
Abstract
Purpose
To confirm the exact break-point of a novel long-range deletion discovered in one female parathyroid carcinoma (PC) patient who has a strong family history suggesting familial hyperparathyroidism, and to investigate the expression of parafibromin in the patient’s affected lesion.
Methods
Clinical information of one female patient as well as five of her relatives was collected. Their genomic DNA extracted from peripheral blood went through Sanger sequencing and multiplex ligation-dependent probe amplification (MLPA). After completing whole genome sequencing (WGS), clone sequencing was also performed, whose result was aligned with standard human genome database after Sanger sequencing.
Results
The medical history of recurrent hypercalcemia after parathyroidectomy and histopathological investigation confirmed that the female patient was diagnosed with PC. WGS displayed a novel 130 kb long-range deletion spanning UCHL5 to CDC73 that was later confirmed by clone sequencing. MLPA showed similar results in four of her five relatives, suggesting these people to be carriers of the same long-range deletion, and three among them had a history of primary hyperparathyroidism (PHPT) ahead of the proband’s first visit.
Conclusions
We discovered a novel 130 kb long-range deletion spanning CDC73 in a family of 5 persons, and the existence of the deletion was related to PHPT and PC. Our discovery validated the role of CDC73 mutation in the occurrence of PHPT and PC, which provided new information to the genetic studies of PC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary hyperparathyroidism (PHPT), as the third most frequent endocrine disorder in western countries, is a metabolic bone disease caused by autonomous excessive parathyroid hormone (PTH). Parathyroid carcinoma (PC) is a rare pathologic type that comprises 0.5–5% of patients with PHPT [1,2,3]. Since there are no specific clinical, biochemical, or radiological characteristics of PC, it has been a challenge for clinicians to distinguish PC from parathyroid adenoma (PA) or parathyroid hyperplasia (PH). Therefore, the diagnosis of PC is often made after surgery based on histopathological examinations, and sometimes after recurrence or metastasis.
PC is commonly sporadic, but it can also be a part of specific syndromes in familial PHPT including multiple endocrine neoplasia type 1 (MEN1) and 2A (MEN2A) [4], particularly, the hyperparathyroidism-jaw tumor syndrome (HPT-JT) [5, 6]. Germline mutations of the CDC73 (located in 1q32.1, initially named HRPT2) gene was initially associated with HPT-JT [6, 7]. Till now, in 15–70% of sporadic PC cases, CDC73 mutations were found to be somatic, and in one-third of them, such mutations are found as germline [8].
The transcription product of tumor suppressor gene CDC73 is a 531-amino acid protein parafibromin, which is responsible for cell cycle arrest by acting through both transcriptional and post-transcriptional control pathways [9]. Parafibromin has been proved to play a role in Wnt signaling pathway [10] and interact with polymerase II-associated factor 1 transcription elongation complex [11]. Its expression is detectable in PH and PA tissues through immunohistochemical methods, while in PCs, it is missing, indicating that loss of parafibromin expression is a marker of PC.
Loss-of-function mutations of CDC73 associated with PC result in loss of expression of parafibromin [12], abnormal parafibromin subcellular localization [13], or depleted anti-proliferative ability [14]. Till now, about 100 cases of CDC73 germline loss-of-function mutation have been reported, most of which involve frameshift mutation or nonsense mutation [15]. Intragenic mutation or deletion of CDC73 have also been discovered in a few cases of PC and related syndromes [16,17,18,19].
In this study, we reported the clinical feature of a PC patient who had a family history of PHPT and the genetic inspections of the patient’s family of seven people covering three generations. The clinical data and novel genetic findings of the long-range deletion spanning part of CDC73 and several upstream genes will be of help in further understanding of the disease.
Subjects and methods
The patient was diagnosed and monitored at the Department of Endocrinology, Peking Union Medical College Hospital (PUMCH) voluntarily. The Ethics Committee of PUMCH approved this study protocol. Collection of peripheral blood and genetic analysis were performed after acquiring the patient and her family’s full consent.
Diagnosis of PHPT was established based on serum calcium (SCa) or albumin-corrected calcium (CCa, CCa = SCa + [0.02 × (40 – albumin)]) levels greater than 2.70 mmol/L and/or plasma ionized calcium higher than 1.28 mmol/L, combing with elevated serum PTH levels in the absence of other causes of hypercalcemia [20]. Familial or syndromic PHPT such as MEN1, MEN2A, MEN4, and HPT-JT were screened by full personal and family history with laboratory and imaging evaluation of relative endocrine glands, urinary system, and jaws [21, 22].
Clinical data
The clinical data were retrospectively collected from medical records maintained at PUMCH, including general characteristics, clinical manifestations (e.g., bone involvement, urinary system damage, gastrointestinal symptoms, and hypercalcemia crisis), treatment strategies, pathological features, and follow-up (postoperative recurrence and metastasis).
Serum and urinary biochemical markers were measured at the Department of Clinical Laboratory of PUMCH described previously [23]. Serum PTH was measured via chemiluminescence (Siemens ADVIA Centaur, Erlangen, Germany) with the normal range of 12–68 pg/mL. Serum 25-dihydroxy vitamin D was measured by electrochemiluminescence immunoassay (Roche Cobas E601, Germany). Histopathological classification of parathyroid lesions was determined by the pathologists at the Department of Pathology, PUMCH. Diagnostic criteria base on the WHO pathological criteria of PC [24] including vascular invasion, perineural-space invasion, capsular penetration, and/or documented metastases [25]. Parafibromin immunohistochemistry (IHC) staining of parathyroid tissue collected through parathyroidectomy of the proband and the proband’s brother was conducted as previously described [26] with a PA tissue from a previous study as a positive control.
Genetic analysis
Genomic DNA was extracted from peripheral blood leukocytes using a commercial kit (Blood DNA Midi Kit D3494, Omega Bio-Tek, Georgia, USA) following instructions given by the manufacturer. Sanger sequencing and results’ analyzing of exons and splice regions of CDC73 and MEN1 of the proband were performed as previously described [27].
Copy number analysis was conducted by multiplex ligation-dependent probe amplification (MLPA) using the P466-A1 kit (MRC-Holland, Amsterdam, Netherlands) following manufacturers’ protocol. Results of the MLPA products were obtained and analyzed on an ABI3130XL genetic analyzer (Applied Biosystems) using GeneMapper software (Applied Biosystems). Analysis of MLPA results was conducted with Coffalyser.Net software (Version 04, MRC-Holland, Amsterdam, The Netherlands) by comparing the sample’s peak height of each electrophoresis fragment with those of the same length of standard comparisons. A ratio of 0.7–1.3 would be considered a normal copy number of the locus, while a ratio below 0.7 would be defined as half-reduced copy number and a ratio above 1.3 would be defined as an increased copy number of the locus.
Approximately 1 ng of the patient’s genomic DNA sample was sent to WuXiAppTec to perform whole genome sequencing (WGS). According to the manufacturer’s instructions, Agilent liquid capture system (Agilent SureSelect Human All Exon V6) was used to enrich the exome sequence from the sample DNA. The qualified genomic DNA sample was randomly fragmented using a Covaris S220 sonicator into an average size of 180–280 bp. After connecting adaptors to both ends of the resulting fragments by paired ends (Illumina), they are selectively enriched in the PCR reaction. The extracted DNA was amplified using ligation-mediated PCR, AMPureXP system (Beckman Coulter, Beverly, California, USA), and quantified using Agilent high sensitivity DNA assay on Agilent Bioanalyzer 2100 system. Finally, the DNA library was sequenced on Illumina for paired-end 150 bp reads.
After receiving the WGS results, primers PC-F and PC-R were designed flanking the deleted sequence using Oligo7 software (Version 7.60, Molecular Biology Insights Inc), after which PCR was done as described previously [27]. The product was sent to SinoGenoMax for Sanger sequencing and clone sequencing (ABI3130XL genetic analyzer, Applied Biosystems). Results of clone sequencing were analyzed by 4Peaks (Version 1.4, Neocleobytes) and Basic Local Alignment Search Tool supported by Genomic Institute of the University of California, Santa Cruz.
Results
Case presentation
In 2013, a 33-year-old woman (proband, III2) presented with bilateral lower limb weakness, light lower limb pain worsening with activity. She underwent examinations in a local hospital with the results of PTH 981 pg/mL (reference range: 11–67 pg/L), serum albumin-corrected Ca 2.80 mmol/L (reference range: 2.08–2.60 mmol/L), P 0.67 mmol/L (reference range: 0.96–1.62 mmol/L), Mg 0.4 mmol/L (reference range: 0.75–1.02 mmol/L), and alkaline phosphatase (ALP) 536 U/L (reference range: 50–135 U/L). X-ray indicated the right mid-femur fracture and osteitis fibrosa cystica in the right lib. Ultrasound investigations revealed right cystic kidney and multiple urolithiasis on both sides. The right inferior parathyroid gland was resected, as the preoperative ultrasound indicated enlarged parathyroid gland (4.5 × 3.5 × 3.0 cm). The pathological examination showed atypical parathyroid adenoma (APA) with Ki-67 of 10%. Her postoperative SCa and PTH levels reverted to normal ranges since the first day after surgery. At the 3-month follow-up, serum PTH was 258 pg/mL and SCa was 1.84 mmol/L. The patient did not follow up regularly; thus, further post-surgical biochemical data were unavailable.
In 2019, she came to our center for recurrent bilateral lower limb weakness with xerostomia. Physical examination revealed an old surgical scar on the neck with unremarkable cervical palpation and stable vital signs. Biochemical evaluation confirmed the recurrence of PHPT: serum albumin-corrected Ca 2.67 mmol/L (reference range: 2.13–2.70 mmol/L), serum P 0.31 mmol/L (reference range: 0.81–1.45 mmol/L), PTH 752.8 pg/mL, plasma ionized Ca 1.56 mmol/L (reference range: 1.08-1.28 mmol/L), ALP 191 U/L, β-CTX 0.323 ng/mL, 24hUCa 4.3 mmol, 24hUP 11.96 mmol, 25OHD 11.5 ng/mL, and 1,25[OH]2D3 87.37 pg/mL. Both neck ultrasound and the parathyroid 99mTc-sestamibi scanning identified lesions on the middle-upper of the left lobe and middle-lower of the right lobe of the thyroid. The BMD, as measured by DXA, was 0.981 g/cm2 in the lumbar spines 1–4 (Z score –1.6), 0.880 g/cm2 in the femur neck (Z score –0.5), and 0.799 g/cm2 in the total hip (Z score −1.5). Serum calcitonin, insulin, proinsulin, and gastrin were all within normal ranges, and there were no abnormalities found in pituitary MRI and pancreatic enhanced MRI. Through the scanning of F18-PET/CT, MR, and ultrasound, no possible metastasis or involvement of uterine and mandible was found. The patient, accordingly, underwent right superior and total left parathyroidectomy + total hemithyroidectomy + resection of surrounding anterior muscle of neck + neck lymphadenectomy. During neck exploration, infiltration of the affected lesion into the adjacent muscle as well as adhesion with surrounding tissue was found. Pathological examinations confirmed the diagnosis of PC, with lesions infiltrating into thyroid tissue, surrounding muscle tissue, and right recurrent laryngeal nerve (Fig. 2a, b). Immunochemical examinations showed parafibromin (–) (Fig. 2d), PTH (+), CgA(+), Calcitonin(–), AE1/AE3(+), Syn(+), TTF-1(–), and Ki-67 index of 20%. Her serum levels of PTH (8.7 pg/mL) and calcium (2.01 mmol/L) significantly decreased postoperatively. She felt some numbness in her limbs without tetany and was treated with calcium carbonate (1.5 g/day) and calcitriol (1 μg/day), which relieved her symptoms gradually. In 2020, after a year of surgery, her SCa level increased to 2.76 mmol/L, while the PTH level was 208 pg/mL. Both the parathyroid 99mTc-sestamibi scanning and ultrasound identified no new lesion, so the patient chose to use cinacalcet instead of surgery. Three months after starting the medication, the patient experienced only occasional nausea. Her SCa level was 2.42 mmol/L at the time of the latest follow-up, while the PTH level ranged between 260 and 400 pg/mL.
Genetic findings
Sanger sequencing showed no point mutations of CDC73 or MEN1 of the proband, while results of MLPA showed a half-reduced copy number variant of exon 1–6 of CDC73 and upper-stream TROVE2 exon 8, GLRX2 exon 3. But copy number of ABCA4 exon 15 was unaltered. This copy number variation had not been described previously.
WGS revealed the long-range missing covered part of UCHL5, TROVE2, GLRX2, and part of CDC73 in the proband, and further clone sequencing confirmed that the 130 kb deletion spanned chr1:193009590-193139118 (Fig. 1C). Considering results of MLPA and tight kinship connection, we presumed that the II4, II3, III3, and IV1 carried the same long-range missing (Table 1 and Fig. 1A).

A Pedigree chart of the patient. Black solid square/circle: person diagnosed with PHPT. Slash line filled square/circle: person carrying CDC73 mutation. Shadowed square/circle: a person without CDC73 mutation. Crossed-out square/circle with single slash line: deceased member. B Changes of serum corrected calcium level and PTH level at different time. Full curve indicates PTH level, while dotted curve indicates serum calcium level. C Peak chart showing results of clone sequencing and respected model picture of an affected genomic region. (Illustrated by 4Peaks and Adobe Photoshop.)
The proband (III2) had a family history of PHPT and renal calculi (results of MLPA confirmed five of the family members, II3, II4, III3, IV1, and IV2, carried the same variation of CDC73 gene) (Table 2 and Fig. 1A). II4 (patient’s father) was diagnosed as PHPT at 36 years old and underwent a single gland parathyroidectomy for PA. No signs of recurrence or metastasis were found on her father in the 20-year follow-up. II3 (sister of the proband’s father) and III3 (proband’s brother) were also diagnosed as PHPT with renal calculi. III3 underwent examinations in our hospital with the results of PTH 224 pg/mL, serum albumin-corrected Ca 2.70 mmol/L, plasma ionized Ca 1.29 mmol/L; the preoperative ultrasound and sestamibi-(99m)Tc indicated two abnormal hyperplasia in the left parathyroid. III3 underwent left superior and inferior parathyroidectomy with the pathological diagnosis of PA (Fig. 2e). Immunochemical examinations showed PTH (+), CgA(+), Calcitonin(–), AE1/AE3(+), Syn(+), TTF-1(–), and Ki-67 index of 1%. IHC staining showed that nuclear parafibromin expression was also absent in III3’s parathyroid sample (Fig. 2f). IV1 (proband’s daughter) and IV2 (son of proband’s brother) showed no clinical manifestation of PHPT with normal serum Ca and PTH levels. The same MLPA results presented in II4 (patient’s father), II3 (sister of the proband’s father), III3 (proband’s brother), and IV1 (proband’s daughter) (Table 2).

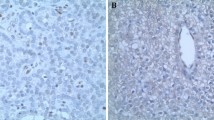
Histological images and immunohistochemistry (IHC) staining of the proband and the proband’s brother (II3). a The image of the proband’s affected parathyroid and adjacent tissue (10×, HE staining). Invasion of parathyroid into sternocleidomastoid muscle can be seen clearly. b The image of hyperplastic tissue of affected parathyroid breaking through the capsule. c The image of the proband’s affected parathyroid tissue (100×, HE staining). d The image of parafibromin IHC of the proband, showing complete loss of expression of parafibromin. e The image of the proband’s brother(II3)’s affected parathyroid tissue (100×, HE staining). f The image of parafibromin IHC of the proband’s brother(II3), showing partial loss of expression of parafibromin. g The image of HE staining of one parathyroid adenoma patient’s affected parathyroid tissue (100×, HE staining). h The image of parafibromin IHC of the PA patient’s affected tissue, showing normal expression of parafibromin
Discussion
In this study, we have discovered and located a novel 130 kb long-range deletion ranging chr1:193009590-193139118 spanning GLRX2 to exon 6 of CDC73 by conducting several genetic analyses. Clinical features of the patient and her family history have also been described. To our knowledge, this is the largest long-range deletion involving CDC73 related to familial PHPT among Chinese population and the second-largest discovered in Asia.
PHPT can present with the form of sporadic or inherited when the latter of which accounted for approximately 10% of all PHPT cases [28]. Among the syndrome-related genes, CDC73 is the second most commonly mutated one. Different investigations have shown that both point mutations and large deletions of CDC73 can be the underlying genetic basis of oncogenesis of familial PHPT. In a previous study conducted at our center, 22 patients from four FIHP/ HPT-JT families were recruited. Genetic analysis revealed three point mutations and one gross deletion (exon 4–6) of CDC73 [16]. In a systematic review of 153 FIHP kindreds that receiving genetic testing, 15% carried mutations of CDC73 [29]. Thirty-seven hereditary PHPT patients from 15 FIHP kindreds were enrolled in the review, and genetic analysis showed one CDC73 germline mutation in three unrelated kindreds, though copy number variant of CDC73 was not analyzed. A British cohort of 30 familial hyperparathyroidism patients revealed one CDC73 point mutations among 30 familial hyperparathyroidism patients. MLPA was also conducted at the same time, though no copy number variant of CDC73 was found [30].
Apart from point mutation revealed by gene screening, there have been six studies reporting different long-range deletions involving CDC73 that resulted in familial PHPT, PC, or HPT-JT. Cascón et al. reported a germline 547 kb long-range deletion spanning CDC73 and TROVE2, GLRX2, B3GALT2, and UCHL5 found in a two-generation HPT-JT family, resulted in PHPT or ossifying jaw fibroma among six individuals of this family [17]. Bricarie et al. discovered 20 patients bearing CDC73 mutations from 250 patients diagnosed with PHPT, HPT-JT, FIHP, non-familial HPT, and PC [31]. Among these 20 patients, 7 unrelated patients carried a germinal gross deletion of CDC73. Four of the seven patients carried intragenic deletion, and three bore whole CDC73 deletion, resulting in PA and PC, respectively, in these two CDC73 differently affected groups. Another study also discovered similar deletion of exon 1–17 in one PC patient [32]. Mahajan reported a novel deletion of CDC73 involving exon 1–13 in a recurrent PC patient [33]. Muscarella’s investigation of one HPT-JT family of nine people revealed a deletion of exon 1–10 of CDC73 among seven of them [34]. The same deletion of CDC73 was also found in a FIHP family consisted of seven deletion carriers [19], among which six people were previously diagnosed with PHPT and had gone through parathyroidectomy, revealing two PCs, one APA, and three PAs. Taking the results of CDC73 screening among PHPT, PC, HPT-JT, FIHP, and case reports on CDC73 deletion together into consideration, we presume that though germline CDC73 mutation is accountable for only part of PHPT, considering the high prevalence of PC, HPT-JT, and FIHP among CDC73 carriers, it is still necessary to perform CDC73 deletion investigation on patients without CDC73 point mutation but with PC or a family history of PHPT.
Aside from CDC73, some other genes were affected by the long-range deletion in our case. One of them is UCHL5, also known as UCH37, which is the coding gene of ubiquitin carboxyl-terminal hydrolase isozyme L5 (UCHL5, also named UCH37). UCHL5 is a member of the ubiquitin C-terminal hydrolases (UCHs) family, which is believed to disassemble Lys48-linked poly-ubiquitin from the distal subunit of the chain in cells selectively [35]. Over-expressed UCHL5 has been observed in hepatocellular carcinoma [36], esophageal squamous cell carcinoma [37], and epithelial ovarian cancer [38] tissues, and it has been associated with poor clinical outcome. In vitro experiments have indicated the relationship between silenced UCHL5 and reduced PAI-1 [39] expression, which are believed to promote tumor migration and invasion through TGF-β dependent pathways. In our case, neither the patient nor her family members have shown clinical manifestations of diseases related to the lack of expression of UCHL5, suggesting that the expression of a single allele is sufficient for the normal functioning of the pathway.
The whole length of TROVE2 is also affected in the long-range deletion. The 12-exon TROVE2 is officially named RO60, and it encodes the protein RO60 that is ubiquitously expressed in brain, prostate, and 25 other tissues. The highly conserved RO60 has a much common name, SSA2, the existence of which has been associated with autoimmune diseases (such as Sjögren syndrome [40] and systemic lupus erythematosus [41,42,43]). Previous studies have indicated the possible relationship between RO60 and poor clinical outcomes in breast cancer, melanoma, non-Hodgkin lymphoma, and T-cell lymphoma [44,45,46]. A recent research discovered the increased expression of RO60 in pancreatic ductal adenocarcinoma tissues while using siRNA to knockdown RO60 exerted decreased cell proliferation and invasion in vitro and repressed subcutaneous tumor growth in vivo [47]. To date, the downregulation of TROVE2 has not been proved to be relevant with PC or any other diseases. The clinical manifestation of our case has not shown the trace of autoimmune diseases, prompting that the deletion of one TROVE2 allele may not be a deciding influential factor.
Results of WGS showed that GLRX2 was also involved in the affected area. GLRX2 is also named GRX2, and the protein it encodes is a member of the glutaredoxin family of proteins that maintain cellular thiol homeostasis. GRX2 plays the role of mitochondrial redox homeostasis and cellular differentiation and transformation. Previous research has shown that over-expressed Grx2 protected the heart from doxorubicin and ischemia-induced cardiac injury in mice, while knocking-out Grx2 induces left ventricular hypertrophy and fibrosis [48]. Another research showed that zebrafish with silenced Grx2 presented with loss of all types of neurons by apoptotic cell death and unable to develop axonal scaffold [49]. Though there has not been evidence concerning the downregulation of GLRX2 and specific diseases in human, attention should be paid for the reason that GRX2 mediates thiol redox signaling who anti-apoptotic function of cell that involves tumorogenesis.
Some limitations of our study should be mentioned. First, there is still no clear evidence indicating whether UCHL5, TROVE2, or GLRX2 is related to parathyroid function or proliferation, which needs further clarifications. Second, all of the five persons detected by MLPA that carried a long-range deletion should be analyzed for structural variation, but in fact, only the proband received a WGS analysis. Furthermore, family member IV1 (8-year-old) who was a carrier of the long-range deletion presented with normal SCa and PTH levels. Considering the association between familial PHPT/PC and inactivating mutation of CDC73, a close follow-up is essential for monitoring the onset of PHPT as early as possible.
Conclusion
We found a novel 130 kb deletion spanning exon 1-6 of CDC73 and the upper-stream UCHL5, TROVE2, and GLRX2 in five patients among a family of three generations. The clinical manifestations of the five deletion carriers were also described, confirming not only the association between inactivating mutation of CDC73 and familial PHPT, but also the necessity of screening for long-range deletion of CDC73 when no point mutation of CDC73 was found in familial PHPT, especially PC patients. Genetic investigations and follow-up should also be performed in the proband’s kindred if possible due to the malignancy of PC and the hereditability of the underlying mutation. Our discovery expanded the genetic mutation spectrum of CDC73-associated familial PHPT, suggesting genetic analysis may be of help for planning surgical extents and genetic counseling.
References
N. Talat, K.M. Schulte, Ann. Surg. Oncol. 17, 2156 (2010)
F. Cetani, E. Pardi, C. Marcocci, J. Endocrinol. Invest. 39, 595 (2016)
A.S. Salcuni, F. Cetani, V. Guarnieri, V. Nicastro, E. Romagnoli, D. de Martino, A. Scillitani, D.E.C. Cole, Best Pract. Res. Clin. Endocrinol. Metab. 32, 877 (2018)
P.J. Jenkins, M.A. Satta, M. Simmgen, W.M. Drake, C. Williamson, D.G. Lowe, K. Britton, S.L. Chew, R.V. Thakker, G.M. Besser, Clin. Endocrinol. (Oxf). 47, 747 (1997)
L. Cinque, F. Pugliese, A.S. Salcuni, A. Scillitani, V. Guarnieri, Best Pract. Res. Clin. Endocrinol. Metab. 32, 891 (2018)
A.J. Gill, Endocr. Pathol. 25, 30 (2014)
J.D. Carpten, C.M. Robbins, A. Villablanca, L. Forsberg, S. Presciuttini, J. Bailey-Wilson, W.F. Simonds, E.M. Gillanders, A.M. Kennedy, J.D. Chen, S.K. Agarwal, R. Sood, M.P. Jones, T.Y. Moses, C. Haven, D. Petillo, P.D. Leotlela, B. Harding, D. Cameron, A.A. Pannett, A. Höög, H. 3rd Heath, L.A. James-Newton, B. Robinson, R.J. Zarbo, B.M. Cavaco, W. Wassif, N.D. Perrier, I.B. Rosen, U. Kristoffersson, P.D. Turnpenny, L.-O. Farnebo, G.M. Besser, C.E. Jackson, H. Morreau, J.M. Trent, R.V. Thakker, S.J. Marx, B.T. Teh, C. Larsson, M.R. Hobbs, Nat. Genet. 32, 676 (2002)
T.M. Shattuck, S. Välimäki, T. Obara, R.D. Gaz, O.H. Clark, D. Shoback, M.E. Wierman, K. Tojo, C.M. Robbins, J.D. Carpten, L.-O. Farnebo, C. Larsson, A. Arnold, N. Engl. J. Med. 349, 1722 (2003)
G.E. Woodard, L. Lin, J.-H. Zhang, S.K. Agarwal, S.J. Marx, W.F. Simonds, Oncogene 24, 1272 (2005).
C. Mosimann, G. Hausmann, K. Basler, Cell 125, 327 (2006)
A. Yart, M. Gstaiger, C. Wirbelauer, M. Pecnik, D. Anastasiou, D. Hess, W. Krek, Mol. Cell. Biol. 25, 5052 (2005)
C. Juhlin, C. Larsson, T. Yakoleva, I. Leibiger, B. Leibiger, A. Alimov, G. Weber, A. Höög, A. Villablanca, Endocr. Relat. Cancer 13, 509 (2006)
L. Lin, M. Czapiga, L. Nini, J.-H. Zhang, W.F. Simonds, Mol. Cancer Res. 5, 183 (2007)
C. Zhang, D. Kong, M.-H. Tan, D.L.J. Pappas, P.-F. Wang, J. Chen, L. Farber, N. Zhang, H.-M. Koo, M. Weinreich, B.O. Williams, B.T. Teh, Biochem. Biophys. Res. Commun. 350, 17 (2006)
K. van der Tuin, C.M.J. Tops, M.A. Adank, J.-M. Cobben, N.A.T. Hamdy, M.C. Jongmans, F.H. Menko, B.P.M. van Nesselrooij, R.T. Netea-Maier, J.C. Oosterwijk, G.D. Valk, B.H.R. Wolffenbuttel, F.J. Hes, H. Morreau, J. Clin. Endocrinol. Metab. 102, 4534 (2017)
J. Kong, O. Wang, M. Nie, J. Shi, Y. Hu, Y. Jiang, M. Li, W. Xia, X. Meng, X. Xing, Clin. Endocrinol. (Oxf). 81, 222 (2014)
A. Cascón, C.V. Huarte-Mendicoa, L. Javier Leandro-García, R. Letón, J. Suela, A. Santana, M.B. Costa, I. Comino-Méndez, I. Landa, L. Sánchez, C. Rodríguez-Antona, J.C. Cigudosa, M. Robledo, Genes. Chromosomes Cancer 50, 922 (2011)
A. Dutta, R. Pal, N. Jain, P. Dutta, A. Rai, A. Bhansali, A. Behera, U.N. Saikia, V. Vishwajeet, D. Collier, H. Boon, M. Korbonits, S.K. Bhadada, J. Endocr. Soc. 3, 2224 (2019)
E. Korpi-Hyövälti, T. Cranston, E. Ryhänen, J. Arola, K. Aittomäki, T. Sane, R.V. Thakker, C. Schalin-Jäntti, J. Clin. Endocrinol. Metab. 99, 3044 (2014)
S.M. Wilhelm, T.S. Wang, D.T. Ruan, J.A. Lee, S.L. Asa, Q.Y. Duh, G.M. Doherty, M.F. Herrera, J.L. Pasieka, N.D. Perrier, S.J. Silverberg, C.C. Solórzano, C. Sturgeon, M.E. Tublin, R. Udelsman, S.E. Carty, JAMA Surg. 151, 959 (2016)
J. Kong, O. Wang, M. Nie, J. Shi, Y. Hu, Y. Jiang, M. Li, W. Xia, X. Meng, X. Xing, PLoS ONE 11, (2016)
W. Wang, J. Kong, M. Nie, Y. Jiang, M. Li, W. Xia, X. Meng, X. Xing, O. Wang, Clin. Endocrinol. (Oxf). 87, 865 (2017)
A. Song, Y. Yang, Y. Wang, S. Liu, M. Nie, Y. Jiang, M. Li, W. Xia, O. Wang, X. Xing, Endocr. Pract. 26, 1093 (2020)
R. Lloyd, R. Osamura, G. Klöppel, WHO Classification of Tumours of Endocrine Organs (IARC, Lyon, France, 2017)
L.D. Ritchie, E.B. Fung, B.P. Halloran, J.R. Turnlund, M.D. Van Loan, C.E. Cann, J.C. King, Am. J. Clin. Nutr. 67, 693 (1998)
O. Wang, C. Wang, M. Nie, Q. Cui, H. Guan, Y. Jiang, M. Li, W. Xia, X. Meng, X. Xing, PLoS ONE 7, (2012)
Y. Yang, X. Chu, M. Nie, A. Song, Y. Jiang, M. Li, W. Xia, X. Xing, O. Wang, Endocrine 69, 212 (2020)
W.D. Fraser, Lancet 374, 145 (2009)
E. Pardi, S. Borsari, F. Saponaro, F. Bogazzi, C. Urbani, S. Mariotti, F. Pigliaru, C. Satta, F. Pani, G. Materazzi, P. Miccoli, L. Grantaliano, C. Marcocci, F. Cetani, PLoS ONE 12, 1 (2017)
S. Mariathasan, K.A. Andrews, E. Thompson, B.G. Challis, S. Wilcox, H. Pierce, J. Hale, S. Spiden, G. Fuller, H.L. Simpson, B. Fish, P. Jani, I. Seetho, R. Armstrong, L. Izatt, M. Joshi, A. Velusamy, S.M. Park, R.T. Casey, Clin. Endocrinol. (Oxf). 93, 409 (2020)
L. Bricaire, M.F. Odou, C. Cardot-Bauters, B. Delemer, M.O. North, S. Salenave, D. Vezzosi, J.M. Kuhn, A. Murat, P. Caron, J.L. Sadoul, C. Silve, P. Chanson, A. Barlier, E. Clauser, N. Porchet, L. Groussin, J. Clin. Endocrinol. Metab. 98, 403 (2013)
P.S.A. Wijewickrama, N.P. Somasundaram, Case Rep. Endocrinol. 2020, 0 (2020)
G. Mahajan, A. Sacerdote, BMJ Case Rep. 11, (2018)
L.A. Muscarella, D. Turchetti, A. Fontana, F. Baorda, O. Palumbo, A. la Torre, D. de Martino, R. Franco, N.S. Losito, A. Repaci, U. Pagotto, L. Cinque, M. Copetti, M.G. Chiofalo, L. Pezzullo, P. Graziano, A. Scillitani, V. Guarnieri, Oncotarget 9, 20721 (2018)
Y.A. Lam, G.N. DeMartino, C.M. Pickart, R.E. Cohen, J. Biol. Chem. 272, 28438 (1997)
Y. Fang, D. Fu, W. Tang, Y. Cai, D. Ma, H. Wang, R. Xue, T. Liu, X. Huang, L. Dong, H. Wu, X. Shen, Biochim. Biophys. Acta - Mol. Cell Res. 1833, 559 (2013)
Y. Chen, D. Fu, J. Xi, Z. Ji, T. Liu, Y. Ma, Y. Zhao, L. Dong, Q. Wang, X. Shen, Dig. Dis. Sci. 57, 2310 (2012)
L. Wang, Y.J. Chen, K. Xu, Y.Y. Wang, X.Z. Shen, R.Q. Tu, Tumor Biol. 35, 11427 (2014)
A.J. Cutts, S.M. Soond, S. Powell, A. Chantry, Int. J. Biochem. Cell Biol. 43, 604 (2011)
M.A. Alspaugh, E.M. Tan, J. Clin. Invest. 55, 1067 (1975)
R.L. Loch, B.B. Beaulieu Jr, E.J.T. Chrystal, P. Goldman, Science 211, 400 (1981)
S.L. Deutscher, J.B. Harley, J.D. Keene, Proc. Natl. Acad. Sci. U. S. A. 85, 9479 (1988)
E. Ben-Chetrit, B.J. Gandy, E.M. Tan, K.F. Sullivan, J. Clin. Invest. 83, 1284 (1989)
B.C. Böckle, G. Stanarevic, G. Ratzinger, N.T. Sepp, Br. J. Dermatol. 167, 1067 (2012)
S. Bernatsky, R. Ramsey-Goldman, J. Labrecque, L. Joseph, J.F. Boivin, M. Petri, A. Zoma, S. Manzi, M.B. Urowitz, D. Gladman, P.R. Fortin, E. Ginzler, E. Yelin, S.C. Bae, D.J. Wallace, S. Edworthy, S. Jacobsen, C. Gordon, M.A. Dooley, C.A. Peschken, J.G. Hanly, G.S. Alarcón, O. Nived, G. Ruiz-Irastorza, D. Isenberg, A. Rahman, T. Witte, C. Aranow, D.L. Kamen, K. Steinsson, A. Askanase, S. Barr, L.A. Criswell, G. Sturfelt, N.M. Patel, J.L. Senécal, M. Zummer, J.E. Pope, S. Ensworth, H. El-Gabalawy, T. McCarthy, L. Dreyer, J. Sibley, Y. St. Pierre, A.E. Clarke, J. Autoimmun. 42, 130 (2013)
J.G. Routsias, J.D. Goules, G. Charalampakis, S. Tzima, A. Papageorgiou, M. Voulgarelis, Semin. Arthritis Rheum. 43, 178 (2013)
D. Liu, W. Qian, D. Li, L. Kong, Biochem. Biophys. Res. Commun. 495, 2519 (2018)
R.J. Mailloux, J.Y. Xuan, S. McBride, W. Maharsy, S. Thorn, C.E. Holterman, C.R.J. Kennedy, P. Rippstein, R. DeKemp, J. Da Silva, M. Nemer, M. Lou, M.E. Harper, J. Biol. Chem. 289, 14812 (2014)
L. Bräutigam, L.D. Schütte, J.R. Godoy, T. Prozorovski, M. Gellert, G. Hauptmann, A. Holmgren, C.H. Lillig, C. Berndt, Proc. Natl. Acad. Sci. USA 108, 20532 (2011)
Acknowledgements
We thank the patient and the family for permitting us to publish certain information.
Funding
This work was supported by the Chinese Academy of Medical Sciences (CAMS) Initiative for Innovative Medicine (CAMS- I2M) (No. 2017-I2M-1–001) and the National Natural Science Foundation of China (No. 82070817).
Author information
Authors and Affiliations
Contributions
All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
This study was approved by the Ethics Committee of Peking Union Medical College Hospital and conducted under the instructions of the principles in the Declaration of Helsinki.
Consent to participate
Written informed consent to participate was obtained from all participants.
Consent for publication
Written informed consent for publication was obtained from all participants.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, Y., Song, A., Nie, M. et al. A novel long-range deletion spanning CDC73 and upper-stream genes discovered in a kindred of familial primary hyperparathyroidism. Endocrine 75, 907–915 (2022). https://doi.org/10.1007/s12020-021-02917-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-021-02917-5