
Abstract
Neurodegenerative disorders such as Alzheimer’s, Parkinson’s and Huntington’s diseases have high prevalence among the elderly. Many strategies have been established to alleviate the symptoms experienced by affected individuals. Recent studies have shown that exercise helps patients with neurological disorders to regain lost physical abilities. PGC1α/FNDC5/BDNF has emerged recently as a critical pathway for neuroprotection. PGC1α is a highly conserved co-activator of transcription factors that preserves and protects neurons against destruction. PGC1α regulates FNDC5 and its processed and secreted peptide Irisin, which has been proposed to play a critical role in energy expenditure and to promote neural differentiation of mouse embryonic stem cells. FNDC5 may also increase the expression of the neurotrophic factor BDNF, a neuroprotective agent, in the hippocampus. BDNF is secreted from hippocampus, amygdala, cerebral cortex and hypothalamus neurons and initiates intracellular signaling pathways through TrkB receptors. These pathways have positive feedback on CREB activities and lead to enhancement in PGC1α expression in neurons. Therefore, FNDC5 could behave as a key regulator in neuronal survival and development. This review presents recent findings on the PGC1α/FNDC5/BDNF pathway and its role in neuroprotection, and discusses the controversial promise of irisin as a mediator of the positive benefits of exercise.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The prevalence of neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD) increases by age worldwide. Because life expectancy is steadily lengthening so has the prevalence of these diseases and the critical need for research directed at finding cures.
Neurodegenerative disorders result in progressive loss of neuronal structure and/or function (Przedborski et al. 2003; Vila and Przedborski 2003). These disorders are characterized by protein misfolding and aggregation, mitochondrial dysfunction and oxidative stress, and programmed cell death of neurons, but what causes the disorders remains a mystery. Physical exercise has been shown to be neuroprotective in the context of several neurodegenerative diseases, including AD and PD. However, the mechanisms by which physical exercise prevent neurodegenerative diseases or improve symptoms in patients with neurodegenerative diseases are currently unclear. Monteiro-Junior et al. (2015) recently put forward two hypotheses in the context of Parkinson’s disease. One is that exercise protects neurons from oxidative stress. The other is that exercise increases expression of neuronal trophic factors, such as BDNF, and other factors that promote survival, maintenance and function of neuronal progenitors and of neuronal tissues in general.
Mitochondria and Peroxisomes and Oxidative Stress
The pathology of neurodegenerative diseases including AD, PD and HD have close correlation with oxidative stress and the impairment of mitochondrial energy metabolism (Tretter et al. 2004; Guo et al. 2013). Oxidative stress can occur when production of reactive oxygen species (ROS), which include the superoxide anion radical (O2–), hydrogen peroxide (H2O2) and the hydroxyl radical (–OH), as well as reactive nitrogen species (RNS), which include the nitric oxide radical (–NO), the nitrogen dioxide radical (–NO2), nitrite (NO2–) and peroxynitrite (ONOO–), outpaces a cell’s ability to inactivate ROS/RNS and to repair damage. ROS/RNS can damage all of the major biomolecules and initiate cell death (Rinnerthaler et al. 2015). Mitochondria and peroxisomes are important sites of ROS/RNS production. Mitochondria are the major source of ATP by oxidative phosphorylation (OXPHOS). During oxidative phosphorylation, electrons are transferred from electron donors to electron acceptors in redox reactions. These redox reactions are carried out by the electron transport chain (ETC), which consists of four discrete complexes (I–IV) that are embedded in the inner mitochondrial membrane. These complexes generate an electrochemical gradient that is used to drive ATP production. The last complex (complex IV) in the ETC transfers two electrons and two protons to O2, to form water.
The main type of ROS produced during oxidative phosphorylation is superoxide (O2–), which forms when an electron is transferred prematurely to O2, chiefly by complexes I and III, though other mechanisms exist (Mailloux 2015). Mitochondrial superoxide dismutases (SODs) of the manganese (SOD2) types process superoxide into H2O2, a less potent ROS, but still capable of causing oxidative damage. Another source of mitochondrial H2O2 that is especially important in neurons is monoamine oxidases (MAOs), which catalyze the turnover of monoamine neurotransmitters (serotonin, norepinephrine, dopamine) (Edmondson 2014). The presence of superoxide and H2O2 can also lead to production of other ROS/RNS. For instance, superoxide can react with FeS-containing proteins (e.g., complex I) to produce Fe2+, which can react via the Fenton reaction with H2O2 to form the hydroxyl radical (–OH) (Winterbourn 1995). The presence of –OH can initiate a cascade of reactions that result in the formation of lipid radicals and lipid peroxide radicals (Ayala et al. 2014). In addition, a number of mitochondrial enzymes produce nitric oxide (–NO), which can combine in the mitochondrial matrix with superoxide (O2–) to form peroxynitrite (ONOO–) (Murphy 2009). The functions of animal peroxisomes include β-oxidation of very long chain fatty acids and synthesis of ether-linked phospholipids, both of which have relevance for oxidative stress (Antonenkov et al. 2010; Fransen et al. 2012; Lodhi and Semenkovich 2014). β-Oxidation of fatty acids also occurs in mitochondria in mammals, where electrons produced by the first step of β-oxidation are transferred to FADH to generate FADH2 and subsequently fed into the electron transport chain to produce ATP. In contrast, in peroxisome β-oxidation, FADH2 transfers electrons directly to O2, generating H2O2. β-Oxidation in peroxisomes is thought to shorten very long (>26 carbon) chain fatty acids, which can then be further oxidized in mitochondria. In addition to the H2O2 produced by β-oxidation and other processes, peroxisomes also contain enzymes capable of producing –NO and O2–, and so it is likely that peroxisomes also contain ONOO–.
Although mitochondria and peroxisomes are major sources of ROS/RNS, they are also rich in antioxidants that inactivate ROS/RNS. They can thus serve as either as net sources or as net sinks of ROS/RNS (Fransen et al. 2012; Andreyev et al. 2015). The mechanisms that exist for inactivating ROS/RNS in mitochondria and peroxisomes include both enzyme and non-enzyme-based systems. The enzyme-based systems include the aforementioned SODs, which processes superoxide to form H2O2, as well as catalase, glutathione peroxidase (GPx) and peroxiredoxins, which convert H2O2 to H2O. Non-enzyme-based systems include glutathione (GSH)-based systems (Andreyev et al. 2015).
Another way that peroxisome function may protect cells against oxidative stress is via production of ether phospholipids, which contain a fatty alcohol instead of a fatty acid at the sn-1 position. Members of the plasmalogen family of ether phospholipids are modified to contain a vinyl ether bond, and are enriched at the sn-2 position in the polyunsaturated fatty acids docosahexaenoic acid (DHA), C22:6 ϖ-3 or arachidonic acid C20:4 ϖ-6(Braverman and Moser 2012; Lodhi and Semenkovich 2014). The presence of the fatty alcohol results in changes in membrane structure in plasmalogen-enriched membranes that may affect signaling, ion transport and trafficking within the endomembrane system. The vinyl ether bond is more reduced relative to the acyl bond in a fatty acid, and thus, plasmalogens have been suggested to act as antioxidants, though this is controversial. In addition, DHA is a precursor of resolvins, docosatrienes and neuroprotections, which regulate inflammatory responses. Interestingly, plasmalogens are abundant in brain and enriched in myelin. Accordingly, reduction in plasmalogen levels is correlated with AD severity (Bennett et al. 2013).
Importantly, ROS/RNS production by mitochondria and peroxisomes is affected by the metabolic status of the cell. For instance, ROS production from the ETC increases if, e.g., glucose and fatty acid levels are high (lots of NADH and FADH2 being produced), but energy levels are low (Ruetenik and Barrientos 2015). Accordingly, although the acute effect of exercise is an increase in ROS production and oxidative stress, regular exercise leads to induction of antioxidants, DNA repair and protein-degrading enzymes, thus shifting the balance and decreasing the probability of oxidative stress (Radak et al. 2005).
In addition to mitochondrial superoxide, cytosolic NADPH oxidase is another major site for superoxide production in the cell. NADPH oxidase is mainly located on the cytosolic face of phagocyte membranes and plays a critical role in immune defense. Microglia, which are the phagocytes of the central nervous system (CNS), have high NADPH oxidase activity especially in many neurodegenerative diseases. Therefore, activated microglia produce oxidants that can negatively affect the function of the CNS (Bordt and Polster 2014).
Although any part of a cell can be affected by oxidative stress, oxidative damage in the mitochondria is likely to be particularly devastating because it potentially results in a feedback loop: The oxidative damage can decrease mitochondrial function in ways that result in higher production of ROS/RNS, leading to even more damage. Because the mitochondrial genome (mtDNA) contains 24 genes encoding tRNAs and rRNAs important for mitochondrial protein synthesis, and 13 encoding components of the ETC, much mitochondrial dysfunction is likely to occur as a result of damage to mitochondria DNA (mtDNA) (Keogh and Chinnery 2015).
It is likely that mtDNA is particularly susceptible to oxidative damage in part because it lies in the mitochondrial matrix in close proximity with the electron transport chain, which as mentioned above is a major source of ROS. Conditions of cellular stress that impact ROS/RNS production and/or antioxidant efficiency could result in an increase in mtDNA mutations that could further impact mitochondrial function (Ciccone et al. 2013; Farrar et al. 2013; Guo et al. 2013; Keogh and Chinnery 2015; Payne and Chinnery 2015).
The fact that cells contain 100–10,000 mitochondria, each of which contains 2–10 mtDNA copies, is likely to counteract the effects of inactivating mutations in any one mtDNA (a phenomenon known as inter-mitochondrial complementation). In fact, recent high-throughput sequencing analysis indicates that essentially all mitochondria are heteroplasmic (containing both mutant and wild-type mtDNA genomes). On the other hand, replication of mtDNA occurs throughout the life of the cell with the potential for new mutations arising. Thus, the composition of heteroplasmic mitochondria inherited at birth coupled with the generation of new mtDNA mutations over the course of time and with genetic drift can all result in particular cells containing a large proportion of defective mitochondria genes (Keogh and Chinnery 2015; Yang et al. 2015).
In summary, any dysfunction in OXPHOS affects mitochondrial function and can lead to excessive ROS production, which results in oxidative lipid, DNA and protein damage. Living cells constantly generate ROS for physiological functions; however, excess ROS production results in pathophysiological conditions such as ischemia–reperfusion injury, neurodegenerative diseases and aging (Balaban et al. 2005; West et al. 2011; Kotiadis et al. 2014).
Mitochondrial Function is Particularly Important for Neuronal Function
Mitochondrial function has a pivotal role in neuronal health for a number of reasons. One reason is that post-mitotic neurons depend on mitochondrial metabolism exclusively (in contrast to, e.g., replicating cells that use mostly glycolysis). They are also large and highly polarized cells with many processes that have high demand for ATP consumption. For instance, the function of synapses, which are located at the tips of axons and dendrites that are often far from the cell body, requires a great deal of energy and the ability to buffer Ca2+, both of which are supplied by mitochondria. Thus, mitochondria are enriched in and have to be transported to (often distant) synapses (requiring even more energy for transport), and damaged mitochondria have to be transported all the way back to the cell body in order to fuse with lysosomes during mitophagy (Amadoro et al. 2014; Picone et al. 2014; Lin and Sheng 2015).
Consequently, mitochondrial impairment leads to necrosis and apoptosis of neurons (Green and Kroemer 2004; Nagley et al. 2010). Mitochondrial disorders have been categorized into three general subclasses: (a) primary disorders due to mitochondrial gene mutations; (b) disorders relating to nuclear gene mutations affecting mitochondrial function; and (c) secondary disorders that result in the accumulation of mitochondrial damage over the time. Neurodegenerative disorders typically belong to the latter category. Diseases based on mitochondrial dysfunction vary from well-known diseases such as glaucoma, inflammation, neurodegenerative diseases, type 2 diabetes and cancers, to rare ones such as Friedreich’s ataxia (FA), Kearns–Sayre syndrome (KSS) and mitochondrial encephalopathy lactic acidosis and strokes (MELSAS) (Haas et al. 2008; Chaturvedi and Beal 2013).
Given the importance of mitochondrial function for neuronal health and the long life of neurons, there is an especially great need in neurons for mitochondrial quality control (QC). In addition to the antioxidants described above, mitochondrial quality control mechanisms include mitochondrial proteases, which degrade damaged proteins (damaged, e.g., by oxidation) in the matrix and inter-membrane space; ubiquitin–proteasome systems that remove damaged proteins located in the outer membrane or nuclear-coded mitochondrial proteins that are not correctly imported; and vesicle-based removal of oxidated proteins/lipids for targeting to lysosomes or peroxisomes. When mitochondrial damage is too great, mitophagy, a form of autophagy, occurs, and if that is not sufficient to repair cell function, cells undergo apoptosis (Amadoro et al. 2014).
Mitochondrial dynamics (transport along the cytoskeleton, regulation of mitochondrial architecture and connectivity mediated by tethering, fusion and fission) also play a role in mitochondrial QC. For instance, mitochondrial fission is required to fragment mitochondria into smaller pieces as required for mitophagy. Hyperfusion occurs as a result of several different types of cellular stress, which is likely to facilitate inter-mitochondrial complementation and (likely due to larger-sized mitochondria) protects cells against mitophagy and apoptosis. Moreover, mitofusin2, a protein involved in mitochondrial fusion, is also important for tethering mitochondria to the endoplasmic reticulum, and participates in the unfolded protein response (Shutt and McBride 2013; Zorzano and Claret 2015). Removing mitofusin2 from the mouse cerebellum leads to reduced ETC activity, aberrant mitochondrial ultrastructure and altered mitochondrial distribution, and result in degeneration of Purkinje cells (Chen et al. 2007).
Mutations in genes encoding proteins, such as pink1, parkin, DJ-1 and α-synuclein in PD, amyloid beta (Aβ) in AD, superoxide dismutase (SOD) in amyotrophic lateral sclerosis (ALS) and huntingtin in HD, not only disrupt mitochondrial membranes but also alter OXPHOS, thereby enhancing excessive ROS production (Reddy 2009; Guo et al. 2013). These molecular aspects of neurodegenerative disorders illuminate the fact that cellular organelles, especially mitochondria and peroxisomes, play vital roles in the progression of these diseases.
For instance, PD is associated with loss of dopaminergic neurons in the substantia nigra pars compacta. The resulting loss of dopamine in the striatum causes the characteristic PD motor symptoms, which include tremor, rigidity, slowness in the execution of movement and postural instability (Blesa et al. 2015; Hwang 2013; Exner et al. 2012; Trempe and Fon 2013; Amadoro et al. 2014). On a cellular level, both sporadic and familial PD are strongly linked to oxidative stress, mitochondrial dysfunction (in particular inhibition of ETC complex I) and impaired clearance of misfolded proteins and damaged organelles (Exner et al. 2012; Han et al. 2014; Blesa et al. 2015).
Dopaminergic neurons appear to be particularly prone to oxidative stress. Dopamine is itself a potential cause of oxidative stress as it is a target of MAO and can also auto-oxidize, resulting in modification of a number of PD-related proteins. Accordingly, there is evidence for oxidative stress in the substantia nigra of PD patients: oxidized lipids, proteins and DNA are all found at increased levels, and reduced glutathione is decreased (Hwang 2013).
Accordingly, genes associated with familial PD have been implicated in the response to oxidative stress and to mitochondrial QC. These include the parkin gene, which encodes an E3 ubiquitin ligase, and the PINK1 gene, which encodes a kinase (Exner et al. 2012; Tempe and Fon 2013; Han et al. 2014). Parkin is recruited to defective mitochondria in a PINK1-dependent manner, resulting in mitophagy. In addition, both Parkin and PINK1 have been linked to mitochondrial fusion and fission. In particular, the mitochondrial fusion proteins mitofusin1 and mitofusin2 are ubiquitinated by Parkin, targeting them for destruction by the proteasome, and are reduced in cells undergoing mitophagy. Parkin-induced inhibition of mitochondrial fusion may be important to allow fission, which is a prerequisite for mitophagy (Trempe and Fon 2013). Interestingly, Parkin is cysteine rich and therefore can be inactivated by misfolding under conditions of oxidative stress. Oxidatively modified, misfolded Parkin has been found in the brains of PD patients (Exner et al. 2012; Blesa et al. 2015).
In summary, neurodegenerative diseases are associated with oxidative stress and mitochondrial dysfunction. A strategy of integrative therapies should be considered, combining medical treatment with other therapies such as physical activities for patients with certain neurodegenerative diseases, especially therapies that lead to increased mitochondrial density or mitigation of mitochondrial functions.
Oxidative Stress and Neurodegeneration
Neurons have specific membrane potential features, which are required for neurotransmission and polarization. Na+-K+ ATPase plays a vital role in maintenance of neuronal membrane potential at resting state (Yu 2003). Therefore, mitochondrial dysfunction or neuronal stressors such as ROS can disrupt polarization and maintenance of plasma membrane potential. When this occurs, cell can fall into a rigid condition (Mochel and Haller 2011). Depolarization of the cellular membrane leads to changes in the activity of N-methyl-d-aspartate (NMDA) receptors. High activity of NMDA receptors triggers calcium influx into the cell, thereby resulting in neuronal death (Song et al. 2011). Calcium clearing, in physiologic and pathological conditions, is undertaken by mitochondrial uptake, and mitochondrial malfunction also abates calcium uptake and mitochondrial membrane potential (Tillement et al. 2011). Calcium overload in the cytosol induces ROS generation via many signaling pathways, and this process could create a positive correlation between ROS and free calcium in cells (Moreira et al. 2007). In contrast to necrosis, apoptosis is an active form of cell death, which can be induced by ATP depletion in cells (Witte et al. 2010). Apoptotic pathways are integrated with the mitochondrion, due to its central role in apoptosis (Rossignol and Frye 2012). Thus, defective mitochondria can trigger neurodegenerative disorders.
Elevated ROS is correlated with several neurodegenerative diseases. For example, it has been shown that mitochondrial complex I activity decreases in the substantia nigra in PD, resulting in disrupted calcium homeostasis and elevated ROS generation (Legros et al. 2004; Eichner and Giguère 2011). Moreover, PD can be chemically induced by inhibitors of complex I activity (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), paraquat and rotenone) (Schon et al. 2010). Rotenone, paraquat and MPTP elevate ROS generation, cytochrome c release from mitochondria, and DNA damage in neurons (Nicholls 2008). Similarly, low aerobic glucose utilization in basal ganglia and the cerebral cortex of Huntington’s disease (HD) patients (Herben-Dekker et al. 2014) leads to anaerobic metabolism with high lactate production. This phenomenon leads to enhancement in oxidative damage to neurons. In addition, complex II, III, IV deficiencies were reported in HD patients’ postmortem brains (Lezi and Swerdlow 2012). Mutant huntingtin protein (mHtt) also impairs the mitochondrial membrane potential and calcium homeostasis by influencing mitochondrial activities (Jin and Johnson 2010). Inhibition of complex II showed similar behavioral and pathophysiological features to HD in rodents (Wallace and Fan 2009). In addition to energy imbalance, calcium homeostasis and oxidant generation are also detected in AD patients’ tissues in correlation with complex IV inhibition (Ferreiro et al. 2012). Taken together, neurological disorders from ischemia to neurodegeneration have close correlation with mitochondrial activities. Hence, dysfunction in mitochondrial activities can cause chronic damage and also trigger apoptotic or necrotic pathways in neurons and is a major pathogenic cause of neurodegenerative diseases (Obulesu and Lakshmi 2014).
PGC1α in Organelle Biogenesis and Neuroprotection
Mitochondria
Ligand-dependent nuclear receptor transcription factors have critical roles in many biological and cellular activities, including mitochondrial metabolism. They respond to a variety of ligands and are controlled by many co-activators or co-repressors (Evans 2005). Peroxisome proliferator-activated receptor γ co-activator α (PGC1α) was first identified as an interactor of nuclear receptor transcription factor PPARγ (Puigserver et al. 1998) and functions as a highly conserved transcriptional co-activator with important roles in mitochondrial oxidative metabolism, maintenance of glucose and lipid, and energy homeostasis (Corona and Duchen 2015; Villena 2015). PGC1α belongs to the PGC1 protein family and shares high sequence similarity with its paralog, PGC1β (Lin et al. 2002a). PGC1α regulates mitochondrial maintenance through expression of mitochondrial biogenesis factors (Kressler et al. 2002; Wenz 2009). Nuclear respiratory factors 1 and 2 (NRF1 and NRF2) (Wu et al. 1999), peroxisome proliferator receptor alpha (PPARα) (Vega et al. 2000), estrogen-related receptor alpha (ERRα) (Schreiber et al. 2004) and thyroid receptor (TR) (Zhang et al. 2004) are among the transcriptional factors involved in mitochondria biogenesis that utilize PGC1α as a co-regulator. NRF1 and NRF2 are targets of PGC1α, and they regulate nuclear OXPHOS genes such as cytochrome c and mitochondrial transcription factor A (TFAM) (Kelly and Scarpulla 2004). Increases in PGC1α activity, which is induced by exercise, result in elevation of mitochondrial mass (Lin et al. 2002). Thus, PGC1α is the key regulator of mitochondrial biogenesis (Scarpulla 2008; Kotiadis et al. 2014).
PGC1α and AD
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by neurofibrillary tangles (NFTs), which are generated by tau hyperphosphorylation and Aβ deposition in the brain. AD has been correlation with PGC1α expression (Katsouri et al. 2011). Low expression of PGC1α in the brain of AD patients causes Aβ formation because activity of the beta secretase (BACE1) enzyme, which generates Aβ, is regulated by PGC1α (Wang et al. 2013). In addition, mitochondrial biogenesis and control of oxidative stress depend on PGC1α expression and any disruption in this condition causes neuronal death (Qin et al. 2009).
PGC1α and HD
The first hints of a connection between PGC1α, impaired mitochondrial function and HD pathogenesis came from mice lacking PGC1α (McGill and Beal 2006). These mice have neurodegenerative lesions in the striatum and are hyperactive, which are hallmarks of HD in humans (Lin et al. 2004; McGill and Beal 2006). Further studies showed that PGC1α is required in 10T1/2 cells for induction of ROS-detoxifying enzymes such as copper/zinc superoxide dismutase (SOD1), manganese SOD (SOD2), catalase and GPx in response to H2O2 treatment, that PGC1α null mice are more sensitive to oxidative stress and that increasing PGC1α can protect neural cells in culture from oxidative stress-mediated death. These changes depend upon cAMP-responsive element binding protein (CREB) as a potent PGC1α co-activator (St-Pierre et al. 2006; Wu et al. 2006).
Other data indicate that mHtt can reduce PGC1α expression by interfering with the CREB/transcription initiation factor 4 (TAF4) complex (Cui et al. 2006). Cui et al. 2006 showed a 30 % reduction of PGC1α mRNA in the caudate nucleus of striatum (the first region affected by HD) of presymptomatic HD patients, but not in cerebellum or hippocampus. PGC1α reduction in HD abates mitochondrial cytochrome c and cytochrome oxidase IV expression (Martin et al. 2011). In this regard, previous studies have shown that PGC1α overexpression has potential to protect striatal neurons against expression of polyglutamine expanded forms of huntingtin. In addition, postmortem analysis of HD patient brains showed low expression levels of PGC1α target genes (Tsunemi et al. 2012). In other types of neurodegenerative disorders such as PD, PGC1α can act as a protective agent, as PGC1α-deficient mice were more vulnerable to MPTP treatment. Therefore, elevated PGC1α expression has a neuroprotective property against MPTP toxicity (Mudò et al. 2012). Overall, it appears that PGC1α expression is neuroprotective in neurodegenerative diseases and aging.
Posttranslational modifications of PGC1α, such as deacetylation or phosphorylation, promote PGC1α activity (Outeiro et al. 2008). Deacetylation of PGC1α by sirtuin-1 (SIRT1) enhances PGC1α activity. This may be a key basis for the neuroprotective role of SIRT1 in AD, PD, HD and ALS (Qin et al. 2006; Zheng et al. 2010). Recent studies have shown that parkin-interacting substrate (PARIS) is a new partner protein in PD onset that has the potential to repress PGC1α activity and enhance degeneration of dopaminergic neurons (Shin et al. 2011). In Alzheimer’s disease, PGC1α expression is also abated (Sheng et al. 2012) and its overexpression in animal models of AD attenuated neurological phenotypes (Qin et al. 2009). In addition, ectopic expression of PGC1α decreased Aβ secretion and increased its soluble form (Katsouri et al. 2011). This phenomenon indicated a link between quality control of mitochondria and neurodegenerative disorders, suggesting that amelioration in neurological disorders was mediated by stimulating mitochondria biogenesis. In this regard, single-nucleotide polymorphisms (SNPs) have been detected in the gene encoding PGC1α in HD and PD individuals (Clark et al. 2011). Expression of SOD1 and SOD2, critical scavengers of ROS, are regulated by PGC1α (Johnson et al. 2005; Weydt et al. 2009). This may account for increased survival of SOD1 mutant ALS mice when PGC1α was overexpressed. In this model, overexpression of PGC1α increased antioxidant capacity by elevating expression of other antioxidant enzymes, offsetting the loss of SOD1 (Zhao et al. 2011). In accordance with these observations, it has been reported that PGC1α silencing suppresses the expression of ROS-detoxifying enzymes (Mudò et al. 2012). Based on these studies, enhancing the PGC1α pathway might be a promising therapeutic strategy in neurological disorders (Handschin 2009; Wenz 2011).
Peroxisomes
Scavenger enzymes and proteins such as catalase, glutathione peroxidase, PMP20, peroxiredoxin 1 and epoxide hydrolase protect peroxisomes and the cell from H2O2 (van der Valk et al. 1985). Oxidative stress induces morphological changes in peroxisomes, which varies depending on ROS levels (Lopez-Huertas et al. 2000). Over a threshold level, ROS leads to peroxisome destruction, while below this threshold, it induces elongation of peroxisomes, stimulates peroxisome proliferation and subsequently results in high density of peroxisomes with high antioxidant capacity to decompose ROS (del Río et al. 2002). In hippocampal neurons, peroxisome proliferation protects neurons from ROS and degeneration (Santos et al. 2005). Therefore, dysfunction of peroxisomes leads to accumulation of toxic metabolites which predispose cells to aging and apoptosis (Sheikh et al. 1998; Wood et al. 2006).
Peroxisome dysfunction causes neurological disruption, incomplete neuronal migration, dysmyelination, and defects in neuronal development (Steinberg et al. 2006; Kassmann et al. 2007). Disorders effecting peroxisome function are subdivided into two groups: the peroxisome biogenesis disorders (PBDs) and the peroxisomal enzyme deficiencies (PEDs) (Wanders and Waterham 2006; Baes and Aubourg 2009). PBDs are specified by the absence of functional peroxisomes due to deficiencies in the peroxisomal transport system (Wanders and Waterham 2005). Mutations in peroxin (Pex) genes impact peroxisome biogenesis and assembly, leading to PBDs (Baes and Van Veldhoven 2006). Biochemical outputs of these mutations are altered lipid metabolism, oxidative stress and mitochondria dysfunction (Wanders and Waterham 2005). These types of changes are also associated with PD. α-Synuclein accumulation, an important hallmark of PD, occurs due to alteration in posttranslational modifications of α-synuclein such as phosphorylation, nitration and oligomerization (Fujiwara et al. 2002; Sharon et al. 2003a). Yakunin et al. (2010) also reported that mutations in PEX2, PEX5 and PEX13 correlated with α-synuclein deposition in neurons. Accumulation of α-synuclein is associated with changes in metabolism of long-chain polyunsaturated fatty acids (PUFAs), and postmortem brains of patients with PD had high amounts of cytosolic PUFA (Sharon et al. 2003b). Together, these findings suggest an impact on peroxisome function in PD. Moreover, an imbalance of metabolites related to peroxisomal dysfunction might account for AD initiation (Nixon et al. 2000; Lin and Beal 2006), suggesting further the importance of optimal peroxisome function in multiple neurodegenerative diseases. In this context, it is interesting to note that plasmalogens and docosahexaenoicacid (DHA) (Calon et al. 2004) are among the lipids that are synthesized in peroxisomes that have antioxidant features and may delay AD onset (Ginsberg et al. 1995; Kalmijn et al. 1997). Plasmalogen synthesis is increased by peroxisome proliferation, while DHA, a polyunsaturated fatty acid, is synthesized in minute amounts by glial and endothelial peroxisomes in the final step of β-oxidation from eicosapentaenoic acid or linolenic acid (Conquer et al. 2000). Considering the fact that DHA levels are decreased in AD patients’ brains and plasma on one hand, and disrupted production of very long chain fatty acids in the neurons of the patients with AD on the other hand, this suggests that peroxisome proliferation can be targeted for therapeutic strategies in neurodegenerative disorders related to excessive ROS generation (Kou et al. 2011).
Taking into account the role of PGC1α in regulating mitochondrial and peroxisomal biogenesis, remodeling and proliferation (Koepke et al. 2007; Ivashchenko et al. 2011), the PGC1α pathway shows promise as a potential pharmacological target to overcome neurological disorders associated with peroxisome and mitochondria dysfunction.
PGC1α/FNDC5/BDNF Pathway
Recently, there has been much interest, but also much controversy, surrounding the discovery of irisin, a potential myokine reported to elicit beneficial effects of exercise (Boström et al. 2012).
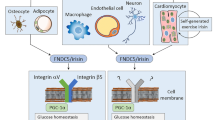
Fibronectin type III domain-containing 5 (FNDC5), formerly known as peroxisomal protein (PeP), is a transmembrane precursor protein that is processed into a soluble secretory peptide termed “irisin” (Fig. 1), which is highly conserved among mammals. FNDC5 has an N-terminal 29-amino acid signal peptide followed by a fibronectin domain that is flanked by a proteolytic cleavage site. After cleavage of the signal peptide and the cleavage site flanking the fibronectin domain, the secreted irisin glycopeptide of 112 amino acids is produced. C-terminal to the irisin peptide region is a transmembrane domain and a cytoplasmic domain (Fig. 1) (Ferrer-Martínez et al. 2002; Ostadsharif et al. 2011; Lee et al. 2014). Spiegelmann’s group in 2012 reported that mouse transgenics with elevated PGC1α expression in muscle show “browning” of subcutaneous white adipose tissue. This occurs via a secreted factor, because conditioned media from myocytes expressing PGC1α were also capable of inducing “browning” in primary subcutaneous adipocytes. They further showed that PGC1α regulates FNDC5 expression and that FNDC5 is expressed in human muscle tissue in response to exercise. Secretion of irisin also increases in response to exercise in both mice and humans, and reducing irisin levels in conditioned media from myocytes reduces their “browning” effects on fat cell culture. Exogenous irisin expression in obese and insulin-resistant mice resulted in increased irisin was a myokine responsible for browning of white adipose tissue through enhancing oxygen consumption and induction of expression of thermogenin (uncoupling protein 1) mRNA and other mitochondrial genes, as well as an increase in oxygen consumption induction (Boström et al. 2012), and elevated levels of irisin in the blood were reported to help mice resist against insulin and increased body weight. PGC1α regulates production of irisin in muscles in cooperation with PPARα (Spiegelman 2013). The specific role of FNDC5/irisin in energy expenditure and differentiation of muscle and cardiac cells has also been described (Aydin et al. 2014; Rabiee et al. 2014).

Irisin is processed from FNDC5 and its proposed role in neuroprotection. a Schematic of FNDC5 structure showing the cleavage site that results in the processed form Irisin, which is proposed to have a wide spectrum of functions in various tissues. b FNDC5 and its proposed role in a pathway that promotes neuroprotection
Exercise has also been shown to have beneficial effects on brain performance (Cotman et al. 2007; Mattson 2012), and has been reported to result in improved outcomes in Alzheimer’s disease and Parkinson’s disease (Ahlskog 2011). Considering the brain’s dependence on glucose, it seemed plausible that PGC1α/FNDC5/irisin could also serve an essential role in mediating the effects of exercise on brain performance (Wrann et al. 2013). As described above, PGC1α has already been implicated in neurodegeneration (Lin et al. 2004; Zheng et al. 2010; Castillo-Quan 2011). Cerebellar purkinje cells in rat and mice also express irisin (Dun et al. 2013). Furthermore, the results from our group showed that FNDC5 plays an important role in neural specification of embryonic stem cells (Hashemi et al. 2013; Ghahrizjani et al. 2015). In this regard, it has been shown that neurogenesis in the hippocampus is regulated by irisin, and increased numbers of mouse hippocampal neurons are produced in the presence of irisin (Moon et al. 2013). Our work also indicated that overexpression of FNDC5 could enhance the overall rate of neural differentiation of embryonic stem cells (Forouzanfar et al. 2015). In 2013, Wrann et al. (2013) reported that FNDC5 expression was induced in the hippocampus by exercising through the FNDC5/PGC1α/BDNF pathway. Hippocampal neurons have a critical role in AD. Thus, these findings suggest that FNDC5 could serve as a neurogenic factor in the hippocampus and delay AD onset as well as other neurodegenerative disorders through exercise, so further studies are needed to validate links between exercise and irisin, before we consider its potential benefit to AD.
Brain-derived neurotrophic factor (BDNF) is known to promote brain development (Greenberg et al. 2009). BDNF also appears to be a critical mediator of the benefits of exercise on brain function, since exercise induces its expression in a number of brain regions, including the hippocampus (Cotman et al. 2007), which is an important site for memory and learning in the brain (Petzold et al. 2015).
How FNDC5 influences neuronal health and the beneficial effects of exercise remains to be elucidated. However, recent findings indicate that irisin triggers expression of BDNF (Wrann et al. 2013). The neuroprotective effects of BDNF through physical activity have been established (Mattson 2012; Phillips et al. 2014). BDNF, along with the other neurotrophins (NGF, neurotrophin-3, neurotrophin-4/5, neurotrophin-6 and neurotrophin-7), exerts its action through binding to tyrosine kinase receptors (Fiore et al. 2009). BDNF is expressed in the central and peripheral nervous system during fetal and postnatal development, and plays an important role in serotonergic, dopaminergic, noradrenergic and cholinergic neuronal specification (Dwivedi 2009). In addition, BDNF influences synaptic plasticity by intensification of long-term potentiation (LTP) (McAllister 2001). One of the protective mechanisms of neurons against toxicity is enhanced expression of BDNF, acting through tyrosine kinase receptor B (TrkB) (Binder and Scharfman 2004). TrkB is abundant in the hippocampus, amygdala and prefrontal cortex, where high expression of the BDNF is observed (Tapia-Arancibia et al. 2004). Activation of TrkB triggers various intracellular cascades including mitogen-activated protein kinase (MAPK), phosphatidylinositol-3-kinase (PI3K) or phospholipase C-γ (PLCγ) pathways (Huang and Reichardt 2001). The PI3K pathway has an important role in synaptic plasticity while MAPK triggers differentiation and neurogenesis in the nervous system (Reichardt 2006). Impairment of synaptic plasticity is a clinical feature of AD, HD and PD. The numbers of synapses decline in AD, and their degeneration has a close correlation with disease progression (Selkoe and Schenk 2003). Loss of synapses leads to impairment in hippocampal memory (Chapman et al. 1999). In HD, mHtt aggregation causes synaptic degeneration in the striatum and worsening of cognitive performance (Milnerwood and Raymond 2010). In addition, loss of motor and cognitive functions in PD patients has a correlation with synaptic decline in the striatum (Picconi et al. 2012). In the patients’ brains of these neurodegenerative disorders, BDNF levels and its transcripts are significantly reduced (Tapia-Arancibia et al. 2008). Incidentally, in stroke BDNF secretion is altered, and elevation of BDNF was detected in epilepsy and autism (Connolly et al. 2006; Tsai 2006). The presence of the TrkB in cortical neurons decreases in AD, and this phenomenon suppresses the effectiveness of BDNF (Tong et al. 2004). On the other hand, recent studies have shown that BDNF treatment protected striatal neurons against neurotoxin, 6-hydroxy dopamine [6-OHDA and MPTP in PD models (Sun et al. 2005)]. A neuroprotective feature of BDNF in neurodegenerative disorders includes Bcl2/Bax regulation. BDNF acts as an anti-apoptotic protein by up-regulating activity of the anti-apoptotic protein Bcl2, while decreasing the pro-apoptotic Bax protein, both of which act at mitochondria (Schäbitz et al. 2000).
In addition to the neuroprotective roles associated with BDNF levels, SNPs in the coding sequence of its gene are correlated with neurological disorders. A Val to Met residue substitution at position 66 in the BDNF gene leads to vulnerability of neurons to disorders such as Alzheimer’s disease (Ventriglia et al. 2002), depression (Sen et al. 2003) and PD (Momose et al. 2002). In addition to SNPs in BDNF, considering the fact that BDNF is produced as a precursor protein, SNPs related to trafficking and secretion of BDNF in neurons (Chen et al. 2004) leads to cognitive impairments due to synaptic degeneration (Desai et al. 1999). As mentioned above, one of the pathways downstream of BDNF/TrkB is MAPK, which in turn phosphorylates CREB as its target (Pizzorusso et al. 2000). Phosphorylated CREB is a transcription factor and exerts important roles in neuroprotection, neurotransmission and learning and memory (Carlezon et al. 2005). CREB also controls PGC1α expression (Volakakis et al. 2010). Such a positive feedback has been reported in the neocortex and striatum (Giampà et al. 2013). This appears to be the molecular mechanism underlying improvement of neurological disorders following exercise in depression, epilepsy, stroke and Alzheimer’s disease (Kobilo et al. 2011).
Irisin was discovered 3 years ago as a factor that is induced by exercise and converts white adipose tissue to brown adipose in mice (Boström et al. 2012). However, concern over the accurate detection of irisin peptide arose immediately (Erickson 2013), and a recent report challenges the conservation of the physiological effects of irisin in humans (Albrecht et al. 2015). Recent studies showed that the start codon in human FNDC5 is a non-canonical AUA codon (Erickson 2013; Raschke et al. 2013), which occurs at a low frequency in the human genome and is less efficient at translation (Ivanov et al. 2011). The result is a much lower level of expression of FNDC5 in humans compared to mice (Raschke et al. 2013). Moreover, the first ATG codon in human FNDC5 is located 76 codons 3′ to this start codon and, if utilized, generates a frameshift and premature truncation that would preclude synthesis of irisin (Raschke et al. 2013). In addition, they showed that recombinant irisin could not differentiate preadipocyte to browning fat (Raschke et al. 2013). Moreover, the antibody previously used for irisin detection was specific to the C terminus of FNDC5, which is not present in the cleaved and secreted irisin (Erickson 2013). In a more recent report, it was revealed that the commonly used commercial ELISA kit used for irisin detection had polyclonal antibodies that did not detect synthetic irisin and instead detected other non-specific serum proteins, producing false-positive signals and casting further doubts on recent findings (Albrecht et al. 2015). This issue is certainly not resolved, as another study used a different antibody combined with mass spectrometry to detect irisin in human serum and showed that irisin levels increased after exercise or cold exposure (Lee et al. 2014). With these recent reports raising questions about the roles of irisin, it is imperative to further validate and reach a consensus regarding this potentially important myokine.
Prospective Studies
Recent studies have shown that exercise can significantly benefit patients with neurological disorders to regain lost activities. Whether FNDC5/irisin plays a critical role in this phenomenon remains an important issue to resolve. It is important to establish whether irisin, having emerged very recently as a key regulator of neuronal survival and development, sits in an established pathway for cellular activities involving PGC1 and BDNF that center around neuronal health and the positive impact that exercise plays on aging-related pathologies. Moreover, our group recently showed that FNDC5 promotes neural development and differentiation of embryonic stem cells (Hashemi et al. 2013), a perhaps separate role from the controversial one proposed for irisin in neuronal metabolic health. Also, numerous studies have shown that exercise protects the brain against neurodegeneration and the progression and onset of neurological disorders such as AD and PD. Since PGC1α/FNDC5/BDNF was discerned as a critical pathway in neuroprotection elicited by exercise, irisin could serve as a protective factor by its activation of BDNF expression. Based on previous reports, BDNF is secreted from neurons and initiates many intracellular signaling events through TrkB receptors. These pathways have positive feedback on CREB activities and lead to enhancement in PGC1α expression. The net result is improvement in brain performance. Further evaluation will establish whether the PGC1α/FNDC5/BDNF pathway will be an effective target for therapeutic interventions in neurological diseases and aging. If the promise of irisin holds, it has the therapeutic potential to ameliorate the effects of aging and CNS disorders.
Abbreviations
- 6-OHDA:
-
6-Hydroxy dopamine
- Aβ:
-
Amyloid beta
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- ATP:
-
Adenosine triphosphate
- BDNF:
-
Brain-derived neurotrophic factor
- CNS:
-
Central nervous system
- CREB:
-
cAMP-responsive element binding protein
- DHA:
-
Docosahexaenoicacid
- ERRα:
-
Estrogen-related receptor alpha
- ETC:
-
Electron transport chain
- FA:
-
Friedreich’s ataxia
- FNDC5:
-
Fibronectin type III domain-containing 5
- GPx:
-
Glutathione peroxidase
- HD:
-
Huntington’s disease
- KSS:
-
Kearns–Sayre syndrome
- LTP:
-
Long-term potentiation
- MAO:
-
Monoamine oxidase
- MAPK:
-
Mitogen-activated protein kinase
- MELSAS:
-
Mitochondrial encephalopathy lactic acidosis and strokes
- mHtt:
-
Mutant huntingtin protein
- Mn-SOD:
-
Manganese superoxide dismutase
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NMDA:
-
N-Methyl-d-aspartate
- NRF:
-
Nuclear respiratory factor
- OXPHOS:
-
Oxidative phosphorylation system
- PARIS:
-
Parkin-interacting substrate
- PBDs:
-
Peroxisome biogenesis disorders
- PD:
-
Parkinson’s disease
- PEDs:
-
Peroxisomal enzyme deficiencies
- PEP:
-
Peroxisomal protein
- PGC1α:
-
Peroxisome proliferator-activated receptor γ co-activator α
- PI3K:
-
Phosphatidyl inositol-3-kinase
- PLCγ:
-
Phospholipase C-γ
- PPARα:
-
Peroxisome proliferator receptor alpha
- PUFAs:
-
Polyunsaturated fatty acids
- ROS:
-
Reactive oxygen species
- SIRT1:
-
Sirtunin1
- SNP:
-
Single-nucleotide polymorphism
- SOD:
-
Superoxide dismutase
- TAF4:
-
Transcription initiation factor 4
- TFAM:
-
Mitochondrial transcription factor A
- TR:
-
Thyroid receptor
- TrkB:
-
Tyrosine kinase receptor B
- VDAC:
-
Voltage-dependent anion channels
References
Ahlskog, J. E. (2011). Cheaper, simpler, and better: Tips for treating seniors with Parkinson disease. Mayo Clinic Proceedings, 86, 1211–1216.
Albrecht, E., Norheim, F., Thiede, B., Holen, T., Ohashi, T., Schering, L., et al. (2015). Irisin—A myth rather than an exercise-inducible myokine. Scientific reports, 5, 8889.
Amadoro, G., Corsetti, V., Florenzano, F., Atlante, A., Bobba, A., Nicolin, V., et al. (2014). Morphological and bioenergetic demands underlying the mitophagy in post-mitotic neurons: the pink-parkin pathway. Front Aging Neurosci, 6, 18.
Andreyev, A. Y., Kushnareva, Y. E., Murphy, A. N., & Starkov, A. A. (2015). Mitochondrial ROS metabolism: 10 years later. Biochemistry (Mosc), 80, 517–531.
Antonenkov, V. D., Grunau, S., Ohlmeier, S., & Hiltunen, J. K. (2010). Peroxisomes are oxidative organelles. Antioxidants and Redox Signaling, 13, 525–537.
Ayala, A., Muñoz, M. F., & Argüelles, S. (2014). Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Medicine and Cellular Longevity, 2014, 360438.
Aydin, S., Kuloglu, T., Eren, M. N., Celik, A., Yilmaz, M., Kalayci, M., et al. (2014). Cardiac, skeletal muscle and serum irisin responses to with or without water exercise in young and old male rats: Cardiac muscle produces more irisin than skeletal muscle. Peptides, 52, 68–73.
Baes, M., & Aubourg, P. (2009). Peroxisomes, myelination, and axonal integrity in the CNS. Neuroscientist, 15, 367–379.
Baes, M., & Van Veldhoven, P. P. (2006). Generalised and conditional inactivation of Pex genes in mice. Biochimica et Biophysica Acta, 1763, 1785–1793.
Baines, C. P. (2010). Role of the mitochondrion in programmed necrosis. Frontiers in Physiology, 1, 156.
Balaban, R. S., Nemoto, S., & Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell, 120, 483–495.
Bennett, S. A., Valenzuela, N., Xu, H., Franko, B., Fai, S., & Figeys, D. (2013). Using neurolipidomics to identify phospholipid mediators of synaptic (dys)function in Alzheimer’s Disease. Frontiers in Physiology, 4, 168.
Binder, D. K., & Scharfman, H. E. (2004). Brain-derived neurotrophic factor. Growth Factors, 22, 123–131.
Blesa, J., Trigo-Damas, I., Quiroga-Varela, A., Jackson-Lewis, V.R. (2015). Oxidative stress and Parkinson’s disease. Front Neuroanat, 9, 91.
Boström, P., Wu, J., Jedrychowski, M. P., Korde, A., Ye, L., Lo, J. C., et al. (2012). A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature, 481, 463–468.
Bordt, E.A.,& Polster, B.M. (2014). NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: a bipartisan affair? Free Radic Biol Med, 76, 34–46.
Braverman, N. E., & Moser, A. B. (2012). Functions of plasmalogen lipids in health and disease. Biochimica et Biophysica Acta, 1822, 1442–1452.
Calon, F., Lim, G. P., Yang, F., Morihara, T., Teter, B., Ubeda, O., et al. (2004). Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron, 43, 633–645.
Carlezon, W. A., Duman, R. S., & Nestler, E. J. (2005). The many faces of CREB. Trends in Neurosciences, 28, 436–445.
Castillo-Quan, J. I. (2011). Parkin’ control: Regulation of PGC-1α through PARIS in Parkinson’s disease. Disease Models and Mechanisms, 4, 427–429.
Chapman, P. F., White, G. L., Jones, M. W., Cooper-Blacketer, D., Marshall, V. J., Irizarry, M., et al. (1999). Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nature Neuroscience, 2, 271–276.
Chaturvedi, R. K., & Beal, M. F. (2013). Mitochondrial diseases of the brain. Free Radical Biology and Medicine, 63, 1–29.
Chen, H., McCaffery, J. M., & Chan, D. C. (2007). Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell, 130, 548–562.
Chen, Z. Y., Patel, P. D., Sant, G., Meng, C. X., Teng, K. K., Hempstead, B. L., & Lee, F. S. (2004). Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. Journal of Neuroscience, 24, 4401–4411.
Ciccone, S., Maiani, E., Bellusci, G., Diederich, M., & Gonfloni, S. (2013). Parkinson’s disease: A complex interplay of mitochondrial DNA alterations and oxidative stress. International Journal of Molecular Sciences, 14, 2388–2409.
Clark, J., Reddy, S., Zheng, K., Betensky, R. A., & Simon, D. K. (2011). Association of PGC-1alpha polymorphisms with age of onset and risk of Parkinson’s disease. BMC Medical Genetics, 12, 69.
Connolly, A. M., Chez, M., Streif, E. M., Keeling, R. M., Golumbek, P. T., Kwon, J. M., et al. (2006). Brain-derived neurotrophic factor and autoantibodies to neural antigens in sera of children with autistic spectrum disorders, Landau–Kleffner syndrome, and epilepsy. Biological Psychiatry, 59, 354–363.
Conquer, J. A., Tierney, M. C., Zecevic, J., Bettger, W. J., & Fisher, R. H. (2000). Fatty acid analysis of blood plasma of patients with Alzheimer’s disease, other types of dementia, and cognitive impairment. Lipids, 35, 1305–1312.
Corona, J. C., & Duchen, M. R. (2015). PPARγ and PGC-1α as therapeutic targets in Parkinson’s. Neurochemical Research, 40, 308–316.
Cotman, C. W., Berchtold, N. C., & Christie, L. A. (2007). Exercise builds brain health: Key roles of growth factor cascades and inflammation. Trends in Neurosciences, 30, 464–472.
Cui, L., Jeong, H., Borovecki, F., Parkhurst, C. N., Tanese, N., & Krainc, D. (2006). Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell, 127, 59–69.
del Río, L. A., Corpas, F. J., Sandalio, L. M., Palma, J. M., Gómez, M., & Barroso, J. B. (2002). Reactive oxygen species, antioxidant systems and nitric oxide in peroxisomes. Journal of Experimental Botany, 53, 1255–1272.
Desai, N. S., Rutherford, L. C., & Turrigiano, G. G. (1999). BDNF regulates the intrinsic excitability of cortical neurons. Learning and Memory, 6, 284–291.
Dun, S. L., Lyu, R. M., Chen, Y. H., Chang, J. K., Luo, J. J., & Dun, N. J. (2013). Irisin-immunoreactivity in neural and non-neural cells of the rodent. Neuroscience, 240, 155–162.
Dwivedi, Y. (2009). Brain-derived neurotrophic factor: Role in depression and suicide. Neuropsychiatric Disease and Treatment, 5, 433–449.
Edmondson, D. E. (2014). Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: biological implications. Current Pharmaceutical Design, 20, 155–160.
Eichner, L. J., & Giguère, V. (2011). Estrogen related receptors (ERRs): A new dawn in transcriptional control of mitochondrial gene networks. Mitochondrion, 11, 544–552.
Erickson, H. P. (2013). Irisin and FNDC5 in retrospect: An exercise hormone or a transmembrane receptor? Adipocyte, 2, 289–293.
Evans, R. M. (2005). The nuclear receptor superfamily: A rosetta stone for physiology. Molecular Endocrinology, 19, 1429–1438.
Exner, N., Lutz, A.K., Haass, C., Winklhofer, K.F. (2012). Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J, 31(14), 3038–3062.
Farrar, G. J., Chadderton, N., Kenna, P. F., & Millington-Ward, S. (2013). Mitochondrial disorders: Aetiologies, models systems, and candidate therapies. Trends in Genetics, 29, 488–497.
Ferreiro, E., Baldeiras, I., Ferreira, I. L., Costa, R. O., Rego, A. C., Pereira, C. F., & Oliveira, C. R. (2012). Mitochondrial- and endoplasmic reticulum-associated oxidative stress in Alzheimer’s disease: From pathogenesis to biomarkers. International Journal of Cell Biology, 2012, 735206.
Ferrer-Martínez, A., Ruiz-Lozano, P., & Chien, K. R. (2002). Mouse PeP: A novel peroxisomal protein linked to myoblast differentiation and development. Developmental Dynamics, 224, 154–167.
Fiore, M., Chaldakov, G. N., & Aloe, L. (2009). Nerve growth factor as a signaling molecule for nerve cells and also for the neuroendocrine-immune systems. Reviews in the Neurosciences, 20, 133–145.
Forouzanfar, M., Rabiee, F., Ghaedi, K., Beheshti, S., Tanhaei, S., Shoaraye Nejati, A., et al. (2015). Fndc5 overexpression facilitated neural differentiation of mouse embryonic stem cells. Cell Biology International, 39, 629–637.
Fransen, M., Nordgren, M., Wang, B., & Apanasets, O. (2012). Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochimica et Biophysica Acta, 1822, 1363–1373.
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nature Cell Biology, 4, 160–164.
Ghahrizjani, F. A., Ghaedi, K., Salamian, A., Tanhaei, S., Nejati, A. S., Salehi, H., et al. (2015). Enhanced expression of FNDC5 in human embryonic stem cell-derived neural cells along with relevant embryonic neural tissues. Gene, 557, 123–129.
Giampà, C., Montagna, E., Dato, C., Melone, M. A., Bernardi, G., & Fusco, F. R. (2013). Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PLoS One, 8, e64037.
Ginsberg, L., Rafique, S., Xuereb, J. H., Rapoport, S. I., & Gershfeld, N. L. (1995). Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer’s disease brain. Brain Research, 698, 223–226.
Goncalves, R. L., Rothschild, D. E., Quinlan, C. L., Scott, G. K., Benz, C. C., & Brand, M. D. (2014). Sources of superoxide/H2O2 during mitochondrial proline oxidation. Redox Biology, 2, 901–909.
Green, D. R., & Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science, 305, 626–629.
Greenberg, M. E., Xu, B., Lu, B., & Hempstead, B. L. (2009). New insights in the biology of BDNF synthesis and release: implications in CNS function. Journal of Neuroscience, 29, 12764–12767.
Guo, C., Sun, L., Chen, X., & Zhang, D. (2013). Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regeneration Research, 8, 2003–2014.
Haas, R. H., Parikh, S., Falk, M. J., Saneto, R. P., Wolf, N. I., Darin, N., et al. (2008). The in-depth evaluation of suspected mitochondrial disease. Molecular Genetics and Metabolism, 94, 16–37.
Han, J.Y., Kim, J.S., Son, J.H. (2014). Mitochondrial homeostasis molecules: regulation by a trio of recessive Parkinson's disease genes. Exp Neurobiol, 23(4), 345–351.
Handschin, C. (2009). The biology of PGC-1α and its therapeutic potential. Trends in Pharmacological Sciences, 30, 322–329.
Hashemi, M. S., Ghaedi, K., Salamian, A., Karbalaie, K., Emadi-Baygi, M., Tanhaei, S., et al. (2013). Fndc5 knockdown significantly decreased neural differentiation rate of mouse embryonic stem cells. Neuroscience, 231, 296–304.
Herben-Dekker, M., van Oostrom, J. C., Roos, R. A., Jurgens, C. K., Witjes-Ané, M. N., Kremer, H. P., et al. (2014). Striatal metabolism and psychomotor speed as predictors of motor onset in Huntington’s disease. Journal of Neurology, 261, 1387–1397.
Huang, E. J., & Reichardt, L. F. (2001). Neurotrophins: Roles in neuronal development and function. Annual Review of Neuroscience, 24, 677–736.
Hwang, O. (2013). Role of oxidative stress in Parkinson’s disease. Exp Neurobiol, 22(1), 11–17.
Ivanov, I.P., Firth, A.E., Michel, A.M., Atkins, J.F., Baranov, P.V. (2011). Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res, 39(10), 4220–4234.
Ivashchenko, O., Van Veldhoven, P. P., Brees, C., Ho, Y. S., Terlecky, S. R., & Fransen, M. (2011). Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Molecular Biology of the Cell, 22, 1440–1451.
Jin, Y. N., & Johnson, G. V. (2010). The interrelationship between mitochondrial dysfunction and transcriptional dysregulation in Huntington disease. Journal of Bioenergetics and Biomembranes, 42, 199–205.
Johnson, W. T., Johnson, L. A., & Lukaski, H. C. (2005). Serum superoxide dismutase 3 (extracellular superoxide dismutase) activity is a sensitive indicator of Cu status in rats. Journal of Nutritional Biochemistry, 16, 682–692.
Johri, A., & Beal, M. F. (2012). Mitochondrial dysfunction in neurodegenerative diseases. Journal of Pharmacology and Experimental Therapeutics, 342, 619–630.
Kalmijn, S., Launer, L. J., Ott, A., Witteman, J. C., Hofman, A., & Breteler, M. M. (1997). Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Annals of Neurology, 42, 776–782.
Kassmann, C. M., Lappe-Siefke, C., Baes, M., Brügger, B., Mildner, A., Werner, H. B., et al. (2007). Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nature Genetics, 39, 969–976.
Katsouri, L., Parr, C., Bogdanovic, N., Willem, M., & Sastre, M. (2011). PPARγ co-activator-1α (PGC-1α) reduces amyloid-β generation through a PPARγ-dependent mechanism. Journal of Alzheimer’s Disease, 25, 151–162.
Kelly, D. P., & Scarpulla, R. C. (2004). Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes and Development, 18, 357–368.
Keogh, M.J., Chinnery, P.F. (2015). Mitochondrial DNA mutations in neurodegeneration. Biochim Biophys Acta, 1847(11), 1401–1411.
Kobilo, T., Liu, Q. R., Gandhi, K., Mughal, M., Shaham, Y., & van Praag, H. (2011). Running is the neurogenic and neurotrophic stimulus in environmental enrichment. Learning and Memory, 18, 605–609.
Koepke, J. I., Nakrieko, K. A., Wood, C. S., Boucher, K. K., Terlecky, L. J., Walton, P. A., & Terlecky, S. R. (2007). Restoration of peroxisomal catalase import in a model of human cellular aging. Traffic, 8, 1590–1600.
Kotiadis, V. N., Duchen, M. R., & Osellame, L. D. (2014). Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochimica et Biophysica Acta, 1840, 1254–1265.
Kou, J., Kovacs, G. G., Höftberger, R., Kulik, W., Brodde, A., Forss-Petter, S., et al. (2011). Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathologica, 122, 271–283.
Kressler, D., Schreiber, S. N., Knutti, D., & Kralli, A. (2002). The PGC-1-related protein PERC is a selective coactivator of estrogen receptor alpha. Journal of Biological Chemistry, 277, 13918–13925.
Lee, P., Linderman, J. D., Smith, S., Brychta, R. J., Wang, J., Idelson, C., et al. (2014). Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metabolism, 19, 302–309.
Legros, F., Malka, F., Frachon, P., Lombès, A., & Rojo, M. (2004). Organization and dynamics of human mitochondrial DNA. Journal of Cell Science, 117, 2653–2662.
Lezi, E., & Swerdlow, R. H. (2012). Mitochondria in neurodegeneration. Advances in Experimental Medicine and Biology, 942, 269–286.
Lin, M. T., & Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443, 787–795.
Lin, J., Puigserver, P., Donovan, J., Tarr, P., & Spiegelman, B. M. (2002a). Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. Journal of Biological Chemistry, 277, 1645–1648.
Lin, M.Y., & Sheng, Z.H. (2015). Regulation of mitochondrial transport in neurons. Exp Cell Res, 334(1), 35–44.
Lin, J., Wu, P. H., Tarr, P. T., Lindenberg, K. S., St-Pierre, J., Zhang, C. Y., et al. (2004). Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell, 119, 121–135.
Lin, J., Wu, H., Tarr, P. T., Zhang, C. Y., Wu, Z., Boss, O., et al. (2002b). Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature, 418, 797–801.
Lodhi, I. J., & Semenkovich, C. F. (2014). Peroxisomes: A nexus for lipid metabolism and cellular signaling. Cell Metabolism, 19, 380–392.
Lopez-Huertas, E., Charlton, W. L., Johnson, B., Graham, I. A., & Baker, A. (2000). Stress induces peroxisome biogenesis genes. EMBO Journal, 19, 6770–6777.
Mailloux, R. J. (2015). Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biology, 4, 381–398.
Martin, E., Betuing, S., Pagès, C., Cambon, K., Auregan, G., Deglon, N., et al. (2011). Mitogen- and stress-activated protein kinase 1-induced neuroprotection in Huntington’s disease: role on chromatin remodeling at the PGC-1-alpha promoter. Human Molecular Genetics, 20, 2422–2434.
Mattson, M. P. (2012). Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metabolism, 16, 706–722.
McAllister, A. K. (2001). Neurotrophins and neuronal differentiation in the central nervous system. Cellular and Molecular Life Sciences, 58, 1054–1060.
McGill, J. K., & Beal, M. F. (2006). PGC-1alpha, a new therapeutic target in Huntington’s disease? Cell, 127, 465–468.
Milnerwood, A. J., & Raymond, L. A. (2010). Early synaptic pathophysiology in neurodegeneration: Insights from Huntington’s disease. Trends in Neurosciences, 33, 513–523.
Mochel, F., & Haller, R. G. (2011). Energy deficit in Huntington disease: Why it matters. Journal of Clinical Investigation, 121, 493–499.
Momose, Y., Murata, M., Kobayashi, K., Tachikawa, M., Nakabayashi, Y., Kanazawa, I., & Toda, T. (2002). Association studies of multiple candidate genes for Parkinson’s disease using single nucleotide polymorphisms. Annals of Neurology, 51, 133–136.
Monteiro-Junior, R. S., Cevada, T., Oliveira, B. R., Lattari, E., Portugal, E. M., Carvalho, A., & Deslandes, A. C. (2015). We need to move more: Neurobiological hypotheses of physical exercise as a treatment for Parkinson’s disease. Medical Hypotheses,. doi:10.1016/j.mehy.2015.07.011
Moon, H. S., Dincer, F., & Mantzoros, C. S. (2013). Pharmacological concentrations of irisin increase cell proliferation without influencing markers of neurite outgrowth and synaptogenesis in mouse H19-7 hippocampal cell lines. Metabolism, 62, 1131–1136.
Moreira, P. I., Santos, M. S., Seiça, R., & Oliveira, C. R. (2007). Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. Journal of the Neurological Sciences, 257, 206–214.
Mudò, G., Mäkelä, J., Di Liberto, V., Tselykh, T. V., Olivieri, M., Piepponen, P., et al. (2012). Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cellular and Molecular Life Sciences, 69, 1153–1165.
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochemical Journal, 417, 1–13.
Nagley, P., Higgins, G. C., Atkin, J. D., & Beart, P. M. (2010). Multifaceted deaths orchestrated by mitochondria in neurones. Biochimica et Biophysica Acta, 1802, 167–185.
Nicholls, D. G. (2008). Oxidative stress and energy crises in neuronal dysfunction. Annals of the New York Academy of Sciences, 1147, 53–60.
Nixon, R. A., Cataldo, A. M., & Mathews, P. M. (2000). The endosomal–lysosomal system of neurons in Alzheimer’s disease pathogenesis: A review. Neurochemical Research, 25, 1161–1172.
Obulesu, M., & Lakshmi, M. J. (2014). Apoptosis in Alzheimer’s disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochemical Research, 39, 2301–2312.
Ostadsharif, M., Ghaedi, K., Hossein Nasr-Esfahani, M., Mojbafan, M., Tanhaie, S., Karbalaie, K., & Baharvand, H. (2011). The expression of peroxisomal protein transcripts increased by retinoic acid during neural differentiation. Differentiation, 81, 127–132.
Outeiro, T. F., Marques, O., & Kazantsev, A. (2008). Therapeutic role of sirtuins in neurodegenerative disease. Biochimica et Biophysica Acta, 1782, 363–369.
Payne, B. A., & Chinnery, P. F. (2015). Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim Biophys Acta, 1847(11), 1347–1353.
Petzold, A., Psotta, L., Brigadski, T., Endres, T., & Lessmann, V. (2015). Chronic BDNF deficiency leads to an age-dependent impairment in spatial learning. Neurobiology of Learning and Memory, 120, 52–60.
Phillips, C., Baktir, M. A., Srivatsan, M., & Salehi, A. (2014). Neuroprotective effects of physical activity on the brain: a closer look at trophic factor signaling. Frontiers in Cellular Neuroscience, 8, 170.
Picconi, B., Piccoli, G., & Calabresi, P. (2012). Synaptic dysfunction in Parkinson’s disease. Advances in Experimental Medicine and Biology, 970, 553–572.
Picone, P., Nuzzo, D., Caruana, L., Scafidi, V., Di Carlo, M. (2014). Mitochondrial dysfunction: different routes to Alzheimer's disease therapy. Oxid Med Cell Longev, 2014, 780179.
Pizzorusso, T., Ratto, G. M., Putignano, E., & Maffei, L. (2000). Brain-derived neurotrophic factor causes cAMP response element-binding protein phosphorylation in absence of calcium increases in slices and cultured neurons from rat visual cortex. Journal of Neuroscience, 20, 2809–2816.
Przedborski, S., Vila, M., & Jackson-Lewis, V. (2003). Neurodegeneration: What is it and where are we? Journal of Clinical Investigation, 111, 3–10.
Puigserver, P., Wu, Z., Park, C. W., Graves, R., Wright, M., & Spiegelman, B. M. (1998). A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell, 92, 829–839.
Qin, W., Haroutunian, V., Katsel, P., Cardozo, C. P., Ho, L., Buxbaum, J. D., & Pasinetti, G. M. (2009). PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Archives of Neurology, 66, 352–361.
Qin, W., Yang, T., Ho, L., Zhao, Z., Wang, J., Chen, L., et al. (2006). Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. Journal of Biological Chemistry, 281, 21745–21754.
Rabiee, F., Forouzanfar, M., Ghazvini Zadegan, F., Tanhaei, S., Ghaedi, K., Motovali Bashi, M., et al. (2014). Induced expression of Fndc5 significantly increased cardiomyocyte differentiation rate of mouse embryonic stem cells. Gene, 551, 127–137.
Radak, Z., Chung, H. Y., & Goto, S. (2005). Exercise and hormesis: Oxidative stress-related adaptation for successful aging. Biogerontology, 6, 71–75.
Raschke, S., Elsen, M., Gassenhuber, H., Sommerfeld, M., Schwahn, U., Brockmann, B., et al. (2013). Evidence against a beneficial effect of irisin in humans. PLoS ONE, 8, e73680.
Reddy, P. H. (2009). Role of mitochondria in neurodegenerative diseases: Mitochondria as a therapeutic target in Alzheimer’s disease. CNS Spectrums, 14, 8–13. discussion 16–18.
Reichardt, L. F. (2006). Neurotrophin-regulated signalling pathways. Philosophical Transactions of the Royal Society of London. Series B, Biological sciences, 361, 1545–1564.
Rinnerthaler, M., Bischof, J., Streubel, M. K., Trost, A., & Richter, K. (2015). Oxidative stress in aging human skin. Biomolecules, 5, 545–589.
Rossignol, D. A., & Frye, R. E. (2012). A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Molecular Psychiatry, 17, 389–401.
Ruetenik, A., & Barrientos, A. (2015) Dietary restriction, mitochondrial function and aging: From yeast to humans. Biochim Biophys Acta, 1847(11), 1434–1447.
Santos, M. J., Quintanilla, R. A., Toro, A., Grandy, R., Dinamarca, M. C., Godoy, J. A., & Inestrosa, N. C. (2005). Peroxisomal proliferation protects from beta-amyloid neurodegeneration. Journal of Biological Chemistry, 280, 41057–41068.
Scarpulla, R. C. (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiological Reviews, 88, 611–638.
Schäbitz, W. R., Sommer, C., Zoder, W., Kiessling, M., Schwaninger, M., & Schwab, S. (2000). Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke, 31, 2212–2217.
Schon, E. A., DiMauro, S., Hirano, M., & Gilkerson, R. W. (2010). Therapeutic prospects for mitochondrial disease. Trends in Molecular Medicine, 16, 268–276.
Schreiber, S. N., Emter, R., Hock, M. B., Knutti, D., Cardenas, J., Podvinec, M., et al. (2004). The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proceedings of the National Academy of Sciences USA, 101, 6472–6477.
Selkoe, D. J., & Schenk, D. (2003). Alzheimer’s disease: Molecular understanding predicts amyloid-based therapeutics. Annual Review of Pharmacology and Toxicology, 43, 545–584.
Sen, S., Nesse, R. M., Stoltenberg, S. F., Li, S., Gleiberman, L., Chakravarti, A., et al. (2003). A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology, 28, 397–401.
Sharon, R., Bar-Joseph, I., Frosch, M. P., Walsh, D. M., Hamilton, J. A., & Selkoe, D. J. (2003a). The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron, 37, 583–595.
Sharon, R., Bar-Joseph, I., Mirick, G. E., Serhan, C. N., & Selkoe, D. J. (2003b). Altered fatty acid composition of dopaminergic neurons expressing alpha-synuclein and human brains with alpha-synucleinopathies. Journal of Biological Chemistry, 278, 49874–49881.
Sheikh, F. G., Pahan, K., Khan, M., Barbosa, E., & Singh, I. (1998). Abnormality in catalase import into peroxisomes leads to severe neurological disorder. Proceedings of the National Academy of Sciences, 95, 2961–2966.
Sheng, B., Wang, X., Su, B., Lee, H. G., Casadesus, G., Perry, G., & Zhu, X. (2012). Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. Journal of Neurochemistry, 120, 419–429.
Shin, J. H., Ko, H. S., Kang, H., Lee, Y., Lee, Y. I., Pletinkova, O., et al. (2011). PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell, 144, 689–702.
Shutt, T.E.,& McBride, H.M. (2013). Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta, 1833(2), 417–424.
Song, W., Chen, J., Petrilli, A., Liot, G., Klinglmayr, E., Zhou, Y., et al. (2011). Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nature Medicine, 17, 377–382.
Spiegelman, B. M. (2013). Banting Lecture 2012 Regulation of adipogenesis: Toward new therapeutics for metabolic disease. Diabetes, 62, 1774–1782.
Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., & Moser, H. W. (2006). Peroxisome biogenesis disorders. Biochimica et Biophysica Acta, 1763, 1733–1748.
St-Pierre, J., Drori, S., Uldry, M., Silvaggi, J. M., Rhee, J., Jäger, S., et al. (2006). Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell, 127, 397–408.
Sun, M., Kong, L., Wang, X., Lu, X. G., Gao, Q., & Geller, A. I. (2005). Comparison of the capability of GDNF, BDNF, or both, to protect nigrostriatal neurons in a rat model of Parkinson’s disease. Brain Research, 1052, 119–129.
Tapia-Arancibia, L., Aliaga, E., Silhol, M., & Arancibia, S. (2008). New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Research Reviews, 59, 201–220.
Tapia-Arancibia, L., Rage, F., Givalois, L., & Arancibia, S. (2004). Physiology of BDNF: Focus on hypothalamic function. Frontiers in Neuroendocrinology, 25, 77–107.
Tillement, L., Lecanu, L., & Papadopoulos, V. (2011). Alzheimer’s disease: Effects of β-amyloid on mitochondria. Mitochondrion, 11, 13–21.
Tong, L., Balazs, R., Thornton, P. L., & Cotman, C. W. (2004). Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. Journal of Neuroscience, 24, 6799–6809.
Trempe, J.F., & Fon, E.A. (2013). Structure and Function of Parkin, PINK1, and DJ-1, the Three Musketeers of Neuroprotection. Front Neurol, 4, 38.
Tretter, L., Sipos, I., & Adam-Vizi, V. (2004). Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson’s disease. Neurochemical Research, 29, 569–577.
Tsai, S. J. (2006). TrkB partial agonists: potential treatment strategy for epilepsy, mania, and autism. Medical Hypotheses, 66, 173–175.
Tsunemi, T., Ashe, T. D., Morrison, B. E., Soriano, K. R., Au, J., Roque, R. A., et al. (2012). PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Science Translational Medicine, 4, 142ra197.
van der Valk, P., Gille, J. J., Oostra, A. B., Roubos, E. W., Sminia, T., & Joenje, H. (1985). Characterization of an oxygen-tolerant cell line derived from Chinese hamster ovary. Antioxygenic enzyme levels and ultrastructural morphometry of peroxisomes and mitochondria. Cell and Tissue Research, 239, 61–68.
Vega, R. B., Huss, J. M., & Kelly, D. P. (2000). The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Molecular and Cellular Biology, 20, 1868–1876.
Ventriglia, M., Bocchio Chiavetto, L., Benussi, L., Binetti, G., Zanetti, O., Riva, M. A., & Gennarelli, M. (2002). Association between the BDNF 196 A/G polymorphism and sporadic Alzheimer’s disease. Molecular Psychiatry, 7, 136–137.
Vila, M., & Przedborski, S. (2003). Targeting programmed cell death in neurodegenerative diseases. Nature Reviews Neuroscience, 4, 365–375.
Villena, J. A. (2015). New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS Journal, 282, 647–672.
Volakakis, N., Kadkhodaei, B., Joodmardi, E., Wallis, K., Panman, L., Silvaggi, J., et al. (2010). NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proceedings of the National Academy of Sciences USA, 107, 12317–12322.
Wallace, D. C., & Fan, W. (2009). The pathophysiology of mitochondrial disease as modeled in the mouse. Genes and Development, 23, 1714–1736.
Wanders, R. J., & Waterham, H. R. (2005). Peroxisomal disorders I: Biochemistry and genetics of peroxisome biogenesis disorders. Clinical Genetics, 67, 107–133.
Wanders, R. J., & Waterham, H. R. (2006). Peroxisomal disorders: The single peroxisomal enzyme deficiencies. Biochimica et Biophysica Acta, 1763, 1707–1720.
Wang, R., Li, J. J., Diao, S., Kwak, Y. D., Liu, L., Zhi, L., et al. (2013). Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metabolism, 17, 685–694.
Wenz, T. (2009). PGC-1alpha activation as a therapeutic approach in mitochondrial disease. IUBMB Life, 61, 1051–1062.
Wenz, T. (2011). Mitochondria and PGC-1α in aging and age-associated diseases. Journal of Aging Research, 2011, 810619.
West, A. P., Shadel, G. S., & Ghosh, S. (2011). Mitochondria in innate immune responses. Nature Reviews Immunology, 11, 389–402.
Weydt, P., Soyal, S. M., Gellera, C., Didonato, S., Weidinger, C., Oberkofler, H., et al. (2009). The gene coding for PGC-1alpha modifies age at onset in Huntington’s Disease. Molecular neurodegeneration, 4, 3.
Winterbourn, C. C. (1995). Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicology Letters, 82–83, 969–974.
Witte, M. E., Geurts, J. J., de Vries, H. E., van der Valk, P., & van Horssen, J. (2010). Mitochondrial dysfunction: A potential link between neuroinflammation and neurodegeneration? Mitochondrion, 10, 411–418.
Wood, C. S., Koepke, J. I., Teng, H., Boucher, K. K., Katz, S., Chang, P., et al. (2006). Hypocatalasemic fibroblasts accumulate hydrogen peroxide and display age-associated pathologies. Traffic, 7, 97–107.
Wrann, C. D., White, J. P., Salogiannnis, J., Laznik-Bogoslavski, D., Wu, J., Ma, D., et al. (2013). Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metabolism, 18, 649–659.
Wu, Z., Huang, X., Feng, Y., Handschin, C., Gullicksen, P. S., Bare, O., et al. (2006). Transducer of regulated CREB-binding proteins (TORCs) induce PGC-1alpha transcription and mitochondrial biogenesis in muscle cells. Proceedings of the National Academy of Sciences USA, 103, 14379–14384.
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell, 98, 115–124.
Yakunin, E., Moser, A., Loeb, V., Saada, A., Faust, P., Crane, D. I., et al. (2010). alpha-Synuclein abnormalities in mouse models of peroxisome biogenesis disorders. Journal of Neuroscience Research, 88, 866–876.
Yang, L., Long, Q., Liu, J., Tang, H., Li, Y., Bao, F., Qin, D., Pei, D., Liu, X. (2015). Mitochondrial fusion provides an ‘initial metabolic complementation’ controlled by mtDNA. Cell Mol Life Sci, 72(13):2585-2598.
Yu, S. P. (2003). Na(+), K(+)-ATPase: the new face of an old player in pathogenesis and apoptotic/hybrid cell death. Biochemical Pharmacology, 66, 1601–1609.
Zhang, Y., Ma, K., Song, S., Elam, M. B., Cook, G. A., & Park, E. A. (2004). Peroxisomal proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1 alpha) enhances the thyroid hormone induction of carnitine palmitoyltransferase I (CPT-I alpha). Journal of Biological Chemistry, 279, 53963–53971.
Zhao, W., Varghese, M., Yemul, S., Pan, Y., Cheng, A., Marano, P., et al. (2011). Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1α) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Molecular Neurodegeneration, 6, 51.
Zheng, B., Liao, Z., Locascio, J. J., Lesniak, K. A., Roderick, S. S., Watt, M. L., et al. (2010). PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Science Translational Medicine, 2, 52ra73.
Zorzano, A., & Claret, M. (2015). Implications of mitochondrial dynamics on neurodegeneration and on hypothalamic dysfunction. Front Aging Neurosci, 7, 101.
Acknowledgments
We thank all of our colleagues at the Royan Institute for Biotechnology who contributed to the work discussed in this review.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Jodeiri Farshbaf, M., Ghaedi, K., Megraw, T.L. et al. Does PGC1α/FNDC5/BDNF Elicit the Beneficial Effects of Exercise on Neurodegenerative Disorders?. Neuromol Med 18, 1–15 (2016). https://doi.org/10.1007/s12017-015-8370-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-015-8370-x