
Abstract
Uterine decidualization characterized by stromal cell proliferation and differentiation is critical to the establishment of pregnancy in many species. Progesterone is a key factor in regulating endometrial cell decidualization, however, the molecular basis involved in mediating the effects of progesterone during decidualization remains largely unknown. We report here that the DNA replication licensing factor MCM2, one of the conserved set of six-related proteins (MCM complex: MCM2–7) essential for eukaryotic DNA replication, is dynamically expressed in both proliferative and differentiated stromal cells during mouse periimplantation uterus. Applying PR-knockout mouse model and pharmacological strategy, we further found that the expression of Mcm2 is induced by progesterone action in the mouse uterine stroma. Employing a primary cell culture system, we further demonstrated that siRNA-mediated silencing of MCM2 arrests the cell cycle at G1–S transition during stromal cell proliferation. Moreover, the downregulation of Mcm2 could also compromise stromal cell differentiation. Collectively, our studies uncovered the role of a unique DNA replication licensing molecule MCM2 in mediating Progesterone-induced stromal cell decidualization in mouse uterus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Embryo at blastocyst stage initiates the first physical and physiological interactions with uterine endometrium, a process known as implantation [1]. In rodents, following embryo implantation, the uterine stromal cells surrounding the embryo undergo extensive proliferation, and differentiation into specialized cell type with polyploidy, a process termed as decidualization, to accommodate embryonic growth and invasion [2]. The ovarian hormone progesterone through its nuclear receptor progesterone receptor (PR) was well defined as a factor to direct the differentiation of the uterine cells to support embryo development and implantation [3, 4]. There are two isoforms of PR, PRA and PRB, respectively. Ovarian progesterone signaling through PRA is essential for post-implantation uterine development and decidualization in mice [5]. Mice lacking both PRB and p21, a cyclin-dependent kinase (CDK) inhibitor, are infertile with decidualization defects [6]. However, the underlying mechanism by which progesterone induces stromal cell proliferation and differentiation is still largely unknown.
The stromal cell proliferation and polyploidization in response to ovarian hormones is mediated by the induction of specific cell cycle regulators [7]. The cell cycle is tightly regulated at two particular checkpoints, G1–S and G2–M phases. A wealth of data had revealed that a complex interplay of cyclins, cyclin-dependent kinases (cdks) and cdk inhibitors (CKIs) participated in operating these phases [6]. For example, during G1 phase, the D-type cyclins, also known as G1 cyclins, accumulate and associate with cdk4 or cdk6 to form holoenzymes that facilitate cell entry into the S-phase. Cyclins A and B are involved in the progression from S through G2–M phase. Polyploidization is a hallmark of decidualization that utilizes a specialized cell cycle progression [6]. Among those regulators orchestrating polyploidization in the uterine stroma cells, Cyclin D3 is one of the best-known factors, and its deficiency in mice leads to defective decidualization [8]. Moreover, a body of studies has identified several other regulators crucial for decidualization executing their function through cyclin D3, including Hoxa10, IL-11Ra, and DEDD [9–11]. However, the mechanisms regarding the onset of stromal cell proliferation and differentiation at different phases of cell cycle in response to ovarian hormones are still largely unknown.
Mini-chromosome maintenance (Mcm) 2–7 is a hexameric complex which recruits to the chromatin early in the G1-phase of the cell cycle to form part of the pre-replicative complex (preRC) at origins of DNA replication and is required for replication origin activation to occur as cells enter the S-phase [12]. Activation of MCM complex by cdks leads to the initiation of DNA synthesis and MCM proteins also act as replicative helicase to unwind DNA at replication forks during DNA synthesis [13–15]. MCM proteins are expressed in cycling cells but are down-regulated and dissociated from chromatin in quiescent cells [15]. Previous studies had shown that Progesterone blocks estrogen-induced DNA synthesis through the inhibition of replication licensing MCMs. In the uterine epithelium, E 2 induces uterine epithelial cell proliferation through stimulating the expression of the MCMs and of the loading factor CDT1 at the G1/S-phase transition, while P 4 inhibits the transcript abundance, activity, and cellular localization of MCM 2–6, thus blocks the E 2-induced epithelial cell proliferation [16]. A recent study demonstrated the Kruppel-like transcription 15 (Klf15), a downstream physiological mediator of progesterone’s cell cycle inhibitory action in the uterine epithelium. Klf15 bind to the Mcm2 promoter under the regulation of P 4 + E 2 [17]. After P 4 E 2 exposure and in contrast to E 2-treated mice, the uterine Mcm2 promoter displays increased histone 3 (H3) methylation and the recruitment of histone deacetylase 1 and 3 with the concomitant deacetylation of H3, thus the decreased expression of Mcm2 [17]. Despite the importance of MCMs in mouse and human uterine epithelial cells, its detailed functions during stromal decidualization are not well understood.
We provide herein evidence that MCM2 required for DNA replication licensing is intensely expressed in decidual cells, and inhibition of P 4–PR signaling could severely downregulate its expression. Silence Mcm expression using siRNA in primary-cultured stromal cells hampered normal DNA synthesis and impaired cell differentiation.
Materials and methods
Mice and treatments
Mice were housed in Institutional Animal Care Facility of Institute of Zoology according to institutional guidelines for laboratory animals. Mice of CD1 background (8 weeks old) were purchased from the Vital River Laboratory Animal Technology Co. Ltd. PR null mutant mice were kindly provided by Dr. Francesco DeMayo Lab [18]. The female mice were mated with the fertile or vasectomized males to induce pregnancy or pseudopregnancy (vaginal plug = day 1 of pregnancy), respectively. Artificial decidualization was induced by intraluminal oil perfusion (25 μl) on day 4 morning in pseudopregnant females. To test the ovarian hormonal influence on uterine Mcm2 expression, wildtype and PR −/− mice were ovariectomized and rested for 7 days, then were injected subcutaneously (s.c.) with oil, estradiol-17β (E 2, 100 ng/mouse), progesterone (P 4, 2 mg/mouse), or a combination of E 2 and P 4. In certain experiments, mice were injected s.c. with vehicle or RU486 (1 mg/kg) on day 3 of pregnancy (5:00 pm) and uteri were collected in the morning of day 4 (9:00 am). At least three mice were used for each treatment [19]. Mice were killed at indicated times, and uteri processed for immunostaining and mRNA analysis by quantitative RT-PCR.
Primary uterine stromal cell culture
Murine primary uterine endometrial stromal cells (mESCs) were isolated and cultured as previously described with some modifications [20]. The uteri of pseudopregnant mouse on day 4 were cut into small pieces (3–4 mm). Those tissue pieces were first digested in Hanks’ balanced salt solution (HBSS) containing 6 mg/ml dispase (Gibco) and 25 mg/ml trypsin (Sigma), and then incubated in HBSS containing 0.5 mg/ml collagenase (Gibco). The digested cells were passed through a 70-µm filter to obtain the stromal cells. Cells were plated at 5 × 105 cells per 60 mm dish or the corresponding numbers according to dish area, cultured with phenol red-free Dulbecco’s Modified Eagle’s Medium and Ham’s F-12 nutrient mixture (1:1, DMEM/F12, Gibco) containing 10 % charcoal-stripped fetal bovine serum (C-FBS, Biological Industries) and antibiotics. 2 h later, the medium was replaced with fresh medium and was cultured overnight. On next morning, the medium was replaced with DMEM/F12 containing 1 % C-FBS, E 2 (10 nM), and P 4 (1 µM) and antibiotic to induce decidualization. The experiment with CellTiter 96® AQueous One Solution Cell Proliferation Assay was performed following the protocol of the manual. Briefly, stromal cells were plated in 96-well plate and starved overnight, and then culture medium was replaced by DMEM/F12 containing 1 % C-FBS and antibiotic to recover cell proliferation. At 24, 48, and 96 h, reagent (MTS) was added to culture medium and the plate was incubated at incubator for 3 h, then the supernatant was pipetted to detect its absorbance at 490 nm.
Flow cytometry
Flow cytometric analysis was performed as previously described [20]. Cells were digested and harvested. Cell sediments were suspended in staining solution (PBS containing 30 mg/ml propidium iodide and 0.3 mg/ml DNase-free RNase A). Samples were incubated for 30 min at 37 °C in the dark. They were then returned to room temperature and transferred to flow cytometer for measuring. Maximum excitation of PI bound to DNA is at 536 nm, and emission is 617 nm. Emission is measured using the long-pass 600–610 nm filter. Three independent experiments with different samples were conducted.
Stealth RNA transfection
Stealth RNA corresponding to mouse Mcm2 (AUAAUUUACCACCAAACUCUCACGG) was designed and synthesized by Invitrogen™. The stealth RNA or control-siRNA was transfected into mESCs following the protocol of Lipofectamine™ RNAiMAX (Invitrogen) as previously described [21].
Western blotting
Protein extracts of cells and Western blotting were performed as previously described [22]. In brief, stromal cells were dissolved in RIPA lysis buffer containing proteinase inhibitor. After determining the protein concentration, protein lysates at 20 µg per lane were separated by SDS-PAGE, and then were transferred to PVDF membranes, blocked with 5 % skimmed milk. Antibodies to Mcm2 (1:1000, Santa Cruz), Cyclin B1 (1:1000, Abcam), cleaved-caspase 3 (1:1000 ABclonal), cleaved-PARP (1:5000, Bioworld), Cyclin D3 (1:1000, Cell Signaling Technology) were used, respectively. Beta-actin (Sigma, A2228) is used as a loading control in Western blot analysis. Bands on the membranes were detected using SuperSignal West Pico (Thermo Scientific) according to the manufacturer’s instructions. The intensity of bands was determined using Quantity One software, and the quantitative analyses of gray-scale value of each target protein vs that of individual β-actin were performed.
In situ hybridization
In situ hybridization with isotopes was performed as previously described [22]. Mouse-specific cRNA probe for Mcm2 was used for hybridization. Section hybridized with the sense probe served as a negative control.
Immunostaining
Immunohistochemistry was performed in 5 µm paraffin-embedded sections. Tissues were fixed at 4 % paraformaldehyde, dehydrated via graded ethanol solutions, and embedded in paraffin. For immunohistochemistry analysis, antibody specific to Mcm2 (1:100, Santa Cruz) was used. For immunofluorescence, Immunofluorescence cells on coverslips that were fixed by 4 % paraformaldehyde, after washing with phosphate buffer saline (PBS) and punching with Triton X-100-PBS, were blocked in 0.5 % bovine serum albumin and then incubated with primary antibodies Ki-67 (1:100, Santa Cruz), Mcm2 (1:100, Santa Cruz), Cyclin B1 (1:100, Abcam) overnight. On next day, cells were incubated with Cy3-labeled secondary antibodies and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). For Edu staining, the Cell-Light™ EdU assay kit (C10310, Ribobio) was used according to manufacturer’s instruction. The number of Ki-6 and Edu-positive cells was determined by counting at least 200 cells in each of five separate regions of the culture. For semi-quantitative histologic scoring (HSCORE), uteri from 5 mice at each group were used for paraffin slides and Mcm2 immunohistochemistry, and all these slides were used to conduct semi-quantitative histologic scoring (HSCORE) analysis as previously described [23].
RNA extraction and real-time polymerase chain reaction (RT-PCR)
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed as described [20]. Total RNA was isolated from uterine tissues or cells with TRIzol reagent following the manufacturer’s protocol, and 1–3 µg of RNA was used to synthesize cDNA. The expression levels of different genes were validated by real-time PCR analysis using SYBR green method in ABI 7500 according to manufacturer’s instructions (Applied Biosystems). All primers for real-time PCR are listed in Supplemental Table 1.
Alkaline phosphatase staining
Frozen uterine tissue slides were rinsed with PBS, fixed in 0.2 % glutaraldehyde for 10 min, then washed in PBS, and incubated with chromogenic substrates NBT/BCIP (Sigma) for alkaline phosphatase according to manufacturer’s instruction. The development of a purple color is indicative of alkaline phosphatase activity. The quantification of ALP activity was performed as previously described [24]. In detail, the decidualized stroma cells were lysis in 0.2 % Triton/PBS by repeated freeze and thaw in liquid nitrogen. The activity of ALP enzyme was determined with the ALP Yellow (pNPP) Liquid Substrate System (Sigma) in a 24-well plate. Absorbance at 405 nm was measured using a spectrophotometer and the protein concentration in cell lysis was determined by BCA assay.
Statistical analysis
All data are presented as mean ± SEM. Each experiment included at least three independent samples. Comparison between two groups was made by unpaired Student’s two-tailed t test, small sample size was analyzed by non-parametric equivalent test, and differences between time points for the various parameters studied were analyzed by one-way ANOVA test. P < 0.05 was considered to indicate a significant result.
Results
MCM2 is dynamically expressed in periimplantation uterine stromal cells
To investigate the underlying regulatory molecules governing stromal cell proliferation and differentiation, we analyzed gene expression profiles of uterine implantation sites and inter-implantation sites at day 5 and day 6 of pregnancy and found that the expression of MCM2–7 was highly up-regulated in the implantation sites in comparison with inter-implantation sites (Fig. S1). To reveal the function of MCMs in early pregnancy events, we selected and checked the uterine expression pattern of Mcm2 by in situ hybridization and immunohistochemistry analysis. As shown in Fig. 1a, Mcm2 mRNA was expressed in subepithelial stromal cells on day 1 and day 4 uteri prior to embryo attachment. With the onset of implantation on day 5, the expression expanded to the entire stromal bed. Accompanied by the progression of decidualization, Mcm2 was intensely expressed in the decidualizing cells on day 6 to day 8 at the implantation sites. Using immunohistochemistry staining, we found that the Mcm2 were mainly located in the nuclei of stromal cells and showed similar patterns with its RNA expression. Notably, Mcm2 was not only in the proliferating stromal cells but also intensely expressed in the decidualized stromal cells exhibiting large or binuclei, indicating it might be involved in both the stromal cell proliferation and differentiation process during periimplantation uteri (Fig. 1b).

MCM2 is dynamically expressed in periimplantation uterine stromal cells. a In situ hybridization showing the spatiotemporal expression of Mcm2 in WT uteri on days 1, 4, 5, 6, and 8 of pregnancy. b Immunohistochemistry staining of Mcm2 in WT uteri on days 5, 6, and 8 periimplantation. Arrow indicates the embryo and arrowhead indicates polyploidy decidual cells. Bl blastocyst, Le luminal epithelium, Pdz primary decidual zone, Sdz secondary decidual zone, S stroma. Scale bars indicate 20 μm
Expression of Mcm2 is associated with uterine decidualization independent of embryo
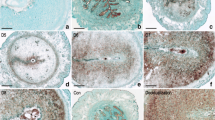
Under normal condition, the stimulus for decidualization is the implanting blastocyst. However, a similar process termed deciduoma can also be experimentally induced in the pseudopregnant or hormonally prepared rodent uterus by intraluminal infusion of oil [25]. To examine whether the dynamic expression of Mcm2 during periimplantation uterus is dependent on the living embryos or not, we analyzed the Mcm2 expression in the artificial uterine decidualization model induced by oil infusion. The alkaline phosphatase staining (ALP) was used as an indicator of stromal cell differentiation, and its staining was gradually increased in stromal cells surrounding the oil drops. The protein expression of MCM2 is expressed in stromal cells before differentiation (Fig. 2, oil 2 and 8 h). Notably, with artificially induced decidualization, Mcm2 is also expressed in decidualized cells with nuclear localization (Fig. 2). These results suggest that the decidual expression of Mcm2 was not dependent on the presence of embryos.

Expression of Mcm2 during artificially induced decidualization in vivo. a ALP staining was used to assess the differentiation of stromal cells at various time points after oil induced in day 4 pseudopregnant uterus. b Immunohistochemistry staining of Mcm2 at various time points after oil induced in day 4 pseudopregnant uterus. Arrows indicate polyploidy cells. Bars indicate 20 μm
PR induces MCM2 expression in mouse uterus during decidualization
Previous studies had demonstrated that the progesterone acts through its receptor PR is essential for decidualization [26–28]. To address the influence of progesterone on uterine Mcm2 expression, we employed ovariectomized mouse model injected with progesterone (2 mg) and collected tissues at different time points. Interestingly, we found that the stromal Mcm2 was progressively induced in progesterone alone treated uterus in a time-dependent manner (Fig. 3a). This facilitative activity of progesterone on stromal Mcm2 expression largely depended on nuclear PR receptors, since progesterone failed to induce Mcm2 expression in PR null mutant stromal cells (Fig. 3a, b). To further identify the PR effects on MCM2 expression, we examined the alterations in uterine Mcm2 in response to the PR antagonist RU486. Consistently, we found that the expression of Mcm2 was seriously blocked in the stromal cells, while its expression in the epithelium was slightly changed (Fig. 3c). This result reinforces the notion that MCM2 is a downstream player for progesterone-PR network during periimplantation. Collectively, this tightly regulated Mcm2 expression in stromal bed point towards its potential role in stromal-decidual transformation during periimplantation uterine development.

The expression of Mcm2 is increased by P 4 treatment in the mouse uterine stromal cells. a Immunohistochemistry staining of uteri sections collected from ovariectomized wildtype and PRKO mice after 0, 6, and 24 h of P 4 administration. b HSCORE analysis of stromal nuclear Mcm2 staining at various time points of after P 4 treatment in ovariectomized wildtype and PRKO mouse. Data shown represent the mean ± SEM, n = 5. *P < 0.05, **P < 0.01. c Pregnant mice were treated without (left) or with (right) RU486 on day 3 of gestation. The uteri were collected 12 h later, on day 4, and subjected to IHC using anti-Mcm2 antibody. Le luminal epithelium, Ge glandular epithelium, S stroma. Bars indicate 50 μm
MCM2 promotes proliferation of endometrial stromal cells cultured in vitro
To investigate the role of MCM2 signaling in uterine function, we established a mouse primary cell culture system in which undifferentiated stromal cells were isolated from day 4 pseudopregnant uteri. The identification of stromal cells was verified by vimentin staining, a type III intermediate filament protein that is expressed in mesenchymal cells (Fig. 4a) [29, 30]. Moreover, decidual prolactin-related protein (dPRP) expression, a marker of decidualization, was successfully induced in the cultured stromal cells in response to E 2 and P 4 (Fig. 4b). siRNA was then transfected into the primary stromal cells to silence Mcm2. Through RT-PCR and Western blot assay, we demonstrated that both mRNA and protein levels of Mcm2 could be effectively down-regulated upon 24 h after siRNA transfection, and the inhibition effect was maintained at 96 h (Fig. 4c). Both the immunohistochemistry and immunoblot analysis revealed a similar reduced Mcm2 protein expression following siRNA transfection (Figs. 4d, e, S2a, b). Previous studies had shown that the six of Mcm2–7 is essential for DNA licensing, and downregulation of any one MCM member could also reduce the levels of other MCM subunits, therefore, we examined other members of Mcm family by RT-PCR after Mcm2 siRNA transfection and found that the levels of Mcm2–7 were all reduced from 48 h after Mcm2 siRNA treatment (Fig. 4f). Taken together, these results suggested that the Mcm2 siRNA could efficiently knockdown the Mcm2 expression both in mRNA and protein level.

Mcm2 is effectively down-regulated using Mcm2 siRNA. a Stromal cells were fixed and subjected to immunofluorescence using anti-vimentin antibody as described previously. Bar indicates 20 μm. b qPCR analysis to monitor PRP mRNA expression in the stromal cells cultured up to 96 h. The relative fold induction of PRP mRNA expression at each time point compared with that of 24 h sample is shown. Data shown represent the mean ± SEM, n = 3. c qPCR analysis of Mcm2 mRNA using RNA collected from stromal cells transfected with control-siRNA (screamed siRNA) and Mcm2 siRNA. The values are normalized to the GAPDH expression level and indicated as the mean ± SEM, n = 3. **P < 0.01, ***P < 0.001. d Immunocytochemistry staining of Mcm2 protein in stromal cells at 48 h after control and siRNA transfection. Scale bar indicates 20 μm. e Western blot analysis of Mcm2 protein in stromal cells at 48, 96 h after control and siRNA transfection. f qPCR analysis of Mcm 3–7 mRNA using RNA collected from stromal cells transfected with control-siRNA (screamed siRNA) and Mcm2 siRNA. The values are normalized to the GAPDH expression level and indicated as the mean ± SEM, n = 3. *P < 0.05
MTS assay was then performed to determine the stromal cell proliferation status upon Mcm2 silencing. As shown in Fig. 5a, stromal cell proliferation activity in Mcm2 siRNA-treated group was significantly attenuated compared with the control one. Consistent with this observation, Ki67-positive cells were dramatically reduced after Mcm2 siRNA treatment (Fig. 5b, c). To clarify the cell cycle status upon Mcm2 inhibition during stromal cell proliferation, we performed flow cytometric analysis. Stromal cells were serum starved to synchronize the cell cycle at the G0/G1 stage, which then were released by cultured medium containing serum. As shown in Figs. 5d and e, there were no apparent differences in the 24 h after transfected with control or Mcm2 siRNA. However, a significantly decreased progression of siRNA-treated stromal cells was found in S-phase from 48 to 96 h post-Mcm2 siRNA treatment. These results indicated that the downregulation of Mcm2 derails the entry of stromal cells into the S-phase and thus the subsequent cell cycle progression. A time course analysis of EdU incorporation assay revealed massively reduced stromal cells in the S-phase during proliferation at 48 h post-transfected with Mcm2 siRNA (Figs. 5f, S3a). In line with this observation, the G2/M transition checkpoint indicator cyclin B1 was significantly reduced in siRNA-treated group (Figs. 5g, S3b). These findings clearly demonstrated that MCM is essential for normal stromal cell proliferation, whereas its inhibition strikingly inhibits the consecutive progression of cell cycle through the S-phase and entering the G2–M phase.

Silencing Mcm2 leads to stromal cell proliferation defects by S-phase blockage. a Measurements of cell proliferation by MTS assay after 24, 48, 72, and 96 h transfection. The values represent the mean ± SEM of six replicates from three independent experiments. *P < 0.05, **P < 0.01. b Immunofluorescence staining of Ki-67 on stromal cells at 48 h after treated with control and Mcm2 siRNA. Scale bars indicate 50 μm. c Ratio of positive Mcm2 staining cells per area in control and Mcm2 siRNA-treated stromal cells at 48 h. **P < 0.01. d FACS analysis of stromal cells treated with control or Mcm2 siRNA at 24, 48, 72, and 96 h. e Edu staining of stromal cells at 48 and 72 h after control or Mcm2 siRNA transfection. f Immunofluorescence of Cyclin B1 in stromal cells treated with control or Mcm2 siRNA at 48 and 72 h
Mcm2 is required for stromal cell differentiation
Since Mcm2 was also intensely expressed in polyploidy stromal cells in vivo (Figs. 1b, 2), we also explored the influence of Mcm2 silence on primary stromal cell decidualization in culture. In response to E 2 plus P 4 treatment, the stromal cells undergo differentiation in vitro. Through siRNA interference, we found that the expression of dPRP was significantly down-regulated from 48 to 96 h after Mcm2 silencing (Fig. 6a), suggesting that the decidualization process was impaired in Mcm2-silenced stromal cells. To further confirm this differentiate defect, we detected the alkaline phosphatase activity on control or Mcm2 siRNA-transfected stromal cells at different time points from 24 to 72 h. As shown in Fig. 6b, we observed that the ALP activity was reduced in MCM2 down-regulated stromal cells after 48 h differentiation. Consistently, the expression of Cyclin D3 was significantly lower in Mcm2-silenced cells (Figs. 6c, S4). We next investigated the functional consequences of this blockade of Mcm2 expression during stromal differentiation. As shown in Fig. 6d, siRNA-mediated downregulation of Mcm2 in the stromal cells led to a significant reduction in the expression of differentiation markers such as Wnt4, C/EBPβ, and DEDD. These results indicated that Mcm2 expression is critical for successful progression of the stromal differentiation program during decidualization.

Mcm2 is required for stromal cell differentiation. Stromal cells transfected with control-siRNA or Mcm2 siRNA and 24 h later undergo differentiation in the E 2 and P 4 treatment. a qPCR analysis to monitor PRP mRNA expression in the differentiated stromal cells transfected control-siRNA or Mcm2 siRNA. The relative fold induction of PRP mRNA expression at each time point compared with that of 24 h sample is shown. Data shown represent the mean ± SEM, n = 3. b Alkaline phosphatase activity. For each condition in panel B, alkaline phosphatase is measured as signal at 405 nm in assays of replicate cultures. Mcm2 siRNA significantly reduced alkaline phosphatase (n = 3, *P < 0.05; **P < 0.01). c Western blots analysis of Mcm2, Cyclin D3 with β-actin as a loading control in differentiated stromal cells transfected with control or Mcm2 siRNA. e Relative expression level of decidualization markers including Wnt4, Cyclin D3, DEDD, C/EBPβ in stromal cells undergo differentiation 48 h after transfected with control or Mcm2 siRNA. Data shown represent the mean ± SEM, n = 3. *P < 0.05, **P < 0.01
Discussion
The uterus provides a unique and dynamic physiological model in which cellular proliferation, differentiation with polyploidization, and apoptosis occur in a temporal and cell-specific manner during pregnancy [31]. Although there is evidence to suggest that cell cycle regulatory molecules play potential roles in the uterus during steroid hormone stimulation and reproductive cycle, for example, the estrogen through the stromal ER receptor to induce the proliferation of uterine epithelium by the paracrine factors from the stroma compartment [32–34], which is countered by the P4 through both the epithelial and stromal PR receptor [35, 36]. However, very limited information is available regarding the onset of stromal cell proliferation and decidualization. In the present study, we found that the Mcm2 required for DNA replication licensing was dynamically expressed in stromal cells and decidual cells of periimplantation uterus. Further studies revealed that the P 4–PR signaling could effectively induce the expression of MCM2 in uterine stromal cells. Using a primary stromal cell culture model and siRNA interference strategy, we demonstrated that Mcm2 was required for normal DNA synthesis in the G1–S-phase, and its silence impaired the differentiation of stromal cells in response to E 2 and P 4. These findings identified MCM as a novel mediator for progesterone to control stromal cell proliferation and differentiation.
Among the MCM proteins, MCM2 has been studied in a wide range of human organs and its overexpression has been identified in various types of tumors as well as tumor-like lesions of the oral mucosa, breast, ovary, kidney, and soft tissue [37–40]. In addition, MCM2 has a typical “licensing” behavior since it binds to chromatins during G1 phase, dissociates from chromatins once S-phase has started, and again binds to chromatins at the end of mitosis [41]. Therefore, we concentrated on the expression and function of MCM2 in stromal cell proliferation and differentiation. Indeed, it was assumed that the stability of the Mcm2–7 complex is compromised to different extents by the downregulation of any of its individual components [42]. In our study, we also found a global reduction in Mcm2–7 levels from 48 h after treatment with siRNA molecules targeting a single subunit Mcm2, which further reinforces the notion that the six Mcm2–7 genes are essential for DNA licensing, and the knockout of any of them in mammalian cells is expected to be lethal as well [42]. Interestingly, the reduction of MCM levels did not significantly affect the progression of cell replication and cell proliferation within 48 h (Fig. 4). However, cells proliferating under limited licensing conditions progressively accumulated DNA damage during S-phase and displayed chromosome instability. These results fit into the notion that there are excess chromatin-bound Mcm2–7 complexes over the minimum amount compatible with cell survival [42, 43].
Progesterone induces proliferation of the stroma through species–specific mechanisms. In rhesus monkeys treated for 20 days with RU 486, the cell cycle was blocked at the boundary of G2–M phase, while the immunostaining of markers during G1–S-phase including Ki-67, proliferating cell nuclear antigen (PCNA), and cyclin B1 was still maintained [44]. In primate uterus, progestins generally suppress endometrial proliferation during G0–G1 phases of the cell cycle [45]. We found herein that progesterone could induce MCM2 expression in the stromal cells in a time-dependent manner, while this induced expression could not occur in PR-knockout ovariectomized mouse. Using RU486, we further demonstrated that Mcm2 was a target of P 4–PR signaling since RU486 could severely block the expression of Mcm2. These results pointing towards Mcm family as a novel factor in mediating the P 4–PR signaling for stromal cell differentiation. However, it seems that PR could not directly regulate Mcm2 at the transcription level since there was no PR enrichment in the Mcm2 promoter in the uterus responsive to P4 [46]. Other regulatory pathways participating in the comprehensive signaling cascade of progesterone in uterine stromal cells need future investigations.
It was recently mentioned that several members of MCM family including Mcm2–5 and Mcm7 were up-regulated in polyploidy decidual cells, suggesting an important role of Mcm in the decidualization [47]. In the present study, we found an intense expression of Mcm2 in polyploidy decidual cells, and knockdown of the expression of Mcm2 under differentiation culture condition leads to significant defects manifested by aberrant expression of several decidual markers including Cyclin D3, Wnt4, and C/EBPβ, which demonstrated that the MCMs were involved in the decidualization process in mice. Previous studies showed that labeling of stromal cells in vivo results in the appearance of 3H thymidine in decidual cells conclusively demonstrating that they are the progenitors of decidual cells [48]. In this regard, it is possible to assume that the onset of decidualization comprise caused by Mcm2 silence may originate from S-phase entry defects in proliferative progenitor cells. However, the underlying mechanisms of how Mcm2 participates in the regulation of polyploidy cell formation still warrants further investigation.
Nonetheless, we herein provide novel evidence showing that the replicative license of MCMs is one of the early gene targets for P 4–PR signal controlled stromal cell proliferation and differentiation. Since impaired progesterone signaling is associated with aberrant endometrial cell growth, our study may raise the possibility for targeting the MCM for diagnosis and treatment of endometrial-related diseases such as endometrial hypotrophy or endometrial carcinoma in women.
References
H. Wang, S.K. Dey, Roadmap to embryo implantation: clues from mouse models. Nat. Rev. Genet. 7, 185–199 (2006)
S.K. Dey, Focus on implantation. Reproduction 128, 655–656 (2004)
M.J. Large, F.J. DeMayo, The regulation of embryo implantation and endometrial decidualization by progesterone receptor signaling. Mol. Cell Endocrinol. 358, 155–165 (2012)
J. Cha, X. Sun, S.K. Dey, Mechanisms of implantation: strategies for successful pregnancy. Nat. Med. 18, 1754–1767 (2012)
O.M. Conneely et al., Reproductive functions of progesterone receptors. Recent Prog. Horm. Res. 57, 339–355 (2002)
S.K. Das, Cell cycle regulatory control for uterine stromal cell decidualization in implantation. Reproduction 137, 889–899 (2009)
S. Zhang et al., Deciphering the molecular basis of uterine receptivity. Mol. Reprod. Dev. 80, 8–21 (2013)
S.K. Das et al., Cyclin D3 in the mouse uterus is associated with the decidualization process during early pregnancy. J. Mol. Endocrinol. 22, 91–101 (1999)
J.M. Sroga et al., Overexpression of cyclin D3 improves decidualization defects in Hoxa-10(−/−) mice. Endocrinology 153, 5575–5586 (2012)
P. Bilinski, D. Roopenian, A. Gossler, Maternal IL-11Ralpha function is required for normal decidua and fetoplacental development in mice. Genes Dev. 12, 2234–2243 (1998)
M. Mori et al., Death effector domain-containing protein (DEDD) is required for uterine decidualization during early pregnancy in mice. J. Clin. Investig. 121, 318–327 (2011)
J.J. Blow, A. Dutta, Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell Biol. 6, 476–486 (2005)
M. Moritani, Y. Ishimi, Inhibition of DNA binding of MCM2-7 complex by phosphorylation with cyclin-dependent kinases. J. Biochem. 154, 363–372 (2013)
L.C. Chuang et al., Phosphorylation of Mcm2 by Cdc7 promotes pre-replication complex assembly during cell-cycle re-entry. Mol. Cell 35, 206–216 (2009)
Q. Wei et al., Phosphorylation of minichromosome maintenance protein 7 (MCM7) by cyclin/cyclin-dependent kinase affects its function in cell cycle regulation. J. Biol. Chem. 288, 19715–19725 (2013)
H. Pan, Y. Deng, J.W. Pollard, Progesterone blocks estrogen-induced DNA synthesis through the inhibition of replication licensing. Proc. Natl. Acad. Sci. USA 103, 14021–14026 (2006)
S. Ray, J.W. Pollard, KLF15 negatively regulates estrogen-induced epithelial cell proliferation by inhibition of DNA replication licensing. Proc. Natl. Acad. Sci. USA 109, E1334–E1343 (2012)
J.P. Lydon et al., Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 9, 2266–2278 (1995)
G.A. Surveyor et al., Expression and steroid hormonal control of Muc-1 in the mouse uterus. Endocrinology 136, 3639–3647 (1995)
Q. Wang et al., Wnt6 is essential for stromal cell proliferation during decidualization in mice. Biol. Reprod. 88, 5 (2013)
J. Lu et al., A positive feedback loop involving Gcm1 and Fzd5 directs chorionic branching morphogenesis in the placenta. PLoS Biol. 11, e1001536 (2013)
S. Zhang et al., Uterine Rbpj is required for embryonic-uterine orientation and decidual remodeling via Notch pathway-independent and -dependent mechanisms. Cell Res. 24, 925–942 (2014)
B.J. Plante et al., Cyclic regulation of transcription factor C/EBP beta in human endometrium. Reprod. Biol. Endocrinol. 7, 15 (2009)
Q. Li et al., Bone morphogenetic protein 2 functions via a conserved signaling pathway involving Wnt4 to regulate uterine decidualization in the mouse and the human. J. Biol. Chem. 282, 31725–31732 (2007)
L. Loeb, The production of Deciduomata. JAMA 50, 1897 (1908)
B.C. Paria et al., Uterine decidual response occurs in estrogen receptor-alpha-deficient mice. Endocrinology 140, 2704–2710 (1999)
A. Das et al., De novo synthesis of estrogen in pregnant uterus is critical for stromal decidualization and angiogenesis. Proc. Natl. Acad. Sci. USA 106, 12542–12547 (2009)
S.W. Curtis et al., Disruption of estrogen signaling does not prevent progesterone action in the estrogen receptor alpha knockout mouse uterus. Proc. Natl. Acad. Sci. USA 96, 3646–3651 (1999)
S.A. Mani et al., The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008)
S.R. Glasser, J. Julian, Intermediate filament protein as a marker of uterine stromal cell decidualization. Biol. Reprod. 35, 463–474 (1986)
J. Tan et al., Evidence for coordinated interaction of cyclin D3 with p21 and cdk6 in directing the development of uterine stromal cell decidualization and polyploidy during implantation. Mech. Dev. 111, 99–113 (2002)
P.S. Cooke et al., Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc. Natl. Acad. Sci. USA 94, 6535–6540 (1997)
L. Zhu, J.W. Pollard, Estradiol-17beta regulates mouse uterine epithelial cell proliferation through insulin-like growth factor 1 signaling. Proc. Natl. Acad. Sci. USA 104, 15847–15851 (2007)
W. Winuthayanon et al., Uterine epithelial estrogen receptor alpha is dispensable for proliferation but essential for complete biological and biochemical responses. Proc. Natl. Acad. Sci. USA 107, 19272–19277 (2010)
T. Kurita et al., Stromal progesterone receptors mediate the inhibitory effects of progesterone on estrogen-induced uterine epithelial cell deoxyribonucleic acid synthesis. Endocrinology 139, 4708–4713 (1998)
H.L. Franco et al., Epithelial progesterone receptor exhibits pleiotropic roles in uterine development and function. FASEB J. 26, 1218–1227 (2012)
S. Suzuki et al., Overexpression of MCM2 in myelodysplastic syndromes: association with bone marrow cell apoptosis and peripheral cytopenia. Exp. Mol. Pathol. 92, 160–166 (2012)
K.M. Barton, E.M. Levine, Expression patterns and cell cycle profiles of PCNA, MCM6, cyclin D1, cyclin A2, cyclin B1, and phosphorylated histone H3 in the developing mouse retina. Dev. Dyn. 237, 672–682 (2008)
S.S. Li et al., Replicative MCM7 protein as a proliferation marker in endometrial carcinoma: a tissue microarray and clinicopathological analysis. Histopathology 46, 307–313 (2005)
J.M. Bailis, S.L. Forsburg, MCM proteins: DNA damage, mutagenesis and repair. Curr. Opin. Genet. Dev. 14, 17–21 (2004)
K. Kato et al., Expression of replication-licensing factors MCM2 and MCM3 in normal, hyperplastic, and carcinomatous endometrium: correlation with expression of Ki-67 and estrogen and progesterone receptors. Int. J. Gynecol. Pathol. 22, 334–340 (2003)
A. Ibarra, E. Schwob, J. Mendez, Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 105, 8956–8961 (2008)
X.Q. Ge, D.A. Jackson, J.J. Blow, Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 21, 3331–3341 (2007)
O.D. Slayden et al., Chronic treatment of cycling rhesus monkeys with low doses of the antiprogestin ZK 137 316: morphometric assessment of the uterus and oviduct. Hum. Reprod. 13, 269–277 (1998)
O. Heikinheimo et al., Endometrial effects of RU486 in primates–antiproliferative action despite signs of estrogen action and increased cyclin-B expression. J. Steroid Biochem. Mol. Biol. 59, 179–190 (1996)
C.A. Rubel et al., Research resource: genome-wide profiling of progesterone receptor binding in the mouse uterus. Mol. Endocrinol. 26, 1428–1442 (2012)
J.M. Sroga, X. Ma, S.K. Das, Developmental regulation of decidual cell polyploidy at the site of implantation. Front. Biosci. (Sch. Ed.). 4, 1475–1486 (2012)
D.D. Carson, Embryo implantation: molecular, cellular and clinical aspects (Springer Science & Business Media, New York, 2013)
Acknowledgments
This work was supported in parts by the National Basic Research Program of China (2011CB944400 to H.W.) and the National Natural Science Foundation (81130009 and 81330017 to H.W. and to 31471106 to S.Z.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no conflict of interests.
Additional information
Shuangbo Kong and Xue Han have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kong, S., Han, X., Cui, T. et al. MCM2 mediates progesterone-induced endometrial stromal cell proliferation and differentiation in mice. Endocrine 53, 595–606 (2016). https://doi.org/10.1007/s12020-016-0894-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-016-0894-9