
Abstract
We aimed to describe a case of acute kidney injury (AKI) with an uncommon case. We described a previously health 24 years old male that presented acute kidney injury associated with neurological and respiratory symptoms. He was initially admitted at the hospital with nausea, vomiting, blurred vision, and reduced urine output. The patient’s condition got worse approximately in one week. Laboratory tests revealed high levels of nitrogenous waste, hyponatremia, metabolic acidosis with an increased anion gap, and the presence of proteinuria and hematuria. The patient experienced paresthesia, seizures, respiratory alterations, and altered consciousness. The initial diagnostic hypothesis of rapidly progressive glomerulonephritis was not confirmed. A deeper investigation of the case exposed that it could have occurred an intentional exogenous poisoning with diethylene glycol (DEG). Renal biopsy unveil findings suggestive of poison-induced nephrotoxicity, which corroborated the suspicion. Despite therapeutic efforts, the patient died due to pulmonary complications. This case report shows the need to consider DEG poisoning as a etiology of AKI, especially in patients with neurological symptoms. Laboratory and histopathological analysis were crucial for the diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diethylene glycol (DEG), a low-cost, colorless, odorless, sweet-tasting substance, finds wide application in various industries, including pharmaceuticals, cosmetics, lubricants, heating fuels, and plasticizers [1,2,3]. Its versatility as a solvent makes it essential for water-insoluble substances, permeating multiple industrial sectors. Even in the production of alcoholic beverages, DEG plays a role by facilitating the cooling process, which amplifies its harmful potential. One of the most concerning aspects of DEG is the low concentration of the compound that leads to toxicity. The minimal toxic dose is of 0.14 mg/kg, while a lethal dose can range from 1 to 1.63 g/kg [1].
DEG poisoning emerges as a potential lethal condition that causes acute kidney injury (AKI) among other complications. AKI is characterized by a rapid deterioration of renal function often accompanied by systemic complications. Despite its inherent toxicity, DEG poisoning is notable for the challenge of early diagnosis, given its nonspecific initial clinical manifestations. Adding complexity to the scenario, DEG is rapidly absorbed through the gastrointestinal tract, limiting opportunities for intervention after ingestion [1].
In this article, we report the case of a young patient who died as a consequence of AKI accompanied by neurological and respiratory deterioration. The investigation revealed DEG intentional ingestion as the underlying cause of death.
Case report
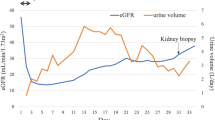
This concerns a 24-year-old male patient who was admitted in a tertiary hospital due to acute kidney injury. He had been referred from a secondary hospital with complaints of nausea, vomiting, blurred vision, and reduced urine output, which had gotten worse over the course of approximately one week. At admission, the patient had a slight increase in systolic blood pressure (140 × 77 mmHg) and displayed some degree of lethargy, although he was lucid (See Fig. 1).

Imaging evaluation at the second day of hospitalization. A) Chest X-Ray showed a discrete infiltrate in pulmonary parenchyma. B, C, D and E) Images of brain computed tomography wihtout structural abnormalities
The patient had no prior comorbidities and was not on any continuous medication. In his medical history, there was a depressive disorder background that had been treated with medication from the ages of 12 to 14 but without specialized medical follow-up after treatment discontinuation.
Laboratory tests revealed high levels of nitrogenous waste products (urea reanging from 136 to 294 and creatinine ranging from 4.72 to 8.62), hyponatremia (sodium ranging from 115 to 125), and metabolic acidosis with an increased anion gap (Table 1). Other laboratory tests, including serologies for HIV, hepatitis B and C, serum complement factor levels (C3: 1.1 g/L, C4: 0.38 g/L), and antinuclear factor, showed no abnormalities. Two consecutive hemocultures were negative.
Urinalysis detected the presence of proteins and red blood cells (proteins + + and > 100 RBCs per field). The renal ultrasound showed topical, enlarged, heterogeneous kidneys with parenchymal changes. There were no signs of calculi or hydronephrosis. Table 1 displays the exams (hemogram, blood gas, glucose levels and lactate), during hospitalization.
On the second day of hospitalization, the patient reported paresthesia on the right side, followed by tonic-clonic seizures on the third day. Imaging studies and analysis of cerebrospinal fluid were conducted to investigate the neurological changes. Study by computed tomography (CT) of the skull showed no demonstrable abnormalities and ruled out intraparenchymal hemorrhage. The lumbar puncture revealed increased protein levels suggestive of an inflammatory process.
On the seventh day of hospitalization, the patient exhibited a change in respiratory pattern, decreased oxygen saturation, and a reduced level of consciousness, necessitating orotracheal intubation. Chest X-ray revealed diffuse bilateral parenchymal infiltrate (Fig. 2), associated with elevated procalcitonin levels, suggesting a secondary lung infection. At this stage, diagnostic hypotheses included pneumonia and sepsis. Broad-spectrum antibiotic therapy was initiated, and cyclophosphamide administration was suspended.

Imaging evalution of the chest at the seventh day of hopitalization. (A) Chest X-Ray showed a diffuse bilateral infiltrate in pulmonary parenchyma. (B) Chest tomography confirmed the bilateral infiltrate of pulmonary parenchyma
The patient was transferred to the intensive care unit (ICU) and underwent renal replacement therapy for uremia, although he maintained a residual urinary volume. The presence of proteinuria and hematuria, along with enlarged kidneys, raised the suspicion of acute glomerulonephritis. Considering the epidemiology, laboratory results, and findings of renal ultrasound, suspicion of Rapidly Progressive Glomerulonephritis (RPGN) was raised. A renal biopsy was performed, and the patient received intravenous corticosteroid pulse therapy (1 g for three days), with a follow-up plan that included the use of cyclophosphamide (1 g/m²). The result of the renal biopsy revealed diffuse tubular vacuolar degeneration, multifocal acute tubular necrosis, and microvascular congestion (Fig. 3).

Renal core biopsy. (A) H&E (10X) micrograph shows diffuse proximal tubular injury, characterized by cellular hydropic vacuolization (see arrows). (B)H&E (20X) micrograph shows diffuse tubular proximal dilatation, with extreme cellular edema, and vacuolization, causing obliteration of the lumen. (C) High magnification of H&E (40X) micrograph shows some tubules with calcium phosphate crystals (see arrows). (D) Interstitial hemorrhage and diffuse tubular injury represented by flattened tubular cells and lost of microvilosity. (E) Transmission Electron Microscopy of fixed renal tissue, buffered 10% formalin, and stained with osmium tetroxide, and ruthenium red that shows diffuse tubular cells edema and vacuolization of the cytoplasm. (F) Details showing proximal tubules with extensive cytoplasmic change and multifocal vacuolization
Renal acute injuries appear to arise mainly from proximal tubular cytoplasm degeneration, markedly presenting as diffuse/severe vacuolation. The morphological findings revealed extensive renal damage, with interstitial hemorrhage and significant proximal tubule edema. Although the renal biopsy findings were considered nonspecific, they suggested nephrotoxicity from an exogenous substance, especially DEG, supported by laboratory and clinical data. Poison toxicity usually presents with lesions in this compartment. Due to suspicion of ingestion or contamination by an exogenous substance, the case was discussed again with the patient’s family. The patient´s brother informed that the patient had recently purchased a bottle of DEG. Based on this finding, it was speculated that the patient ingested DEG as a suicide attempt.
Despite therapy, the patient exhibited progressive deterioration in his clinical condition, mainly due to the secondary lung infection, and died on the fourteenth day of hospitalization.
Discussion
In this case, intentional DEG poisoning triggered a series of complex clinical events. DEG is metabolized predominantly in the liver, resulting in two main metabolites: hydroxyethoxyacetic acid (HEAA) and diglycolic acid (DGA), both considered as toxic agents [4]. HEAA produces renal and neurological injuries. The mechanisms of toxicity include destabilization of the cell membrane, affecting ion channels or phospholipids, and intracellular accumulation of osmotically active metabolites, triggering transcellular fluid shifts. The increase in HEAA concentrations also contributes to metabolic acidosis [5]. DGA produces a significant increase in Ethidium Homodimer (EthD) uptake and lactate dehydrogenase (LDH) release, indicating tissue damage [4]. These mechanisms result in the three distinct phases of DEG poisoning. Initially, gastrointestinal symptoms occur, followed by AKI in the second phase, characterized by reduced urine output and metabolic acidosis with an increased anion gap, and, then, the third phase is marked by neurological alterations [3]. The case reported exhibited this three phase pattern related to DEG toxicity.
AKI is considered a primary marker of DEG intoxication [1, 3]. The precise mechanisms of cellular toxicity have not been fully understood, but the oxidation of the intact DEG molecule is the main responsible for toxic effects. However, in this case, other potential causes for AKI were considered since no information about the etiology was initially available. Therefore, the possibility of Rapidly Progressive Glomerulonephritis (RPGN) was conceived. This assumption may have delayed the correct diagnosis and potentially decreased the patient’s recovery chances. On the other hand, the lack of information in the patients’ history complicated the correct diagnosis.
The patient, young and without significant comorbidities, initially presented gastrointestinal symptoms, progressing to renal involvement and later neurological complications. Initial laboratory evaluation revealed traits of the early phase of DEG poisoning, including elevated nitrogenous loss, hyponatremia, metabolic acidosis, proteinuria, and hematuria. However, these laboratorial alterations are not specific of DEG poisoning. Other causes of AKI may exhibit the same alterations, highlighting the clinical complexity of the diagnose [6]. Clinical cases described in the literature presented similar symptoms and outcomes. An example was a 26-year-old man, without history of disease background or medication use, who ingested an unknown liquid from a bottle, and had manifested intense abdominal pain, nausea, and vomiting two days later. At hospital admission, AKI was diagnosed, and dialysis was initiated [7]. Another case describes a previously healthy 40-year-old man admitted to a hospital with a history of 4-day nausea, general weakness, abdominal pain, diarrhea, and progressive oligo-anuria. Initial laboratory data revealed AKI and metabolic acidosis with an elevated anion gap. Urinalysis indicated mild proteinuria, and renal biopsy found acute and extensive tubular lesions without intratubular oxalate crystals [8].
The renal biopsy was very important for the diagnosis of DEG poisoning. AKI secondary to DEG toxicity normally results from cortical tubular degeneration and proximal tubular necrosis [1]. The kidney tissue had diffuse tubular vacuolar degeneration, multifocal acute tubular necrosis, and microvascular congestion. These findings suggest the possibility of DEG poisoning, which was confirmed by the possibility of suicide with DEG ingestion.
Conclusion
This case report underscores the need to consider DEG poisoning as a potential etiology of AKI, especially in patients with associated neurological symptoms. Laboratory and histopathological analysis played a crucial role in identifying the underlying cause of the patient’s condition.
Despite the severity of DEG poisoning, a gap persists in our understanding of its short and long-term consequences. This gap highlights the critical importance of a meticulous analysis of reported cases, providing a deeper understanding of the extent of tissue damage caused by DEG poisoning.
Key points
-
1.
The possibility of poising should be consider in cases of sudden appearnece of atypical features in previously healthy individuals.
-
2.
A detailed history is fundamental for the diagnosis in cases with complex signs and symptoms. Renal biopsy can help in the suspision of renal toxicity lesions.
References
Gopalakrishnan N, Kamarajan M, Balasubramaniyan T, Sakthirajan R, Dhanapriya J, Dineshkumar T. Diethylene glycol poisoning-induced acute kidney injury. Saudi J Kidney Dis Transpl. 2016;27:1276–9. https://doi.org/10.4103/1319-2442.194692.
Rentz ED, Lewis L, Mujica OJ, Barr DB, Schier JG, Weerasekera G, et al. Outbreak of acute renal failure in Panama in 2006: a case-control study. Bull World Health Organ. 2008;86(10):749–56. https://doi.org/10.2471/blt.07.049965.
de Almeida Araújo S, Faria BCD, Vasconcelos JC, da Cruz AF, de Souza VS, Wanderley DC, et al. Renal toxicity caused by diethylene glycol: an overview. Int Urol Nephrol. 2023;1–9. https://doi.org/10.1007/s11255-023-03604-2.
Landry GM, Martin S, McMartin KE. Diglycolic acid is the nephrotoxic metabolite in diethylene glycol poisoning inducing necrosis in human proximal tubule cells in vitro. Toxicol Sci. 2011;124(1):35–44. https://doi.org/10.1093/toxsci/kfr204.
Schep LJ, Slaughter RJ, Temple WA, Beasley DMG. Diethylene glycol poisoning. Clin Toxicol. 2009;47(6):525–35. https://doi.org/10.1080/15563650903086444.
Yang SY, Chiou TT, Shiao CC, Lin HY, Chan MJ, Wu CH, Sun CY, Wang WJ, Huang YT, Wu VC, Chen YC, Fang JT, Hwang SJ, Pan HC. Nomenclature and diagnostic criteria for acute kidney injury – 2020 consensus of the Taiwan AKI-task force. J Formos Med Assoc. 2022;121(4):749–65. https://doi.org/10.1016/j.jfma.2021.08.005.
Wittschieber D, Heuberger K, Schulz R, Köhler H, Varchmin-Schultheiß K. Fatal poisoning with diethylene glycol in an unusual setting. Forensic Sci Med Pathol. 2019;15:649–52. https://doi.org/10.1007/s12024-019-00123-4.
Morelle J, Kanaan N, Hantson P. The Case∣ cranial nerve palsy and acute renal failure after a ‘special drink’. Kidney Int. 2010;77(6):559–60. https://doi.org/10.1038/ki.2009.514.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
The family of the patient gave signed authorization to report the case. The study of diethylene glycol poising was approved in the Ethics Committee of the Faculdade de Ciências Médicas de Minas Gerais (FCMMG) – Belo Horizonte, Minas Gerais, Brasil.
Conflict of interest
None declared.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Araújo, S.A., Stefanon, I., Wanderley, D.C. et al. A case of suicide secondary to diethylene glycol intentional ingestion. Forensic Sci Med Pathol (2024). https://doi.org/10.1007/s12024-024-00832-5
Accepted:
Published:
DOI: https://doi.org/10.1007/s12024-024-00832-5