spinal and bulbar muscular atrophy. Hum Mol Genet 23:3552–3565
Tokui K, Adachi H, Waza M, et al. (2009) 17-DMAG ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an SBMA model mouse. Hum Mol Genet 18:898–910
Vigh L, Literati PN, Horvath I, et al. (1997) Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat Med 3:1150–1154
Wang AM, Miyata Y, Klinedinst S, et al. (2013) Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol 9:112–118
Waza M, Adachi H, Katsuno M, et al. (2005) 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med 11:1088–1095
Watson LM, Wood MJ (2012) RNA therapy for polyglutamine neurodegenerative diseases. Expert Rev Mol Med 14:e3
Wehling M (2009) Assessing the translatability of drug projects: what needs to be scored to predict success? Nat Rev Drug Discov. 8:541–546
Yang Z, Chang YJ, Yu IC, et al. (2007) ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat Med 13:348–353
Yu Z, Dadgar N, Albertelli M, et al. (2006) Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Invest 116:2663–2672
Yu Z, Wang AM, Adachi H, et al. (2011) Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet 7:e1002321
Zheng CJ, Han LY, Yap CW, Ji ZL, Cao ZW, Chen YZ (2006) Therapeutic targets: progress of their exploration and investigation of their characteristics. Pharmacol Rev 58:259–279
Zoghbi HY, Orr HT (2009) Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J Biol Chem. 284:7425–7429
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Rinaldi, C., Malik, B. & Greensmith, L. Targeted Molecular Therapies for SBMA. J Mol Neurosci 58, 335–342 (2016). https://doi.org/10.1007/s12031-015-0676-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-015-0676-5