
Abstract
Objective
Ipilimumab is a human monoclonal antibody directed against cytotoxic T-lymphocyte antigen-4, that has been shown to significantly improve survival in patients with metastatic melanoma. Blocking cytotoxic T-lymphocyte antigen-4 elicits T cell activation, proliferation and anti-tumor response, but can also trigger immune-related adverse events. Among immune-related endocrinopathies, hypophysitis represents the most frequent, with an incidence up to 17% in patients treated with ipilimumab.
Design and methods
We report nine cases of ipilimumab-induced hypophysitis in a cohort of 273 patients treated with ipilimumab between 2006 and 2015, as part of clinical trials or after its marketing. Thyroid function tests were scheduled at screening and during follow up (every 21 days) in all patients. Cortisol, adrenocorticotropic hormone, follicle-stimulating hormone, luteinizing hormone, and estradiol (for females) or testosterone (for males), prolactin, growth hormone, insulin-like growth factor 1 were measured only in case of clinical suspicion.
Results
The incidence of hypophysitis was 3.3%. The most frequent pituitary failure was adrenocorticotropic hormone and thyroid stimulating hormone secretion with a complete recovery of thyroid stimulating hormone, but not of adrenocorticotropic hormone during follow up. All patients had negative pituitary antibodies. The main symptoms at diagnosis were fatigue and headache.
Conclusion
Clinicians should be aware about the risk of hypophysitis during treatment with immune check-point inhibitors and the necessity of investigating pituitary function during therapy. Pituitary magnetic resonance imaging does not seem pivotal for a definite diagnosis if not performed at the onset of disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ipilimumab is a humanized monoclonal antibody directed against cytotoxic T-lymhocyte antigen-4 (CTLA-4), an inhibitory receptor expressed on the membrane of activated T cells and regulatory T lymphocytes. Blocking CTLA-4 by ipilimumab sustains T cell activation thereby enhancing anti-tumor immune response, including tumor-specific T cells. Ipilimumab was approved in 2011 by the food and drug administration and European medicines agency for treatment of unresectable or metastatic melanoma based on results obtained in a phase III trial in which ipilimumab achieved disease control rate of 28.5% and improved overall survival [1].
However, the increased T cell activity promoted by ipilimumab can result in the development of autoimmune phenomena. In patients treated with ipilimumab the most frequent endocrinological adverse event is pituitary failure with a frequency ranging between 0.7 and 17% [1–6], followed by thyroid and, more rarely, adrenal failure [2, 7].
Ipilimumab-induced hypophysitis is an inflammatory disease of the pituitary that it seems pathologically similar to the classic primitive autoimmune form, but the exact mechanism underlying the pathogenesis of CTLA-4-related hypophysitis is still undefined. In murine models, repeated injections of CTLA-4 blocking antibodies induce hypophysitis characterized by focal lymphocytic infiltrate and induces the development of anterior pituitary antibodies (mainly towards prolactin- and ACTH-secreting cells) [8].
The manufacture recommend high dose corticosteroids in patients developing hypophysitis. However, the real benefit of this approach is not fully documented. Indeed high dose steroid regimen could mitigate inflammatory symptoms such as headache and vision changes, but do not have an impact on recovery of pituitary function [5].
The purpose of this paper is to characterized ipilimumab-induced hypophysitis in a large cohort of patients treated with ipilimumab monotherapy with a long follow-up.
Subjects and methods
Our study analysis was performed retrospectively by collecting data from medical records and database authorized by our Institutional review board in patients treated with ipilimumab from 2006 to 2015 at the Division of Medical Oncology and Immunotherapy of the University Hospital of Siena; all patients were not previously treated with other immune check-point inhibitors. All patients were previously treated with other chemotherapeutic agents, but the wash-out period for each patients was at least 4 weeks.
The study group included 273 patients (108 females and 165 males), 176 within and 97 outside clinical trials (ipilimumab prescribed after its marketing). In addition 30 patients were included in clinical trials, but are still in blinded phase and were excluded from the analysis.
Patients enrolled in clinical trials (7 with metastatic prostate cancer and 169 with metastatic melanoma) received ipilimumab at doses of 3 or 10 mg/Kg, while all patients, who received drug after marketing for metastatic melanoma, were treated with the approved dose of 3 mg/Kg.
All patients received ipilimumab iv infusion every 3 weeks for a total of four doses. In 21 patients, additional infusions as maintenance therapy were continued every 3 months for up to 3 years.
Thyroid function tests were performed in all patients at screening and during follow-up (approximately each 21 days) as requested in clinical trials and in the package insert. Cortisol, adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), luteinizing hormone (LH) and estradiol (for females) or testosterone (for males) were measured in case of clinical suspicion; in these cases serum samples stored in a bio-bank in our Institution were used to test the entire panel of pituitary hormones.
All cases of hypophysitis were studied with a complete hormonal and pituitary antibodies evaluation at baseline and during follow-up.
Serum hormone measurements were performed by chemiluminescence immune assays as follows: cortisol, free thyroxine (fT4), free triiodothyronine (fT3), LH, prolactin (PRL), growth hormone (GH), testosterone (T), FSH (Access, Beckman Coulter, Fullerton, CA, USA); thyroid stimulating hormone (TSH), ACTH and insulin-like growth factor 1 (IGF-1) (Immulite 2000, Siemens Medical Solution Diagnostics, Los Angeles, CA, USA). Pituitary antibodies were detected by indirect immunofluorescence assay (Euroimmun, Lubecca, Germany) using as substrate monkey pituitary gland. [Procedure: apply 25 µl of diluted sample to each reaction field of the reagent tray, then incubate for 30 min at room temperature (+18 to +25 °C). Rinse the BIOCHIP slides with a flush of PBS-Tween using a beaker and immerse them immediately afterwards in a cuvette containing PBS-Tween for at least 5 min and shake with a rotary shaker. Apply 20 µl of fluorescein labeled anti-human globulin to each reaction field of a clean reagent tray. Incubate for 30 min at room temperature (+18 to +25 °C). Rinse the BIOCHIP slides with a flush of PBS-Tween using a beaker and put them into the cuvette filled with the new PBS-Tween for at least 5 min. Play embedding medium onto a cover glass-drops of max 10 µl per reaction field. Use a polystyrene embedding template and read the fluorescence with the microscope].
Hypophysitis was defined as the presence of biochemical evidence of anterior pituitary hormone deficiency with or without evidence of pituitary enlargement associated with mass-effect (i.e., headache, visual disturbances).
In particularly, secondary adrenal insufficiency was defined by the presence of acute onset of symptoms of adrenal insufficiency associated with biochemically proven low (<30 ng/ml) or suppressed morning serum cortisol levels with inappropriately low ACTH levels in the absence of exogenous steroid treatment.
Secondary hypothyroidism was defined by a low fT4 levels with inappropriately low or normal TSH level; when fT3 and fT4 were normal in presence of suppressed TSH level a thyrotropin releasing hormone (TRH) test was performed to confirm the diagnosis.
Hypogonadotropic hypogonadism was diagnosed in male patients by low testosterone levels with inappropriately normal or low gonadropins (pituitary-gonadal axis in patients with metastatic prostate cancer was not evaluable because of hormonal blockage therapy). All females were in the post-menopausal phase so the diagnosis was made based on low gonadotropin levels.
All the previous pituitary deficiencies were defined in absence of confounding factors such as drugs and timing of blood samples.
Dynamic stimulation tests (TRH, corticotropin releasing hormone (CRH), gonadotrophin releasing hormone (GnRH), GH releasing hormone (GHRH) plus arginine stimulation tests) were performed when indicated in selected patients.
Magnetic resonance imaging (MRI) of the brain and diencephalic-pituitary region were retrospectively collected, because not all performed in the same institution. All pituitary MR images had been obtained at a 1.5T magnet, and were 5- to 3-mm-thick, before and after intravenous gadolinium-based contrast medium injection. In most cases, MR study did not include a dynamic contrast enhanced sequence. Pituitary MR images were reviewed by a single neuroradiologist (AC) blinded to both clinical and laboratoristic findings. This review included a qualitative evaluation assessing pituitary volume, pituitary stalk thickness and bright spot of the posterior lobe on T1-weighted images. A more detailed evaluation of pituitary MRI was behind the scope of this retrospective review.
Epidemiological data are presented as the mean ± SD and median when appropriated. The t test for independent data were performed for normal variables. To evaluate significant differences in data frequency we analyzed contingency tables. We used StatView for Windows version 5.0.1 (SAS Institute, Cary, NC) for statistical analysis. We considered p < 0.05 to be statistically significant.
Results
Hypophysitis was diagnosed in 9/273 patients (3.3%), 5/9 (55%) were females and 4/9 (45%) males; all patients had metastatic melanoma; none of these patients had received prior cranial radiotherapy. As shown in Table 1, ACTH deficiency was found in all cases, isolated in 2/9 (22%) and associated with multiple anterior pituitary hormone deficiencies in 7/9. Specifically, gonadotropin and TSH deficiencies were the most frequent failure associated to ACTH deficit (seven and six patients, respectively), GH deficiency was present in one patient and undetectable levels of PRL were found in four patients while no case of hyperprolactinemia were found. None presented posterior pituitary deficiency. Pituitary antibodies were never documented.
The prevalence of hypophysitis was not significantly different among patients treated with 3 or 10 mg/Kg (2.7 vs. 4.3%, respectively) and no gender predilection was demonstrated [4/110 (3.6%) females vs. 5/163 (3.0%) males].
Mean age at diagnosis was 64.13 ± 9.6 years. No age difference was found between the cohort of patients with and without hypophysitis.
The most common presenting symptoms were sudden onset of headache (78%), fatigue (44%) and general discomfort (33%). The follow-up period ranged from 4 to 51 months, with a median length of 23 months.
The clinical and biochemical features of the nine patients with hypophysitis are reported in Table 1. Dynamic hormonal tests were performed in patients 2, 4, 6 and 9, in particular CRH test was performed in patients 4, 6 and 9; TRH test in patients 2 and 4; GHRH + arginine test in patient 3 and GnRH test in patient 2. All patients presented a blunted response to these tests.
Hypophysitis was diagnosed after a median of 7.6 weeks from the first cycle of Ipilimumab (range 5–40 weeks). In particular 4/9 were diagnosed after 4 cycles, 3/9 after 3 cycles and 2/9 after 2 cycles.
In patients 6 the diagnosis of secondary adrenal insufficiency was delayed at 40 weeks from the beginning because before that, he was treated with corticosteroid therapy for brain metastases. However a review of previous MRI, performed for brain metastases 6 months before the clinical diagnosis, showed a pituitary enlargement suspicious for hypophysitis. Follow-up brain MRIs displayed a pituitary reduction with a possible diagnosis of “partial empty sella”.
Recovery of pituitary function was never documented for ACTH. Four patients out of seven recovered normal gonadotropin secretion and all patients with a secondary hypothyroidism restored normal TSH secretion. One patient with GH deficiency and low IGF-1 recovered very quickly (1 month).
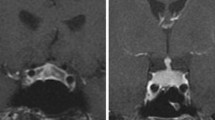
Brain and pituitary MRI was available in seven out of the nine patients with hypophysitis, performed 11–105 days after the clinical diagnosis. Two patients did not undergo brain and pituitary MRI because it was not prescribed by the treating physician. Brain MRI showed pituitary enlargement in four patients without chiasm compression, pituitary contraction in one patients, and no abnormalities in two patients. At MRI follow-up, four patients with pituitary enlargement turned into “empty sella” (Fig. 1). In particular it was documented after 6 months in patient 1, 5 months in patient 3 and 2 months in patients 4 and 6. MRI did not detect intracranial metastases, except in patient # 6 where brain metastases appeared at MRI during follow up and treated with dexamethasone.

Patient #4. Pituitary MRI at unenhanced T1-weighted sagittal images. a Before Ipilimumab treatment (normal morphology) b 40 days after the diagnosis of hypophysitis (pituitary enlargement) and c 2 months later (partial empty sella)
No patients required high-dose corticosteroids for symptoms of intracranial mass-effect, but all patients with secondary adrenal insufficiency received physiological replacement therapy (cortone acetate 37.5 mg/die). Two patients died during follow up, one due to disease progression and one probably due to misdiagnosed adrenal crisis not correctly treated in emergency room.
As shown in Table 1, five patients presented other IRAEs during ipilimumab treatment, namely colitis (grade 1–2 according to CTCAE v.3) in two patients, rash (grade 1–3) in three patients, liver damage (grade 3) and itch (grade 1) in one patient.
Discussion
In our series of ipilimumab treated patients, hypophysitis developed in 3.3% compared to a range between 0.7 and 17% reported in previous studies [1–6]. This wide range of variation is probably due to the lack of uniform criteria in the definition of hypophysitis; this diagnosis may escape recognition, mainly because patients are often treated with corticosteroids (i.e., for AE or brain metastases) or because classical symptoms of hypophysitis could be improperly related to the neoplastic disease.
At variance with other studies which showed a predilection for males [2, 3, 5, 6], we did not observe any gender difference in our series. Moreover, our patients with hypophysitis were not significantly older than the entire group, in contrast to previously reports [3, 6] and no dose-dependency was documented as reported in one study [3] but not in others [2, 6, 9].
The presenting symptoms were the sudden onset of severe headache, fatigue or general discomfort. The median time to disease onset was 7 weeks, similar to previously reports [3, 5, 6].
No patients treated with more than four cycles of ipilimumab developed hypophysitis, suggesting that prolonging the numbers of cycles (maintenance therapy) does not confer a higher risk of developing hypophysitis as reported in other studies [2, 10].
All patients with secondary hypothyroidism recovered a normal TSH secretion during follow up and this observation was in agreement with a recent study by Albarel et al. [6] but not with two other studies [3, 4] where recovery was present in less than 6% of the cases. The recovery of secondary hypothyroidism could be suspected on the basis of TSH increase during the follow up (usually from value below the normal range to normal value); in these cases, to confirm the normalization of thyroid axis, l-thyroxine withdrawal should be planned. On the contrary, in our opinion, corticosteroid withdrawal in patients with secondary hypocorticism is not necessary when morning cortisol levels are low before assuming replacement therapy as it has happened in all our patients.
The pathogenesis of ipilumab-induced hypophysitis is still unknown. A recent study demonstrated that pituitary antibodies, developed in all case of hypophysitis, were directed predominantly to TSH-secreting cells and less frequently to FSH or ACTH-secreting cells [8]. Interestingly, the expression of CTLA-4, the receptor target of ipilimumab, on pituitary cell lines was demonstrated in this study. Thus, the pituitary damage could be caused by a direct effect on pituitary cells from the monoclonal antibody and/or by the action of activated T cells directed toward antigens shared by tumor cells and pituitary cells or by cross-reacting antigens.
Moreover Caturegli et al. confirmed, in a autopsy series, the presence of pituitary CTLA-4 expression in all patients with ipilimumab-induced hypophysitis and its correlation with the wide clinical and morphological heterogeneity; the highest levels were found in the patient who had clinical and pathological evidence of severe hypophysitis. The administration of CTLA-4 blocking antibodies could cause an aggressive form of hypophysitis through type IV (T-cell dependent) and type II (IgG dependent) immune mechanisms [11]. So we are not fully surprised by the clinical heterogeneity of our cases that could be probably due to a different pituitary CTLA-4 expression.
We did not find pituitary antibodies in our cases of hypophysitis, in spite of what was found by Caturegli [8]; maybe it could dependent by the two different pituitary substrates used for the immunofluorescence method (Macaca mulatta vs. human pituitary, respectively). As reported by Ricciuti et al. [12] the human substrate had superior sensitivity and specificity but still now only animal substrates are commercially available. Thus, in our experience the measurement of pituitary antibodies was not an additional tool to establish the diagnosis.
As expected in a retrospective study, pituitary MR images were collected at different times from the diagnosis of hypophysitis and were acquired with different techniques. Moreover a baseline brain or pituitary study was not available for all patients, therefore we are not able to make a comparison of the pituitary morphology with the baseline (before starting Ipilimumab) in all cases. All these limitations did not allow a thorough semiquantitative evaluation, which was behind the scope of this paper. No other disease potentially cause of hypopituitarism was documented in any case.
Notably, pituitary morphology could change over time (i.e. pituitary enlargement followed by empty sella), so it is not surprising that a pituitary enlargement was found only in few patients (4/7). Although our study is limited by the small number of patients, as already proposed in recent studies [3, 10], we suggest that MRI should be performed, at disease onset and in all patients with symptoms and signs clearly evoking mass-effects, such as severe headache or visual field defects. On the contrary it could not be crucial in itself to make diagnosis if the images were acquired later; it may instead be useful in excluding pituitary metastases as a differential diagnosis.
As far as treatment is concerned we believe that high dose steroid therapy should be reserved only in cases of persistent and severe mass-effect symptoms, in view of a recent report in which high-dose corticosteroids did not improve the outcome of hypophysitis and the overall patients survival [5].
In conclusion, hypophysitis represents a rare, but potentially life-threatening complication of ipilimumab treatment, that requires a multidisciplinary management crucial for a prompt diagnosis and appropriate treatment.
Considering that no patients in our series showed ACTH recovery during follow-up, while all patients with a secondary hypothyroidism recovered a normal TSH secretion, we believe that it is not necessary to re-evaluate adrenal axis with dynamic test, but it is mandatory to re-evaluate TSH secretion at some point during follow-up after withdrawal or weaning of l-thyroxine replacement therapy, mainly in patients with a TSH rising during follow up.
In view of the disease presentation observed in our series, we suggest that screening for hypophysitis should be done in any patients presenting with suspicious symptoms and in all patients developing TSH deficiency.
Moreover, considering that some adverse event of Ipilimumab as asthenia and nausea are similar to initial symptoms of hypoadrenalism, apart the thyroid function that is routinely monitored, we suggest to evaluate cortisol and ACTH levels before starting treatment and at least every 2–3 cycles to avoid underestimation of adrenal insufficiency.
Lastly, also during follow-up pituitary function tests should be evaluated in case of clinical suspicious because of a possible, even if rare, later onset.
References
F.S. Hodi, S.J. O'Day, D.F. McDermott, R.W. Weber, J.A. Sosman, J.B. Haanen, R. Gonzalez, C. Robert, D. Schadendorf, J.C. Hassel, W. Akerley, A.J. van den Eertwegh, J. Lutzky, P. Lorigan, J.M. Vaubel, G.P. Linette, D. Hogg, C.H. Ottensmeier, C. Lebbé, C. Peschel, I. Quirt, J.I. Clark, J.D. Wolchok, J.S. Weber, J. Tian, M.J. Yellin, G.M. Nichol, A. Hoos, W.J. Urba, Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010)
S.M. Corsello, A. Barnabei, P. Marchetti, L. De Vecchis, R. Salvatori, F. Torino, Endocrine side effects induced by immune checkpoint inhibitors. J. Clin. Endocrinol. Metab. 98, 1361–1375 (2013)
A.T. Faje, R. Sullivan, D. Lawrence, N.A. Tritos, R. Fadden, A. Klibanski, L. Nachtigall, Ipilimumab-induced hypophysitis: a detailed longitudinal analysis in a large cohort of patients with metastatic melanoma. J. Clin. Endocrinol. Metab. 99, 4078–4085 (2014)
M. Ryder, M. Callahan, M.A. Postow, J. Wolchok, J.A. Fagin, Endocrine-related adverse events following ipilimumab in patients with advanced melanoma: a comprehensive retrospective review from a single institution. Endocr. Relat. Cancer. 21, 371–381 (2014)
L. Min, F.S. Hodi, A. Giobbie-Hurder, P.A. Ott, J.J. Luke, H. Donahue, M. Davis, R.S. Carroll, U.B. Kaiser, Systemic high-dose corticosteroid treatment does not improve the outcome of ipilimumab-related hypophysitis: a retrospective cohort study. Clin. Cancer Res. 21, 749–755 (2015)
F. Albarel, C. Gaudy, F. Castinetti, T. Carré, I. Morange, B. Conte-Devolx, J.J. Grob, T. Brue, Long-term follow-up of ipilimumab-induced hypophysitis, a common adverse event of the anti-CTLA-4 antibody in melanoma. Eur. J. Endocrinol. 172, 195–204 (2015)
A.M. Di Giacomo, M. Biagioli, M. Maio, The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin. Oncol. 37, 499–507 (2010)
S. Iwama, A. De Remigis, M.K. Callahan, S.F. Slovin, J.D. Wolchok, P. Caturegli, Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci. Transl. Med. 6, 230ra45 (2014)
A.V. Maker, J.C. Yang, R.M. Sherry, S.L. Topalian, U.S. Kammula, R.E. Royal, M. Hughes, M.J. Yellin, L.R. Haworth, C. Levy, T. Allen, S.A. Mavroukakis, P. Attia, S.A. Rosenberg, Intrapatient dose escalation of anti-CTLA-4 antibody in patients with metastatic melanoma. J. Immunother. 29, 455–463 (2006)
A. Faje, Immunotherapy and hypophysitis: clinical presentation, treatment, and biological insights. Pituitary 19, 82–92 (2016)
P. Caturegli, G. Di Dalmazi, M. Lombardi, F. Grosso, H.B. Larman, T. Larman, G. Taverna, M. Cosottini, I. Lupi, Hypophysitis secondary to cytotoxic T-lymphocyte-associated protein 4 blockade: insights into pathogenesis from an autopsy series. Am. J. Pathol. 186, 3225–3235 (2016)
A. Ricciuti, A. De Remigis, M.A. Landek-Salgado, L. De Vincentiis, F. Guaraldi, I. Lupi, S. Iwama, G.S. Wand, R. Salvatori, P. Caturegli, Detection of pituitary antibodies by immunofluorescence: approach and results in patients with pituitary diseases. J. Clin. Endocrinol. Metab. 99, 1758–1766 (2014)
Acknowledgements
Michele Maio has received research grants and has taken part to advisory boards by Bristol-Myers-Squibb.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Rights and permissions
About this article

Cite this article
Brilli, L., Danielli, R., Ciuoli, C. et al. Prevalence of hypophysitis in a cohort of patients with metastatic melanoma and prostate cancer treated with ipilimumab. Endocrine 58, 535–541 (2017). https://doi.org/10.1007/s12020-017-1289-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-017-1289-2