
Abstract
This study aimed to investigate whether the classic hepatoprotective drug polyene phosphatidylcholine (PPC) regulates macrophage polarization and explores the potential role of TLR-2 in this process. In RAW264.7 macrophages and murine bone marrow-derived macrophages (BMDMs) stimulated by lipopolysaccharide (LPS), PPC significantly inhibited the production of IL-6, TNF-α, and the mRNA expression of M1-type macrophage markers. Consistently, PPC reduced the mRNA expression of several key enzymes in the pathways of glycolysis and lipid synthesis while increasing the expression of key enzymes associated with lipid oxidation. Moreover, blocking the glycolytic pathway using 2-deoxy-d-glucose (2-DG) significantly enhanced the anti-inflammatory effect of PPC. However, inhibition of lipid oxidation using GW9662 (an inhibitor of PPAR-γ) and GW6471 (an inhibitor of PPAR-α) abolished the anti-inflammatory effect of PPC. Interestingly, TLR-2 expression in macrophages was significantly downregulated after exposure to PPC. Moreover, pre-activation of TLR-2 hampered the anti-inflammatory effect of PPC. In addition, PPC did not inhibit the secretion of IL-6 and TNF-α in TLR-2−/− BMDMs that were activated by LPS. This was consistent with the increased expression of M1 markers and glycolytic and lipid synthesis enzymes but decreased lipid oxidation-related enzymes. These results showed that PPC inhibits the differentiation of M1-type macrophages, which was most likely related to TLR-2-mediated metabolic reprogramming.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Macrophages maintain considerable plasticity and heterogeneity and respond to environmental signals by altering effector phenotypes. Macrophages can polarize into two types according to their phenotype and secreted cytokines, classically activated M1 type and alternatively activated M2-type macrophages [1,2,3]. M1 macrophages develop in response to proinflammatory stimuli (LPS or IFN-γ) and are characterized by high levels of CD16/32, TNF-α, IL-12, iNOS, and chemokines such as CXCL9, CXCL10, and CXCL11. M2 macrophages are induced by IL-4 or chitin and are characterized by high expression of CD206, Arg-1, IL-10, and chemokines such as CCL2, CCL17, and CCL22 [2, 4,5,6,7,8,9]. Interestingly, macrophages can switch between M1 and M2 phenotypes, and specific alternations in macrophage phenotype have been indicated in various pathogenic conditions, such as tumors, obesity, and liver and kidney disease [10, 11]. In particular, the polarization of macrophages to the M2 phenotype is beneficial for controlling inflammation and other pathological states [12,13,14]. Thus, the status adjustment of macrophages may be an important strategy for disease treatment and diagnosis.
Polyene phosphatidylcholine (PPC) is a nontoxic phospholipid. Phosphatidylcholine (PC), the major component of PPC, is a component of organelle membranes and the cell membrane. PPC is rich in polyunsaturated fatty acids such as linolenic acid and oleic acid and is mainly extracted from soybeans [15, 16]. Traditionally, PPC functions to maintain health and beauty. Moreover, PPC is currently widely used to treat various types of liver diseases, such as viral hepatitis liver damage or nonalcoholic steatohepatitis [10, 16, 17], because it can repair cell membranes by improving the function and integrity of biofilm. Interestingly, in recent years, the application of PPC in the medical field has significantly increased. It was reported that several conjugates containing PC improve inflammatory diseases. In our previous study, PPC inhibited the inflammatory response in macrophages and improved the condition in arthritis [18]. Moreover, phosphorylcholine-tuftsin prevents murine colitis induced by dextran sulfate sodium salt [19]. These data suggest that PPC can be used as an anti-inflammatory drug. However, its underlying anti-inflammatory mechanism is largely unclear.
Macrophages express high levels of toll-like receptors (TLRs) after activation. TLRs activate downstream signaling pathways and induce the expression of inflammatory factors, thereby causing an inflammatory response [20,21,22]. TLR-2 is one of the most widely expressed members of the TLR family in macrophages and recognizes the most diverse species of pathogenic microorganisms and their products; TLR-2 is activated by various ligands such as Pam3CSK4, peptidoglycan, apoptotic nucleosomes, and zymosan [22, 23]. Our previous study also showed that TLR-2 was downregulated in PPC-treated macrophages, which suggests that TLR-2 might have an important role in the anti-inflammatory effect of PPC. This study further explored the role of TLR-2 in this process.
In recent years, with the rise of immunometabolism, accumulating evidences have shown that specific metabolic reprogramming events were required for the phenotypic differentiation of macrophages [24,25,26]. A previous study showed the biphasic metabolic kinetics of macrophages during bacterial infection: M1 polarization in the early stages of infection while M2 polarization in chronic infection [27]. Importantly, specific metabolic pathways were indicated in the M1 and M2 polarization of macrophages [26, 27]. Metabolic reprogramming refers to key modulations in the bioenergy pathways in immune cells, including glycolysis, fatty acid oxidation, oxidative phosphorylation, pentose phosphate pathway, and amino acid metabolism. These processes occur in a variety of activated immune cells, which in turn regulate their functional properties. Generally, static and anti-inflammatory cells rely mainly on fatty acid oxidation, while activated and inflammatory cells rely mainly on glycolysis. In macrophages, the conversion of oxidation phosphorylation to glycolysis is a hallmark of activated M1 macrophages, while M2 macrophages rely primarily on the oxidative phosphorylation pathway to provide energy [26, 27]. However, it is still unclear if PPC regulates the inflammatory response through metabolic reprogramming.
The present study investigated whether PPC functions as an anti-inflammatory drug by regulating the polarization of M1-type macrophages and explored the potential role of TLR-2 in this process. The results showed that PPC significantly inhibited M1 differentiation of LPS-activated macrophages through inhibition of the glycolytic pathway and enhancement of lipid oxidation. Moreover, the downregulation of TLR-2 was essential for the anti-inflammatory effect of PPC, which was most likely mediated by triggering this metabolic reprogramming. Overall, this study provides novel insight that clarifies the anti-inflammatory mechanism of PPC.
Materials and methods
Reagents
DMEM (Jiangsu, China); IMDM (Jiangsu, China); Cell Counting Kit-8 (CCK8, Jiangsu, China); fetal bovine serum (FBS, Bioind, Australia); M-CSF (Stem cell, USA); PPC (Chengdu, China); lipopolysaccharide (LPS, O55:B5, Sigma, USA); Pam3CSK4 (InvivoGen, USA); TRIzol (Takara, Japan); PrimeScript™ RT reagent kit with gDNA eraser (Takara, Japan), TB Green™ Premix Ex Taq™ II (Takara, Japan), IL-10, IL-6, and TNF-α ELISA kits (R&D Systems, USA); 2-deoxy-d-glucose (2-DG, Sigma, USA); GW9662 (MedChemExpress, USA); and GW6471 (MedChemExpress, USA).
RAW264.7 cells culture and treatment
RAW264.7 cells (mouse macrophage cell line) were cultured in DMEM supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin and maintained at 37 °C in 5% CO2 humidified air. After stabilization, the cells were stimulated with PPC (20 μg/mL) with or without LPS (100 ng/mL) for 24 h, and the cells and culture supernatants were collected for further analysis.
BMDM preparation, culture, and treatment
Primary bone marrow-derived macrophages (BMDMs) were isolated from female C57BL/6J mice or female TLR2−/− mice at 6–8 weeks of age as described previously [28]. Briefly, the tibias and femurs of the mice were isolated under aseptic conditions, and bone cells were removed with precooled PBS. After the red blood cells were lysed, the cells were resuspended in IMDM containing 10 ng/mL M-CSF, 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin and maintained at 37 °C in 5% CO2 humidified air. On the 3rd and 7th days, the medium was refreshed, respectively. The cells were then stimulated for the indicated times with PPC (20 μg/mL) with or without LPS (100 ng/mL). In some experiments, macrophages were pretreated with 2-DG (10 μM) for 2 h, GW9662 or GW6471 (10 μM) for 3 h, or Pam3CSK4 (10 μg/mL) for 1 h and then stimulated with PPC or PPC plus LPS for an additional 24 h. The culture supernatants were collected for ELISA analysis. For gene expression analysis, the macrophages were stimulated for 4 h with the indicated treatments.
Cell proliferation assay
Cell proliferation was assessed using CCK-8 according to the manufacturer’s instructions. RAW264.7 cells were seeded in 96-well plates at a density of 1 × 104/well in a 100 μL volume. PPC was added to the cells at different concentrations (0, 10, 20, 30, and 50 μg/mL). After 20 h, CCK-8 reagent was added to the medium (10 μL/well). The absorbance of each well was determined at 450 nm after 4 h of incubation at 37 °C in 5% CO2 humidified air.
Enzyme-linked immunosorbent assays
The concentrations of IL-6 and TNF-α in cultured cell supernatants were determined by mouse TNF-α and IL-6 ELISA Ready-SET-Go!® kits (Invitrogen, USA) according to the manufacturer’s protocol.
Real-time quantitative RT-PCR
Total RNA was isolated from RAW264.7 cells or BMDMs using the TRIzol reagent extraction method. The appropriate amounts of RNA were used to synthesize cDNA with a PrimeScript™ RT reagent kit with gDNA Eraser. cDNA was amplified using TB Green™ Premix Ex Taq ™II with gene-specific primers. Quantitative real-time PCR was performed on a Light Cycler 480 II detection system (Roche Applied Science, Penzberg, Germany). The exact thermal cycler conditions were described in a previous study [29]. All primers are listed in Table 1. All experiments were performed in triplicate, and the cycle threshold (Ct) values were normalized to the endogenous reference (β-actin). The relative expression levels of the tested genes in this study were calculated by comparing the Ct values with β-actin by the 2−ΔΔCt method.
Statistical analysis
The data are expressed as mean ± SEM. Experimental graphs were generated using GraphPad Prism software, and all statistical analyses were performed using Statistical Package for the Social Sciences (SPSS) version 19 software. Statistically significant differences between groups were examined by one-way ANOVA. A value of P < 0.05 was considered statistically significant.
Results
PPC inhibited M1 polarization in LPS-activated macrophages
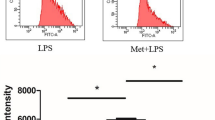
Macrophages play a vital role in the regulation of the inflammatory response. This study used the murine macrophage cell line RAW264.7 and primary BMDMs to evaluate the anti-inflammatory effect of PPC. As shown in Fig. 1a and b, the production and mRNA expression of IL-6 and TNF-α in LPS plus PPC group were significantly reduced compared with those in the LPS-stimulated group (P < 0.05). Moreover, PPC did not show an obvious effect on the production or expression of these cytokines (P > 0.05). In addition, PPC at different concentrations (0, 10, 20, 30, and 50 μg/mL) did not obviously inhibit the proliferation of RAW264.7 cells (P > 0.05, Supplementary Fig. 1). These results indicate that PPC strongly prevents the LPS-induced inflammatory response in macrophage, which did not affect cell viability.

PPC inhibited M1 polarization of LPS-activated macrophages. RAW264.7 cells and BMDMs were stimulated with PPC (20 μg/mL) with or without LPS (100 ng/mL) for 24 h. Cytokine concentrations in the culture supernatants were measured by ELISA, and the relative mRNA expression of cytokines was examined by real-time RT-PCR. a Production and mRNA expression of IL-6 and TNF-α in RAW264.7 cells. b Production and mRNA expression of IL-6 and TNF-α in BMDMs. c mRNA expression of M1 markers (iNOS and CXCL9, CXCL10, and CXCL11) in RAW264.7 cells. The data are presented as mean ± SEM. Comparisons among multiple groups were done using one-way ANOVA. vs PBS group, #P < 0.05, ##P < 0.01, ###P < 0.001; vs LPS group, *P < 0.05, **P < 0.01, ***P < 0.001
We further characterized the phenotype of macrophage following PPC treatment by determining the mRNA expression of specific chemokines and markers. As shown in Fig. 1c, the relative expression of iNOS was significantly downregulated in the PPC plus LPS group compared with that of the LPS group. Consistently, the expression of M1-specific chemokines, such as CXCL9, CXCL10, and CXCL11, was significantly decreased (P < 0.05). Collectively, these results indicate that PPC inhibits M1 polarization of LPS-activated macrophages.
PPC inhibited glycolysis and lipid synthesis but promoted lipid oxidation in LPS-activated macrophages
Specific metabolic reprogramming events have been reported to determine the phenotype of immune cells [26, 27]. Among which, the pathways such as glycolysis and lipid metabolism were extensively investigated. This study investigated whether PPC regulates metabolic reprogramming in macrophages. As shown in Fig. 2a and b, PPC plus LPS co-stimulation inhibited the mRNA expression of two rate-limiting glycolysis enzymes (PFK, phosphofructokinase and PK, pyruvate kinase) and key enzymes related to lipid synthesis (ACC1, acetyl-CoA carboxylase 1; SREBP-1c, sterol-regulatory element binding protein-1c; and FAS, fatty acid synthase) compared with that of LPS stimulation (P < 0.01). However, the mRNA expression levels of lipid oxidation-related enzymes (PPAR-γ, peroxisome proliferator-activated receptor-γ; PPAR-α, peroxisome proliferator-activated receptor-α; MCAD, medium chain acyl-CoA dehydrogenase; and CPT-1a, carnitine palmitoyltransferase-1a) were significantly upregulated (P < 0.05, Fig. 2c). These results show that PPC inhibits glycolysis and lipid synthesis but promotes lipid oxidation, indicating the reprogramming of glucose and lipid metabolism in macrophages.

PPC inhibited glycolysis and lipid synthesis but promoted lipid oxidation in LPS-activated macrophages. RAW264.7 cells were stimulated with PPC (20 μg/mL) with or without LPS (100 ng/mL) for 4 h. The mRNA expressions of key enzymes in the pathways of glycolysis (a), lipid synthesis (b), and lipid oxidation (c) were examined by real-time RT-PCR. The data are presented as mean ± SEM. Comparisons among multiple groups were done using one-way ANOVA. vs PBS group, #P < 0.05, ##P < 0.01, ###P < 0.001; vs LPS group, *P < 0.05, **P < 0.01, ***P < 0.001
The reprogramming of glucose and lipid metabolism was associated with the anti-inflammatory effect of PPC
To explore the possible link between the metabolic reprogramming and the anti-inflammatory effect of PPC, we measured the level of cytokine production after pretreatment with 2-DG (an inhibitor of the glycolytic pathway), GW9662 (an inhibitor of PPAR-γ), or GW6471 (an inhibitor of PPAR-α) in the cell culture system mentioned above. In the presence of LPS, 2-DG plus PPC significantly inhibited the production of IL-6 and TNF-α compared with the PPC alone (P < 0.05, Fig. 3a). However, GW9662 and GW6471 pretreatment abolished the anti-inflammatory effect of PPC in the presence of LPS (P < 0.001, Fig. 3b and c). These results indicate that inhibition of glycolysis and activation of lipid oxidation is associated with the anti-inflammatory effect mediated by PPC.

The reprogramming of glucose and lipid metabolism was associated with the anti-inflammatory effect of PPC. BMDMs were stimulated with PPC (20 μg/mL) and/or LPS (100 ng/mL) in the absence or presence of inhibitors for 24 h. 2-DG, GW9662, and GW6471 were added to the culture system for 2~3 h prior to PPC administration, respectively. The production of IL-6 and TNF-α under the inhibition of 2-DG (a), GW9662 (b), and GW6471 (c) was determined by ELISA. The data are presented as mean ± SEM. Comparisons among multiple groups were done using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001
PPC downregulated TLR-2 expression in LPS-activated macrophages
As one of important pattern recognition receptors (PRRs), TLR-2 is highly expressed in proinflammatory macrophages [20, 21]. This study found that PPC significantly inhibited the mRNA expression of TLR-2 in both RAW264.7 cells and BMDMs regardless of the absence or presence of LPS (P < 0.05, Fig. 4), which suggests that TLR-2 may have an essential role in the anti-inflammatory effect of PPC.

PPC downregulated TLR-2 expression in LPS-activated macrophages. Macrophages were stimulated with PPC (20 μg/mL) with or without LPS (100 ng/mL) for 24 h. a TLR-2 mRNA expression in RAW264.7 cells. b TLR-2 mRNA expression in BMDMs. The data are presented as mean ± SEM. Comparisons among multiple groups were done using one-way ANOVA. vs PBS group, #P < 0.05, ##P < 0.01, ###P < 0.001; vs LPS group, *P < 0.05, **P < 0.01, ***P < 0.001
TLR-2 pre-activation inhibited the anti-inflammatory effect of PPC
Pam3CSK4 is a well-known TLR-2 agonist [22, 23]. To further elaborate the role of TLR-2 on anti-inflammatory effect of PPC, we determined the cytokine levels in macrophages stimulated with Pam3CSK4 for 1 h before the addition of PPC. As shown in Fig. 5, PPC downregulated the mRNA expression of IL-6 and TNF-α in BMDMs activated by Pam3CSK4 (P < 0.05). However, PPC could not downregulate their expression after Pam3CSK4 pre-activation (P < 0.001). Moreover, in the presence with LPS plus PPC, Pam3CSK4 pre-activation even significantly elevated IL-6 and TNF-α expression (P < 0.001, Supplementary Fig. 3). The results suggest that PPC mediates the anti-inflammatory response via downregulating TLR-2 expression.

TLR-2 pre-activation inhibited the anti-inflammatory effect of PPC. BMDMs were stimulated with PPC (20 μg/mL) in the absence or presence of Pam3CSK4 (10 μg/mL) for 24 h. For pre-activation of TLR-2, Pam3CSK4 was added to the culture system for 1 h prior to PPC administration. The mRNA expression of IL-6 and TNF-α was determined by real-time RT-PCR. The data are presented as mean ± SEM. Comparisons among multiple groups were done using one-way ANOVA. vs PBS group, #P < 0.05, ##P < 0.01, ###P < 0.001; vs LPS group, *P < 0.05, **P < 0.01, ***P < 0.001.
The anti-inflammatory effect of PPC was lost in TLR-2−/− BMDMs
To confirm the role of TLR-2 in the anti-inflammatory effect of PPC, we compared the changes of cytokine production and the expression of M1 polarization-associated markers in TLR-2−/− and wild-type (WT) BMDMs after exposure to PPC and/or LPS for 24 h. As shown in Fig. 6a, unlike the trend in WT BMDMs, LPS plus PPC did not inhibit the production of TNF-α and IL-6 in TLR-2−/− BMDMs (P > 0.05). Moreover, in compared with WT BMDM, the mRNA expression of M1 polarization-associated markers (iNOS, CXCL9, CXCL10, and CXCL11) was significantly elevated in TLR-2−/− BMDMs after stimulation by PPC plus LPS (P < 0.05, Fig. 6b and Supplementary Fig. 4A). These results confirm that TLR-2 is required for PPC-mediated inhibition of M1 polarization.

The anti-inflammatory effect of PPC was lost in TLR-2−/− BMDMs. BMDMs isolated from TLR-2−/− and WT mice were stimulated with PPC (20 μg/mL) in the absence or presence of LPS (100 ng/mL) for 4 h. Cytokine production and mRNA expression of genes were detected by ELISA and real-time RT-PCR, respectively. a IL-6 and TNF-α production in the supernatants. b mRNA expression of specific markers for M1 macrophages (iNOS, CXCL9, CXCL10, and CXCL11). The data are presented as mean ± SEM. The results were repeated in triplicate. Comparisons among multiple groups were done using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001
The altered anti-inflammatory potential of PPC was accompanied by the reprogramming of glucose and lipid metabolism in TLR-2−/− BMDMs
We further characterized the effect of PPC on the metabolic profiles in TLR-2−/− and WT BMDMs. Notably, compared with WT BMDMs, the mRNA expression of glycolytic pathway enzymes (PFK and PK) was significantly elevated in TLR-2−/− BMDMs after stimulation by PPC plus LPS and was even almost restored to the level in LPS-activated WT BMDMs (P < 0.05, Fig. 7a). Moreover, the mRNA expression of lipid synthesis enzymes (ACC1, SREBP-1c, and FAS) was significantly upregulated (P < 0.05, Fig. 7b and Supplementary Fig. 4B). In addition, the mRNA expression of lipid oxidation enzymes (PPAR-γ, PPAR-α, MCAD, and CPT-1a) was significantly downregulated (P < 0.05, Fig. 7c). These results suggest that TLR-2-meditated reprogramming of glucose and lipid metabolism participates in the anti-inflammation effect of PPC.

The altered anti-inflammatory potential of PPC was accompanied by the reprogramming of glucose and lipid metabolism in TLR-2−/− BMDMs. BMDMs isolated from TLR-2−/− and WT mice were stimulated with PPC (20 μg/mL) in the absence or presence of LPS (100 ng/mL) for 12 h. The mRNA expression of enzymes in the metabolic pathways was detected by real-time RT-PCR. a mRNA expression of glycolytic enzymes. b mRNA expression of lipid synthesis enzymes. c mRNA expression of lipid oxidation enzymes. The data are presented as mean ± SEM. Comparisons among multiple groups were done using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
Previous studies have reported that PPC downregulates the inflammatory response to improve the condition of arthritis [18] and colitis [19]. However, the underlying mechanism is still unclear. The present study showed that PPC inhibited M1 polarization in LPS-activated macrophages. Mechanistically, the anti-inflammatory effect of PPC was most likely dependent on TLR-2 signaling and was mediated by inhibiting the glycolytic pathway while promoting lipid oxidation. Taken together, these results revealed a novel anti-inflammatory mechanism of PPC.
PPC did not inhibit the proliferation of macrophages, indicating that it will be safe for patients. Moreover, PPC significantly prevented the release of proinflammatory cytokines in LPS-activated macrophages (Supplementary Fig. 2), which was consistent with its preventive effect in mice of arthritis and inflammatory bowel disease [19, 30]. To better understand PPC, this study further explored its regulatory mechanism.
Previous studies have shown that macrophages maintain considerable plasticity and can alter effector phenotypes in response to different environmental signals [2, 6]. It is well known that LPS at a low dose can be a positive stimulator of M1 macrophages. Moreover, elevated levels of LPS have been detected in the sera of patients with various diseases, such as obesity and type 2 diabetes [31, 32]. This study found that even in the presence of LPS, PPC-treated macrophages exhibited reduced expression of M1 macrophage-associated markers, showing that PPC inhibits M1 macrophage polarization. These results suggest that PPC could treat not only inflammatory diseases but also several metabolic diseases.
It is well known that TLR-2 can promote or inhibit the inflammatory response. In LPS-activated macrophages, TLR-2 is upregulated to initiate activation of downstream signaling pathways and the expression of inflammatory factors, ultimately causing a proinflammatory response [20, 21, 33]; however, upregulated expression of TLR-2 in B cells is also necessary to induce the generation of regulatory B cells [34], which inhibit the inflammatory response. Thus, TLR-2 is believed to be an attractive target for drug development [35]. This study confirmed the pivotal role of TLR-2 signaling in the anti-inflammatory effect of PPC. TLR-2 expression in macrophages was significantly downregulated after exposure to PPC. Moreover, PPC did not inhibit the release of proinflammatory cytokines in TLR-2−/− BMDMs in the presence of LPS. Consistent with this, PPC did not show the anti-inflammatory effect after TLR-2 pre-activation using Pam3CSK4. In addition, PPC could inhibit the mRNA expression of M1 polarization-associated markers (iNOS, CXCL9, CXCL10, and CXCL11) in a TLR-2-dependent way. However, it has to be pointed out that the iNOS expression of PPC plus LPS group in TLR-2−/− BMDMs was partially recovered compared with that in WT BMDMs. This suggested that the effect of PPC on iNOS expression may be also regulated by other receptor(s) or factors. In fact, iNOS has diverse biological functions and can be regulated by multiple biological processes such as oxidative stress [36, 37]. Overall, these results indicate the preventive potential of PPC in several TLR-2-mediated diseases such as arthritis and renal inflammation [38].
In recent years, immunometabolism has become a new research hotspot [26, 27]. TLR-mediated metabolic reprogramming determines the phenotype and function of immune cells [27, 39]. M1 macrophages are characterized as enhanced glycolysis, while M2 macrophages exhibit activated lipid oxidation [40, 41]. This study characterized the specific metabolic events related to the anti-inflammatory effect of PPC. Our results showed that PPC inhibited the glycolysis pathway, but activated the lipid oxidation pathway. Such metabolic profiles were consistent with the anti-inflammatory status of macrophages [42,43,44] . 2-DG is a synthetic analogue of glucose, which competitively inhibits the reaction of hexokinase and glucose to inhibit glycolysis [45, 46]. This study further found that inhibition of glycolysis with 2-DG enhanced the anti-inflammatory effect of PPC. PPAR-α and PPAR-γ are ligand-activated transcription factors, which are involved in the transcriptional regulation of lipid metabolism [47, 48]. Inhibition of the two genes using GW9662 and GW6471 both hampered the anti-inflammatory effect of PPC. These results showed that PPC inhibits inflammation by reprogramming glucose and lipid metabolism. In addition, the metabolic phenotype induced by PPC was partially lost in TLR-2−/− BMDMs. Compared with the counterparts in WT BMDMs, the mRNA expression of glycolytic pathway enzymes and lipid synthesis enzymes was significantly elevated, but lipid oxidation associated genes were significantly downregulated in TLR-2−/− BMDMs. Notably, a significantly downregulated expression of SREBP-1c was observed both in TLR-2−/− and WT BMDMs after exposure to PPC, which suggested that this gene was not dependent on the TLR-2, but other receptor(s) or factor(s). Thus, more attention should be paid to investigate the role of TLR-2 in mediating metabolic reprogramming and the anti-inflammatory effect of PPC.
Overall, the present study showed that PPC, a clinically classic hepatoprotective drug, inhibited LPS-induced inflammation in macrophages in vitro. Mechanistically, PPC inhibited M1 polarization of macrophages, which was most likely dependent on TLR-2-medited metabolic reprogramming. These findings revealed a novel anti-inflammatory mechanism of PPC.
References
Feito MJ, Diez-Orejas R, Cicuendez M, Casarrubios L, Rojo JM, Portoles MT. Characterization of M1 and M2 polarization phenotypes in peritoneal macrophages after treatment with graphene oxide nanosheets. Colloids Surf B: Biointerfaces. 2019;176:96–105.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–95.
Huang H, Fletcher A, Niu Y, Wang TT, Yu L. Characterization of lipopolysaccharide-stimulated cytokine expression in macrophages and monocytes. Inflamm Res. 2012;61:1329–38.
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69.
Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12:231–8.
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86.
Barron L, Wynn TA. Macrophage activation governs schistosomiasis-induced inflammation and fibrosis. Eur J Immunol. 2011;41:2509–14.
Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–54.
Montoya D, Mehta M, Ferguson BG, Teles RMB, Krutzik SR, Cruz D, et al. Plasticity of antimicrobial and phagocytic programs in human macrophages. Immunology. 2019;156:164–73.
Zhang QZ, Liu YL, Wang YR, Fu LN, Zhang J, Wang XR, et al. Effects of telmisartan on improving leptin resistance and inhibiting hepatic fibrosis in rats with non-alcoholic fatty liver disease. Exp Ther Med. 2017;14:2689–94.
Imam S, Dar P, Paparodis R, Almotah K, Al-Khudhair A, Hasan SA, et al. Nature of coexisting thyroid autoimmune disease determines success or failure of tumor immunity in thyroid cancer. J Immunother Cancer. 2019;7:3.
Liu RH, Wen Y, Sun HY, Liu CY, Zhang YF, Yang Y, et al. Abdominal paracentesis drainage ameliorates severe acute pancreatitis in rats by regulating the polarization of peritoneal macrophages. World J Gastroenterol. 2018;24:5131–43.
Xie L, Yang Y, Meng J, Wen T, Liu J, Xu H. Cationic polysaccharide spermine-pullulan drives tumor associated macrophage towards M1 phenotype to inhibit tumor progression. Int J Biol Macromol. 2019;123:1012–9.
Li J, Xue H, Li T, Chu X, Xin D, Xiong Y, et al. Exosomes derived from mesenchymal stem cells attenuate the progression of atherosclerosis in ApoE(-/-) mice via miR-let7 mediated infiltration and polarization of M2 macrophage. Biochem Biophys Res Commun. 2019;510:565–72.
Lieber CS, Robins SJ, Li J, DeCarli LM, Mak KM, Fasulo JM, et al. Phosphatidylcholine protects against fibrosis and cirrhosis in the baboon. Gastroenterology. 1994;106:152–9.
Okiyama W, Tanaka N, Nakajima T, Tanaka E, Kiyosawa K, Gonzalez FJ, et al. Polyenephosphatidylcholine prevents alcoholic liver disease in PPARalpha-null mice through attenuation of increases in oxidative stress. J Hepatol. 2009;50:1236–46.
Lieber CS, Leo MA, Aleynik SI, Aleynik MK, DeCarli LM. Polyenylphosphatidylcholine decreases alcohol-induced oxidative stress in the baboon. Alcohol Clin Exp Res. 1997;21:375–9.
Pan W, Hao WT, Xu HW, Qin SP, Li XY, Liu XM, et al. Polyene phosphatidylcholine inhibited the inflammatory response in LPS-stimulated macrophages and ameliorated the adjuvant-induced rat arthritis. Am J Transl Res. 2017;9:4206–16.
Ben-Ami Shor D, Bashi T, Lachnish J, Fridkin M, Bizzaro G, Barshak I, et al. Phosphorylcholine-tuftsin compound prevents development of dextransulfate-sodium-salt induced murine colitis: implications for the treatment of human inflammatory bowel disease. J Autoimmun. 2015;56:111–7.
Zhao H, Dai X, Han X, Liu A, Bao F, Bai R, et al. Borrelia burgdorferi basic membrane protein A initiates proinflammatory chemokine storm in THP 1-derived macrophages via the receptors TLR1 and TLR2. Biomed Pharmacother. 2019;115:108874.
Kumar V. Toll-like receptors in the pathogenesis of neuroinflammation. J Neuroimmunol. 2019;332:16–30.
Zhang J, Diao B, Lin X, Xu J, Tang F. TLR2 and TLR4 mediate an activation of adipose tissue renin-angiotensin system induced by uric acid. Biochimie. 2019;162:125–33.
Jian L, Sun L, Li C, Yu R, Ma Z, Wang X, et al. Interleukin-21 enhances toll-like receptor 2/4-mediated cytokine production via phosphorylation in the STAT3, Akt and p38 MAPK signalling pathways in human monocytic THP-1 cells. Scand J Immunol. 2019;86:e12761.
Patel CH, Leone RD, Horton MR, Powell JD. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat Rev Drug Discov. 2019;18:669–88.
Koelwyn GJ, Corr EM, Erbay E. Regulation of macrophage immunometabolism in atherosclerosis. Nat Immunol. 2018;19:526–37.
Yamada KJ, Kielian T. Biofilm-leukocyte cross-talk: impact on immune polarization and immunometabolism. J Innate Immun. 2019;11(3):280–8.
Shi L, Jiang Q, Bushkin Y, Subbian S, Tyagi S. Biphasic dynamics of macrophage immunometabolism during Mycobacterium tuberculosis infection. mBio. 2019;10(2). https://doi.org/10.1128/mBio.02550-18.
Davis BK. Derivation of macrophages from mouse bone marrow. Methods Mol Biol. 1960;2019:41–55.
Li C, Yang D, Cao X, Wang F, Jiang H, Guo H, et al. LFG-500, a newly synthesized flavonoid, attenuates lipopolysaccharide-induced acute lung injury and inflammation in mice. Biochem Pharmacol. 2016;113:57–69.
Bashi T, Shovman O, Fridkin M, Volkov A, Barshack I, Blank M, et al. Novel therapeutic compound tuftsin-phosphorylcholine attenuates collagen-induced arthritis. Clin Exp Immunol. 2016;184:19–28.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–72.
Moreno-Indias I, Cardona F, Tinahones FJ, Queipo-Ortuno MI. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front Microbiol. 2014;5:190.
Santiago-Tellez A, Castrillon-Rivera LE, Palma-Ramos A, Bello-Lopez JM, Sainz-Espunes T, Contreras-Paredes A, et al. Keratinocyte infection by Actinomadura madurae triggers an inflammatory response. Trans R Soc Trop Med Hyg. 2019;113:392–8.
Pan W, Xu HW, Hao WT, Sun FF, Qin YF, Hao SS, et al. The excretory-secretory products of Echinococcus granulosus protoscoleces stimulated IL-10 production in B cells via TLR-2 signaling. BMC Immunol. 2018;19:29.
Marques CP, Maor Y, de Andrade MS, Rodrigues VP, Benatti BB. Possible evidence of systemic lupus erythematosus and periodontal disease association mediated by toll-like receptors 2 and 4. Clin Exp Immunol. 2016;183:187–92.
Mayhan WG, Arrick DM, Sharpe GM, Sun H. Nitric oxide synthase-dependent responses of the basilar artery during acute infusion of nicotine. Nicotine Tob Res. 2009;11:270–7.
Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009;73:411–8.
Perveen K, Hanif F, Jawed H, Jamall S, Simjee SU. N-(2-hydroxy phenyl) acetamide: a novel suppressor of toll-like receptors (TLR-2 and TLR-4) in adjuvant-induced arthritic rats. Mol Cell Biochem. 2014;394:67–75.
McGarry T, Biniecka M, Gao W, Cluxton D, Canavan M, Wade S, et al. Resolution of TLR2-induced inflammation through manipulation of metabolic pathways in rheumatoid arthritis. Sci Rep. 2017;7:43165.
Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185:605–14.
O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553–65.
Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, et al. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. 2016;539:294–8.
Namgaladze D, Brune B. Fatty acid oxidation is dispensable for human macrophage IL-4-induced polarization. Biochim Biophys Acta. 1841;2014:1329–35.
Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, et al. Fatty acid oxidation in macrophage polarization. Nat Immunol. 2016;17:216–7.
Stafstrom CE, Roopra A, Sutula TP. Seizure suppression via glycolysis inhibition with 2-deoxy-D-glucose (2DG). Epilepsia. 2008;49(Suppl 8):97–100.
Koenig JB, Cantu D, Low C, Sommer M, Noubary F, Croker D, et al. Glycolytic inhibitor 2-deoxyglucose prevents cortical hyperexcitability after traumatic brain injury. JCI Insight. 2019;5. https://doi.org/10.1172/jci.insight.126506.
Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312.
Towfighi A, Ovbiagele B. Partial peroxisome proliferator-activated receptor agonist angiotensin receptor blockers. Potential multipronged strategy in stroke prevention. Cerebrovasc Dis. 2008;26:106–12.
Funding
This work was supported by grants from the Starting Foundation for Talents of Xuzhou Medical College (No. D2015004), the Jiangsu Planned Projects for Postdoctoral Research Funds (No. 2019 K063), and the Jiangsu Qing Lan Project.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: WP, TTF, XYY, and QSL. Performed the experiments: TTF and SSH. Analyzed the data: WP, FFT, and YH. Contributed reagents/materials/analysis tools: WP, QSL, and FFS. Wrote the manuscript: WP, FFT, and XYY.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Disclosure
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOC 574 kb)
Rights and permissions
About this article

Cite this article
Feng, TT., Yang, XY., Hao, SS. et al. TLR-2-mediated metabolic reprogramming participates in polyene phosphatidylcholine-mediated inhibition of M1 macrophage polarization. Immunol Res 68, 28–38 (2020). https://doi.org/10.1007/s12026-020-09125-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-020-09125-9