
Abstract
The newly identified estrogen receptor, G protein-coupled receptor 30 (GPR30), is prevalent in the brain and has been shown to provide significant neuroprotection. Recent studies have linked ferroptosis, a newly characterized form of programmed cell death, closely with cerebral ischemia–reperfusion injury (CIRI), highlighting it as a major contributing factor. Consequently, our research aimed to explore the potential of GPR30 targeting in controlling neuronal ferroptosis and lessening CIRI impacts. Results indicated that GPR30 activation not only improved neurological outcomes and decreased infarct size in a mouse model but also lessened iron accumulation and malondialdehyde formation post-middle cerebral artery occlusion (MCAO). This protective effect extended to increased levels of Nrf2 and GPX4 proteins. Similar protective results were replicated in PC12 cells subjected to Oxygen Glucose Deprivation and Reoxygenation (OGD/R) using the GPR30-specific agonist G1. Importantly, inhibition of Nrf2 with ML385 curtailed the neuroprotective effects of GPR30 activation, suggesting that GPR30 mitigates CIRI primarily through inhibition of neuronal ferroptosis via upregulation of Nrf2 and GPX4.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic stroke remains a significant health challenge, being the major cause of disability and mortality, representing 60% to 70% of all stroke cases (Wu et al., 2019). The optimal intervention for ischemic stroke involves swiftly re-establishing blood flow to the impacted brain tissue (Zhang et al., 2024). However, this reperfusion can paradoxically lead to further brain damage through mechanisms like oxidative stress, cerebral edema, calcium overload, inflammation, and various forms of programmed cell death, all of which exacerbate ischemia–reperfusion injury (Yang et al., 2019).
Recent discoveries have highlighted ferroptosis, a specific type of cell death, as a critical factor in ischemia–reperfusion injuries. Distinguished by iron accumulation, lipid peroxide buildup, and heightened oxidative stress, ferroptosis has been implicated in the pathophysiology of these injuries (Jiang et al., 2021). Experimental studies using middle cerebral artery occlusion (MCAO) models in mice have demonstrated a rise in lipid peroxides and a drop in glutathione levels, signifying the activation of ferroptosis following reperfusion (Chen et al., 2021). The application of Ferrostatin-1, a known ferroptosis inhibitor, has shown potential in alleviating these injuries, indicating that controlling ferroptosis could be pivotal in managing ischemic stroke outcomes.
The excessive buildup of reactive oxygen species (ROS), lipid peroxides, and malondialdehyde (MDA) contributes significantly to neuronal cell death (Li et al., 2023). Strategies targeting ferroptosis have shown potential in reducing the adverse effects associated with cerebral ischemia–reperfusion injury (CIRI) (Xu et al., 2022; Zhang et al., 2021), highlighting the importance of ferroptosis alongside apoptosis and oxidative stress in clinical settings (Han et al., 2022; Liu et al., 2021; Qz et al., 2017). G protein-coupled estrogen receptors (GPER1/GPR30), novel estrogen receptors located in the cell membrane, play pivotal roles across various systems, including neurological, reproductive, immune, endocrine, and cardiovascular through rapid estrogen signaling (Olde & Leeb-Lundberg, 2009).
There is evidence suggesting that GPR30 offers neuroprotection in cerebral ischemia–reperfusion scenarios, and the GPR30-specific agonist G1 could potentially bolster neuronal survival and diminish apoptosis (Hadjimarkou & Vasudevan, 2018; Liu et al., 2012; Roque et al., 2019; Wang et al., 2019). Despite the known benefits of GPR30 activation, its role in mitigating ferroptosis during CIRI remains underexplored. This study will delve into the interaction between GPR30 and ferroptosis through in vivo experiments using normal and GPR30-knockout mice and in vitro tests on PC12 neural cells. Utilizing a mouse model of MCAO and an oxygen–glucose deprivation/reperfusion (OGD/R) model in PC12 cells, we aim to analyze the effects of GPR30 targeting on various pathological and molecular alterations in the brain, such as iron levels, ROS, GSH, MDA, SOD, and cell viability.
Further, this research will examine the modulation of Nrf2/GPX4 protein expression, which may elucidate the mechanisms by which GPR30 targeting could influence ferroptosis and improve outcomes in CIRI.
Materials and Methods
Reagents and Chemicals
The G1 compound (HPLC purity ≥ 98.8%) (T15364) and ML385 (S8790) were sourced from Topscience Technology, Shanghai, China. Dimethyl sulfoxide (DMSO) was procured from Sigma-Aldrich, St. Louis, MO, USA. We utilized various primary antibodies for both western blot (WB) and immunofluorescence (IF) analyses, including Nrf2 (ab62352, 1:1000 for WB), GPX4 (ab125066, Abcam, 1:5000 for WB), GPR30 (ab260033, Abcam, 1:1000 for WB), xCT (ab175186, Abcam, 1:1000 for WB), and Actin (ab8227, Abcam, 1:1000 for WB). Additionally, several assay kits were used, such as the GSH and GSSG Assay Kit (S0053), Cell Counting Kit-8 (C0037), Mitochondrial Membrane Potential Assay Kit with JC-1 (C2006), Lipid Peroxidation MDA Assay Kit (S0131S), and Superoxide Dismutase (SOD) Kit (S0086), all of which were obtained from Beyotime, Shanghai, China.
Cell Culture
PC12 cells were cultured in DMEM/High-Glucose medium enriched with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Subculturing was performed when the cells reached 90% confluence, and experiments were conducted on cells that had undergone three passages. The cell culture density was maintained between 60 and 80% for subsequent experiments.
Cell Viability Assay
Cell viability was assessed using the EnoGeneCellTM Counting Kit-8 (CCK8) (E1CK-000208-5, EnoGene, Nanjing, China). Initially, PC12 cells were plated at a density of 8 × 103 per well in 96-well plates and incubated for 24 h. Subsequent to this period, different treatment groups were established. After a further 24 h of growth under the set conditions, CCK8 reagent was administered as specified by the kit’s protocol. Cells were then incubated at 37 °C for an hour. The absorbance was measured at 450 nm using a fluorescent microplate reader to quantify viability, with results normalized to the control and expressed as a percentage.
Oxygen–Glucose Deprivation/Reoxygenation (OGD/R)
The OGD/R procedure was executed to simulate in vitro CIRI (Duan et al., 2020). PC12 cells were cultured in six-well plates at a concentration of 1 × 105 cells per ml. For the oxygen–glucose deprivation phase, cells were exposed to a glucose-free DMEM and maintained in an anoxic condition (1% O2, 5% CO2, and 94% N2) at 37 °C for 1.5 h. Following this deprivation phase, glucose-free DMEM was replaced with fresh medium, and cells were returned to normoxic conditions (95% air, 5% CO2) for 24 h before harvesting for further analysis.
Animals
C57BL/6J mice were sourced from the Experimental Animal Center of the Fourth Military Medical University, under license number SCXK (Shaanxi) 2021-001. The animals were kept in a controlled environment with temperatures between 21 and 25 °C and a 12-h light/dark cycle, with ad libitum access to food and water during the dark phase. The use of animals in this study was approved by the Experimental Animal Welfare and Ethics Committee of the Fourth Military Medical University, approval number 20201782. Δ3102 GPR30 KO mice were obtained from Beijing Biocytogen Co., Ltd. Both standard GPR30 and Δ3102 GPR30 KO mice were grouped into three categories: sham, MCAO, and MCAO + G1. The administration of G1 followed a method optimized from previous studies (Liu et al., 2012), with the compound dissolved in olive oil at a concentration of 0.2 μg/μL. It was administered subcutaneously in the neck region at 48 and 24 h before reperfusion, and at the time of reperfusion and 24 h afterward, totaling four injections. In contrast, the MCAO control group received 100 μL of olive oil via subcutaneous injection in the neck.
MCAO
To explore the role of GPR30 in vivo, we employed a middle cerebral artery occlusion model in mice. Mice were anesthetized with 2% sodium pentobarbital (0.2 mL/25 mg, obtained from Sigma, St. Louis, MO, USA). A midline incision was made to expose the left common carotid artery (CCA). Ischemia was induced by inserting a 15-mm nylon filament into the external carotid artery to occlude the middle cerebral artery. After maintaining ischemia for 1.5 h, the filament was removed to allow for reperfusion over the next 22.5 h. Blood flow changes were monitored using a laser Doppler flowmeter (Moor Instruments, Devon, UK), with ischemic conditions confirmed when blood flow dropped to approximately 20% of baseline and reperfusion levels reaching 70–80% of baseline values.
Assessment of Neurological Impairment
Neurological deficits were assessed 24-h post-reperfusion using an established scoring system (Li et al., 2000). This scale ranges from 0 (no impairment) to 14 (severe impairment), with scores 1–4 indicating mild impairment, 5–9 suggesting moderate impairment, and 10–14 denoting severe neurological damage.
Infarct Volume Measurement
Brain sections from mice were stained using 2,3,5-triphenyl tetrazolium chloride (TTC) to delineate infarcted tissue. Initially, the brains were sectioned into five slices and then immersed in a 1% solution of TTC, followed by incubation in a darkened oven at 37 °C for 20 min. After incubation, these sections were fixed in 4% paraformaldehyde overnight. Infarcted regions appeared white, contrasting with the red of viable tissue. The area of infarction was quantified using ImageJ software for analysis.
Iron Content Analysis
For in vitro assessment of Fe2+ levels, PC12 cells were plated at a density of 1.2 × 106 cells per well in a six-well plate and allowed to adhere for 24 h. Depending on the experimental conditions, cells were then stained with a 1-μM FerroOrange solution for 30 min in an incubator for live cell imaging. Absorbance intensities were analyzed using ImageJ software. For in vivo analysis, the iron content in mouse serum was determined using an iron assay kit (A039-1-1, Nanjing Jiancheng Bioengineering Institute), with absorbance read at 520 nm.
Detection of ROS
We utilized an ROS Assay Kit for detecting ROS in PC12 cells, employing the DCFH-DA fluorescent probe (E004-1-1, Nanjing Jiancheng Biotechnology, Jiangsu, China). The cells were plated at 1.2 × 106 cells per well in a 6-well plate and incubated for 24 h. The next day, the cells were treated according to their respective experimental groups and incubated for an additional 24 h. After incubation, cells were exposed to the ROS detection probe for 20 min at 37 °C, washed thrice with serum-free medium to remove excess probe, and then treated with an ROS-positive control for 20–30 min to elevate ROS levels significantly. Flow cytometry was then utilized to quantify ROS levels in the cells.
Measurement of Malondialdehyde (MDA) Lipid Peroxidation
To assess lipid peroxidation, we employed an MDA Assay Kit that measures MDA interaction with thiobarbituric acid (TBA) to form a red product, indicative of lipid oxidation levels. PC12 cells were seeded at a density of 1.2 × 106 cells per well in a 6-well plate and cultured for 24 h before being assigned to various treatment groups. After a further 24-h incubation, cells were lysed using 0.1 mL of Beyotime’s Western and IP cell lysis buffer (P0013) per 1.0 × 106 cells. The supernatants were then collected by centrifugation at 10,000–12,000×g for 10 min, and lipid peroxidation was quantified as per the kit’s protocol.
Measurement of Glutathione (GSH/GSSG) Level
The levels of GSH and GSSG were quantified using a GSH and GSSG Assay Kit (Beyotime, China). PC12 cells were plated at a density of 1.2 × 106 cells per well in 6-well plates and allowed to grow for 24 h. Following this, cells underwent various treatments specific to their group assignments. After an additional 24-h incubation, cells were collected, homogenized, and analyzed using the procedures outlined in the kit’s manual.
Measurement of Mitochondrial Membrane Potential
The mitochondrial membrane potential was evaluated using the JC-1 Mitochondrial Membrane Potential Assay Kit (C2006, Beyotime, China), which utilizes the fluorescent probe JC-1 to detect changes in the mitochondrial membrane potential. For this assessment, PC12 cells were again seeded at a density of 1.2 × 106 cells per well and incubated for 24 h. Post-incubation, cells were treated based on their experimental groupings and then processed according to the instructions provided with the JC-1 assay kit.
Western Blot Analysis
Western blot assays were implemented following established protocols (Duan et al., 2020). Protein samples were prepared using a lysis buffer fortified with protease and phosphatase inhibitors. Protein concentrations were determined using a BCA protein assay kit. The proteins were then separated by SDS-PAGE and transferred to PVDF membranes. The membranes were blocked with 5% non-fat milk for one hour at room temperature and then incubated overnight at 4 °C with primary antibodies. After washing with TBST, secondary antibodies were applied, and proteins were visualized using an ECL chemiluminescence system (Seyotin, China). ImageJ software was used for signal quantification.
RNA Interference
GPR30-targeted shRNA was transfected into PC12 cells for 72 h to knock down gene expression, followed by Western blot analysis to confirm transfection efficacy using a GPR30-specific antibody (1:1000 dilution). The shRNA that showed the highest efficiency was subsequently used for further experiments. The sequence used for GPR30 shRNA was 5ʹ-CATGTACAGCAGCGTCTTC-3ʹ.
Statistical Analysis
Various statistical methods were employed to evaluate the significance of the experimental data. Differences in cell viability among treatment groups were analyzed using means, standard errors, analysis of variance (ANOVA), and Tukey’s HSD test for multiple comparisons. Neural functional damage and infarct volumes were assessed using non-parametric tests like the Mann–Whitney U test. For assays measuring iron content, ROS, MDA levels, and GSH/GSSG ratios, as well as mitochondrial membrane potential, t tests or ANOVAs were utilized for group comparisons. Protein expression variations identified in Western blots were also analyzed using t tests or ANOVAs.
Results
Targeting GPR30 Alleviates CIRI and Inhibits Ferroptosis in Normal Mice
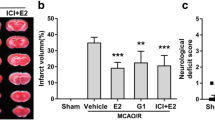
To investigate the potential therapeutic effect of targeting GPR30 in reducing damage caused by cerebral ischemia and reperfusion, we first established an MCAO model in C57BL/6J mice. Use olive oil to dissolve G1 to a concentration of 0.2 μg/μL. Subcutaneous injection into the neck 48 h and 24 h before reperfusion, during reperfusion and 24 h after reperfusion, 25 μL each time (Fig. 1a). We used a laser speckle Doppler imaging system to evaluate cerebral blood flow in experimental mice to verify whether the MCAO model was successful. The results showed that blood flow decreased by approximately 36% after MCAO. After reperfusion, it returned to 83% that indicates that the model is successfully established (Fig. 1b). The extent of neurological damage was evaluated by analyzing TTC staining images and assessing the cerebral infarct size. Cerebral infarction area and neurological score were used as brain injury evaluation indicators, and cerebral infarction volume was calculated using triphenyltetrazolium chloride staining. The model group showed evident cerebral infarction and neurological dysfunction after MCAO, whereas the administration of the GPR30-specific agonist G1 significantly mitigated these impairments (Fig. 1c, d).

GPR30-specific agonist G1 reduces damage caused by cerebral ischemia and reperfusion. a Experimental design schematic for mice, b laser speckle Doppler results indicate blocked cerebral blood flow, with the left side showing the infarct area and the right side showing the non-surgical area, c TTC staining reveals infarct volume, n = 5, d neurological score, n = 5. e–h Concentrations of Fe2+, MDA, GSH/GSSG ratio, and SOD activity in fresh brain tissue from the WT con, WT MCAO, and WT G1 + MCAO groups, n = 5 in each group. ***p ≤ 0.001. I: GPR30, Nrf2, GPX4, and xCT protein expression levels in non-infarcted tissue from the CIRI and sham operation groups. n = 5, *p ≤ 0.05
In order to further explore the potential inhibitory effect of G1 on ferroptosis, different kits were used to detect relevant indicators of ferroptosis in the hippocampus tissue of mice in different groups. G1 treatment resulted in decreased Fe2 + and MDA levels and increased GSH and SOD levels in hippocampal tissue. (Fig. 1e, f, g, h). Additionally, G1 treatment increased the protein expression of GPR30, nuclear factor erythroid 2-related factor 2 (Nrf2), and glutathione peroxidase 4 (GPX4) (Fig. 1i).
The Protective Effect of GPR30 Agonist G1 on CIRI and the Inhibitory Effect on Ferroptosis are Lost in GPR30 Full Knockout Mice
We investigated the impact of targeting GPR30 on ferroptosis by examining the effects of G1 in GPR30-knockout (KO) mice on the MCAO model. The administration method of G1 was the same as that of the normal group of mice. Compared to the MCAO group, no significant improvement was noted in the neurological dysfunction and cerebral infarct size of GPR30 KO mice treated with G1 (Fig. 2a, b).

GPR30 KO mice exhibit larger infarct area and iron metabolism damage than wild-type mice after MCAO. a TTC staining and infarct volume percentage. The red area represents intact tissue, and the white area represents the infarcted tissue, n = 5. b Assessment of neurological deficits in GPR30 KO mice, n = 5. c–f Levels of Fe2+, MDA, GSH/GSSG ratio, and SOD activity in fresh brain tissue from the GPR30 KO con, GPR30 KO MCAO, and GPR30 KO G1 + MCAO groups, n = 5 in each group, ***p ≤ 0.001. g Nrf2, GPX4, and xCT protein expression levels in infarcted tissue of GPR30 KO mice after CIRI and non-infarcted tissue of the same area in the sham operation group, n = 5, *p ≤ 0.05
Furthermore, use different kits for Fe2+, MDA, GSH, and SOD. The results showed that the levels of Fe2+, MDA, GSH, and SOD in GPR30 KO mice given G1 did not show corresponding improvements compared to the MCAO group (Fig. 2c–f). Expression levels of essential proteins associated with Nrf2 and GPX4 were also found to be suppressed (Fig. 2g). These findings indicate that the protective effect of G1 on CIRI is mostly diminished in GPR30 KO mice.
Identification of GPR30 as a Potential Regulator of Ferroptosis in CIRI
We performed bioinformatics correlation analysis to investigate the potential interaction between GPR30 and ferroptosis. Our findings revealed a high expression of GPR30 in CIRI (Fig. 3a). Furthermore, differential gene expression and functional enrichment analyses indicated a close association with ferroptosis (Fig. 3b, c). GSEA enrichment analysis demonstrated a clear correlation between GPR30, CIRI, and ferroptosis (Fig. 3d). Additionally, we analyzed the critical pathways associated with GPR30 (Fig. 3e). Particularly noteworthy, we observed a positive correlation between GPR30, Nrf2, and GPX4 (Fig. 3f–h).

The relationship between GPR30 and ferroptosis in cerebral ischemia–reperfusion injury. a Differential gene expression analysis reveals high expression of GPR30 in cerebral ischemia–reperfusion injury. b and c Functional enrichment analysis identifies the association of GPR30 with programmed death. d GSEA enrichment analysis indicates a positive correlation between GPR30, cerebral ischemia–reperfusion injury, and ferroptosis. e GSEA analysis reveals critical pathways associated with GPR30. f–h Pearson correlation analysis demonstrates the correlation between GPR30, Nrf2, and GPX4
GPR30 Agonist G1 Protects PC12 Cells from OGD/R-induced Damage and Reduces Fe2+ Levels
To determine the relevant conditions of the OGD/R experiment, we conducted CCK8 assays to measure the degree of PC12 cell damage at different time points. The results indicated a significant decrease in cell viability after 1.5 h of hypoxia compared to the control group (Fig. 4a). Therefore, for subsequent in vitro experiments, a hypoxia time of 1.5 h was utilized. Western blot experiments further confirmed that the OGD/R model significantly decreased Nrf2 and GPX4 expression (Fig. 4b). In addition, CCK8 experiments demonstrated that various concentrations of the GPR30 agonist G1 (0.001, 0.01, and 0.1 μM) and E2 (0.001, 0.01, 0.1, and 1 μM) had no detrimental effects on PC12 cells (Fig. 4c, d).

G1 protects PC12 cells against damage induced by OGD/R. a CCK8 assay to determine PC12 cell survival rate at different OGD/R time points, n = 6, **p ≤ 0.01. b Western blot analysis of Nrf2, GPX4, and xCT protein expression levels in normal and OGD/R groups, n = 3, **p ≤ 0.01, ***p ≤ 0.001. c, d CCK8 assay to evaluate the effect of G1 and E2 concentrations on PC12 cell viability, n = 6, *p ≤ 0.05, **p ≤ 0.01. e, f CCK8 assay shows G1 and E2 restore viability in PC12 cells exposed to OGD/R, **p ≤ 0.01 vs control group, #p ≤ 0.05, ns, ##p ≤ 0.01 vs OGD/R group. g GPR30 inhibitor G15 (10 μM) reverses G1’s protective effect, **p ≤ 0.01 vs control group; ns, ##p ≤ 0.01 vs OGD/R group. h Light microscopy analysis shows improved cell morphology in G1-treated PC12 cells. (All data presented as mean ± SEM). i FerroOrange fluorescence intensity measurement reveals G1-treated cells exhibit lowered ferrous iron levels in OGD/R conditions, n = 3, **p ≤ 0.01
To ensure optimal drug efficacy, we selected a concentration of G1 (0.1 μM) for subsequent tests. Upon treating PC12 cells with OGD/R, we observed that both G1 and E2 could rescue the cellular damage caused by OGD/R (Fig. 4e, f). Interestingly, the ferroptosis inhibitor Ferrostain-1 also improved cell viability in the OGD/R model. However, the protective effects of G1 were reversed by the GPR30 inhibitor G15 (Fig. 4g). Additionally, under a light microscope, the cellular morphology of the G1-treated OGD/R group appeared significantly better than that of the OGD/R group (Fig. 4h).
To further evaluate the effect of G1 on PC12 cells under OGD/R, we stained the cells with FerroOrange to quantify the Fe2+ levels. The OGD/R group treated with G1 showed significantly lower Fe2+ levels than the OGD/R group without G1 (Fig. 4i).
Protective Effects of G1 on Oxidative Stress and Mitochondrial Integrity in PC12 Cells Subjected to OGD/R
Then, we focused on the protective effect of G1 on PC12 cells under OGD/R. Firstly, we utilized the ROS Assay Kit and the fluorescent probe DCFH-DA to assess the intracellular levels of ROS. Our experimental results demonstrated that OGD/R induced a significant accumulation of ROS, while treatment with G1 reduced this accumulation significantly (Fig. 5a).

Protective effects of G1 on PC12 cells. a Flow cytometry analysis of ROS levels in different groups. b, c Measurement of cellular GSH/GSSG ratio and MDA contents using commercial kits, (n = 5, *p ≤ 0.05, **p ≤ 0.01). d Western blot analysis of GPR30, NRF2, and GPX4 protein expression levels (n = 3, **p ≤ 0.001). e Evaluation of mitochondrial membrane potential using a mitochondrial membrane potential kit. f CCK8 assay to determine the protective effect of Nrf2 inhibitor on reversing G1. (n = 6, **p ≤ 0.01). g Western blot analysis of GPX4 and xCT with Nrf2 inhibitor treatment. (n = 3, **p ≤ 0.01, ***p ≤ 0.001). (All data are expressed as mean ± SEM)
To further evaluate the impact of G1, we measured the levels of GSH and MDA, which are indicators of lipid peroxidation. G1 treatment elevated GSH levels and improved MDA levels (Fig. 5b, c). Additionally, Western blot analysis revealed that G1 treatment in the OGD/R group enhanced the protein expression of Nrf2 and GPX4 (Fig. 5d).
Furthermore, assessment of mitochondrial membrane potential demonstrated that G1 treatment rescued the damage caused to the mitochondrial membrane potential by OGD/R (Fig. 5e). We employed the Nrf2 inhibitor ML385 (5 μM) to validate our hypothesis. CCK8 experiments confirmed that ML385 reversed the protective effect of G1 on PC12 cells (Fig. 5f). Consistently, Western blot analysis yielded similar results (Fig. 5g).
Inhibition of GPR30 Leads to Impaired Protection Against Ferroptosis and Disrupted Cellular Iron Homeostasis
To elucidate the involvement of GPR30 in lipid peroxidation and ferroptosis, we conducted experiments to evaluate the suppressive impact of G1 on ferroptosis. The PC12 cell line used in this study was transiently transfected with GPR30 shRNA in the experimental group, while the control group received a placebo. Our observations revealed a noteworthy decline in GPR30 protein expression in the GPR30 shRNA group compared to the control group (Fig. 6a).

GPR30-mediated inhibition of ferroptosis in PC12 cells by G1. a Knockdown efficiency of GPR30 in PC12 cells confirmed by western blot analysis (n = 3, ***p ≤ 0.001). b Cell survival rate determined by the Cell Counting Kit-8 (CCK8) assay. c Detection of changes in ferrous iron ions using a cellular ferrous ion fluorescent probe. Enhanced fluorescence intensity was observed in the GPR30 shRNA group. d GSH/GSSG ratio levels (n = 5, **p ≤ 0.01). E Measurement of MDA contents (n = 5, **p ≤ 0.01). F Protein expression levels of Nrf2 and GPX4 determined by western blotting (n = 3, *p ≤ 0.05, **p ≤ 0.01)
Interestingly, the protective ability of G1 against cell death induced by OGD/R was weakened in GPR30 knockdown cell lines (Fig. 6b). We further investigated the effect of G1 on cellular iron levels using FerroOrange staining and found that the inhibitory effect of G1 was almost eliminated in GPR30 knockdown cells (Fig. 6c).
Moreover, the beneficial effects of G1 on GSH and MDA levels were also significantly diminished in GPR30 knockdown cells (Fig. 6d, e). Additionally, we examined the protein expression of key factors involved in ferroptosis regulation and found that the regulatory effects of G1 on GPX4 and Nrf2 were almost abolished in GPR30 knockdown cells (Fig. 6f).
Discussion
Ferroptosis and neuroinflammation are critical factors in CIRI (Jurcau & Simion, 2021; Stockwell et al., 2017). GPR30, identified as a novel estrogen receptor, has been implicated in mitigating CIRI through the attenuation of cellular inflammation and perfusion damage (Zhang et al., 2018). The role of GPR30 in modulating ferroptosis to confer neuroprotection, however, remains to be clarified. Recognized for its role in rapid nongenomic estrogen signaling, GPR30 is abundantly expressed in key brain areas, such as the hippocampus, cortex, and striatum (Hazell et al., 2009). Studies indicate that the GPR30 agonist G1 not only mirrors the neuroprotective attributes of estradiol (E2) in the hippocampus and striatum but also significantly diminishes infarct size post-cerebral ischemia. In experimental settings of global cerebral ischemia due to vascular occlusion, G1 demonstrated a reduction in neuronal death in the hippocampal CA1 area and lowered the secretion of inflammatory cytokines following MCAO/R and OGD/R injuries. The quantification of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 via ELISA 24-h post-reperfusion corroborates G1’s efficacy in decreasing inflammation and enhancing neuroprotection (Duan et al., 2020; Murata et al., 2013; Zhang et al., 2018). The therapeutic potential of activating GPR30 suggests a promising avenue for advancing estrogen replacement strategies in treating cerebral ischemia.
Ferroptosis, a form of cell death distinct from apoptosis, was initially identified by Dixon et al. in 2012 (Stockwell, 2022). Previous studies have highlighted the significant regulatory role of the stable transcription factor Nrf2 in ferroptosis (Wang et al., 2022). Our study aimed to investigate the expression levels of key proteins associated with ferroptosis in mice experiencing cerebral ischemia–reperfusion and the variations in Fe2+, MDA, ROS, and GSH levels. Furthermore, we conducted in vitro experiments employing OGD/R simulation to monitor relevant markers. The results from both the in vivo and in vitro studies confirmed the occurrence of ferroptosis. To further validate our hypothesis, we utilized the GPR30-specific agonist G1 to investigate its potential in mitigating the damage induced by cerebral ischemia–reperfusion through ferroptosis. Our findings illustrated that targeting GPR30 led to an enhancement in nerve injury scores, reduction in infarct volume, mitigation of Fe2+ overload, reduction in lipid peroxide generation, and elevation in the levels of antioxidant transcription factors Nrf2 and GPX4, along with the augmentation of GSH content. Additionally, we observed that targeting GPR30 upregulated Nrf2 expression, while the administration of ML385 reversed the neuroprotective effects associated with G1-mediated GPR30 targeting.
In conclusion, our research employed the GPR30-specific agonist G1 to explore its potential in mitigating CIRI through ferroptosis. The findings revealed that targeting GPR30 could ameliorate the nerve injury score in mice, decrease infarct volume, and enhance the expression of Nrf2 and GPX4 (Fig. 7). Utilizing the OGD/R model, G1 demonstrated the capability to improve the viability of PC12 cells. Consequently, ferroptosis, observable in CIRI and OGD/R, contributes to aggravating brain injury, yet this process can be alleviated through GPR30 targeting.

Schematic diagram of the possible underlying mechanism by which targeting GPR30 exerts neuroprotective effects by inhibiting ferroptosis in cerebral ischemia/reperfusion injury and OGD/R by inhibiting ferroptosis. Activation of GPR30 regulates the Nrf2/GPX4 signaling pathway. GRP30 G protein-coupled receptor 30, CIRI cerebral ischemia–reperfusion, OGD/R Oxygen glucose deprivation/re-oxygenation
Conclusion
Ferroptosis plays a crucial role in CIRI, leading to cell death. The inhibition of Nrf2 and GPX4 proteins by OGD/R contributes to ferroptosis in nerve cells. Targeting GPR30 can alleviate CIRI and rescue cell death induced by OGD/R. This effect is likely due to the activation of the Nrf2/GPX4 pathway. Therefore, GPR30 shows promise as a target for inhibiting ferroptosis, reducing nerve cell damage, and improving stroke prognosis. These findings have significant clinical implications and potential therapeutic strategies for stroke surgery.
Data Availability
No datasets were generated or analyzed during the current study.
References
Chen, J., Yang, L., Geng, L., He, J., Chen, L., Sun, Q., Zhao, J., & Wang, X. (2021). Inhibition of Acyl-CoA synthetase long-chain family member 4 facilitates neurological recovery after stroke by regulation ferroptosis. Frontiers in Cellular Neuroscience, 15, 632354.
Duan, C., Wang, L., Zhang, J., Xiang, X., Wu, Y., Zhang, Z., Li, Q., Tian, K., Xue, M., Liu, L., et al. (2020). Mdivi-1 attenuates oxidative stress and exerts vascular protection in ischemic/hypoxic injury by a mechanism independent of Drp1 GTPase activity. Redox Biology, 37, 101706.
Hadjimarkou, M. M., & Vasudevan, N. (2018). GPER1/GPR30 in the brain: Crosstalk with classical estrogen receptors and implications for behavior. The Journal of Steroid Biochemistry and Molecular Biology, 176, 57–64.
Han, X., Zhang, J., Liu, J., Wang, H., Du, F., Zeng, X., & Guo, C. (2022). Targeting ferroptosis: A novel insight against myocardial infarction and ischemia–reperfusion injuries. Apoptosis, 28(1–2), 108–123.
Hazell, G. G. J., Yao, S. T., Roper, J. A., Prossnitz, E. R., O’Carroll, A.-M., & Lolait, S. J. (2009). Localisation of GPR30, a novel G protein-coupled oestrogen receptor, suggests multiple functions in rodent brain and peripheral tissues. Journal of Endocrinology, 202(2), 223–236.
Jiang, X., Stockwell, B. R., & Conrad, M. (2021). Ferroptosis: Mechanisms, biology and role in disease. Nature Reviews Molecular Cell Biology, 22(4), 266–282.
Jurcau, A., & Simion, A. (2021). Neuroinflammation in cerebral ischemia and ischemia/reperfusion injuries: From pathophysiology to therapeutic strategies. International Journal of Molecular Sciences, 23(1), 78.
Li, C., Wu, Y., Chen, Q., Luo, Y., Liu, P., Zhou, Z., Zhao, Z., Zhang, T., Su, B., Sun, T., et al. (2023). Pleiotropic microenvironment remodeling micelles for cerebral ischemia-reperfusion injury therapy by inhibiting neuronal ferroptosis and glial overactivation. ACS Nano, 17(18), 18164–18177.
Li, Y., Chopp, M., Chen, J., Wang, L., Gautam, S. C., Xu, Y. X., Zhang, Z., et al. (2000). Intrastriatal transplantation of bone marrow nonhematopoietic cells improves functional recovery after stroke in adult mice. Journal of Cerebral Blood Flow and Metabolism, 20(9), 1311–1319.
Liu S-b, Zhang N, Guo Y-y, Zhao R, Shi T-y, Feng S-f, Wang S-q, Yang Q, Li X-q, Wu Y-m et al: G-Protein-Coupled Receptor 30 Mediates Rapid Neuroprotective Effects of Estrogen via Depression of NR2B-Containing NMDA Receptors. The Journal of Neuroscience 2012, 32(14):4887–4900.
Liu, T., Li, X., Cui, Y., Meng, P., Zeng, G., Wang, Q., & Wang, Y. (2021). Bioinformatics analysis identifies potential ferroptosis key genes in the pathogenesis of intracerebral hemorrhage. Frontiers in Neuroscience, 15, 661663.
Murata, T., Dietrich, H. H., Xiang, C., & Dacey, R. G. (2013). G protein-coupled estrogen receptor agonist improves cerebral microvascular function after hypoxia/reoxygenation injury in male and female rats. Stroke, 44(3), 779–785.
Olde, B., & Leeb-Lundberg, L. M. F. (2009). GPR30/GPER1: Searching for a role in estrogen physiology. Trends in Endocrinology & Metabolism, 20(8), 409–416.
Roque, C., Mendes-Oliveira, J., Duarte-Chendo, C., & Baltazar, G. (2019). The role of G protein-coupled estrogen receptor 1 on neurological disorders. Frontiers in Neuroendocrinology, 55, 100786.
Stockwell, B. R. (2022). Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell, 185(14), 2401–2421.
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., Fulda, S., Gascón, S., Hatzios, S. K., Kagan, V. E., et al. (2017). Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell, 171(2), 273–285.
Tuo, Q. Z., Lei, P., Jackman, K. A., Li, X., Xiong, H., Li, X., Liuyang, Z. Y., Roisman, L., Zhang, S. T., Ayton, S., et al. (2017). Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Molecular Psychiatry, 22(11), 1520–1530.
Wang, L., Zhang, X., Xiong, X., Zhu, H., Chen, R., Zhang, S., Chen, G., & Jian, Z. (2022). Nrf2 regulates oxidative stress and its role in cerebral ischemic stroke. Antioxidants, 11(12), 78.
Wang, X. S., Yue, J., Hu, L. N., Tian, Z., Zhang, K., Yang, L., Zhang, H. N., Guo, Y. Y., Feng, B., Liu, H. Y., et al. (2019). Activation of G protein-coupled receptor 30 protects neurons by regulating autophagy in astrocytes. Glia, 68(1), 27–43.
Wu, S., Wu, B., Liu, M., Chen, Z., Wang, W., Anderson, C. S., Sandercock, P., Wang, Y., Huang, Y., Cui, L., et al. (2019). Stroke in China: Advances and challenges in epidemiology, prevention, and management. The Lancet Neurology, 18(4), 394–405.
Xu, Y., Li, K., Zhao, Y., Zhou, L., Liu, Y., & Zhao, J. (2022). Role of ferroptosis in stroke. Cellular and Molecular Neurobiology, 43(1), 205–222.
Yang, S., Wang, H., Yang, Y., Wang, R., Wang, Y., Wu, C., & Du, G. (2019). Baicalein administered in the subacute phase ameliorates ischemia-reperfusion-induced brain injury by reducing neuroinflammation and neuronal damage. Biomedicine & Pharmacotherapy, 117, 109102.
Zhang, Y., Lu, X., Tai, B., Li, W., & Li, T. (2021). Ferroptosis and Its multifaceted roles in cerebral stroke. Frontiers in Cellular Neuroscience, 15, 615372.
Zhang, Y., Ye, P., Zhu, H., Gu, L., Li, Y., Feng, S., Zeng, Z., Chen, Q., Zhou, B., & Xiong, X. (2024). Neutral polysaccharide from Gastrodia elata alleviates cerebral ischemia–reperfusion injury by inhibiting ferroptosis-mediated neuroinflammation via the NRF2/HO-1 signaling pathway. CNS Neuroscience & Therapeutics, 30(3), e14456.
Zhang, Z., Qin, P., Deng, Y., Ma, Z., Guo, H., Guo, H., Hou, Y., Wang, S., Zou, W., Sun, Y., et al. (2018). The novel estrogenic receptor GPR30 alleviates ischemic injury by inhibiting TLR4-mediated microglial inflammation. Journal of Neuroinflammation, 15(1), 78.
Funding
This work was supported by the National Natural Science Foundation of China, (No. 82104158) and Key Research and Development Program in Shaanxi Province of China (fund no. 2022SF-314).
Author information
Authors and Affiliations
Contributions
Y.-Q.Z., T.S., and Z.Z. contributed equally to this study. Y.-Q.Z., T.S., and Z.Z. conceived and designed the study. T.S., J.F., L.Y., and Y.X. performed the experiments. J.-F.Z. and X.-L.T. analyzed the data. Y.-Q.Z., A.L., and M.-G.Z. wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical Approval
All experiments involving mice were approved by the Animal Ethics Committee of premature-maintained room (21–25 °C) under a 12/12-h light/dark cycle. This study was approved by the Laboratory Animal Welfare and Ethics Committee of the Fourth Military Medical University (approval No. 20230702).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Yq., Sun, T., Zhao, Z. et al. Activation of GPR30 Ameliorates Cerebral Ischemia–Reperfusion Injury by Suppressing Ferroptosis Through Nrf2/GPX4 Signaling Pathway. Neuromol Med 26, 33 (2024). https://doi.org/10.1007/s12017-024-08801-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12017-024-08801-5