
Abstract
Painful small fiber neuropathy is a challenging medical condition with no effective treatment. Non-genetic causes can be identified in one half of the subjects. Gain-of-function variants of sodium channels Nav1.7 and Nav1.8 have recently been associated with painful small fiber neuropathy. More recently, mutations of sodium channel Nav1.9 have been linked to human pain disorders, with two gain-of-function mutations found in patients with painful small fiber neuropathy. Here we report a novel Nav1.9 mutation, a glycine 699 substitution by arginine (G699R) in the domain II S4-S5 linker, identified in a patient with painful small fiber neuropathy. In this study, we assayed the mutant channels by voltage-clamp in superior cervical ganglion neurons, which do not produce endogenous Nav1.8 or Nav1.9 currents, and provide a novel platform where Nav1.9 is expressed at relatively high levels. Voltage-clamp analysis showed that the mutation hyperpolarizes (−10.1 mV) channel activation, depolarizes (+6.3 mV) steady-state fast inactivation, slows deactivation, and enhances ramp responses compared with wild-type Nav1.9 channels. Current-clamp analysis showed that the G699R mutant channels render dorsal root ganglion neurons hyperexcitable, via depolarized resting membrane potential, reduced current threshold and increased evoked firing. These observations show that the domain II S4-S5 linker plays an important role in the gating of Nav1.9 and demonstrates that a mutation in this linker is linked to a common pain disorder.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sodium channel Nav1.9 is preferentially expressed within dorsal root ganglions (DRG), trigeminal and myenteric neurons (Dib-Hajj et al. 1998, 2002; Rugiero et al. 2003; Waxman and Zamponi 2014) and their axon terminals in the skin (Persson et al. 2010) and cornea (Fjell et al. 2000). Although it has all the structural motifs of sodium channels, the Nav1.9 channel is the least conserved at the amino acid sequence level relative to other members of the sodium channel family (Dib-Hajj et al. 1998, 1999a, b) and is functionally unique among other sodium channels by its ultra-slow kinetics of inactivation and the broad overlap of the hyperpolarized action and depolarized inactivation, resulting in a large persistent current as well as a substantial window current in both human and rodent DRG neurons (Cummins et al. 1999; Dib-Hajj et al. 1999a).
Painful small fiber neuropathy (SFN), characterized by damage of thinly myelinated and unmyelinated nerve fibers, burning pain, and autonomic dysfunction symptoms (Hoeijmakers et al. 2012a), represents an unmet medical need. In about one half of painful SFN cases, termed idiopathic, no apparent non-genetic cause has been found. Sodium channels Nav1.7, Nav1.8, and Nav1.9 are preferentially expressed in peripheral neurons. Gain-of-function mutations of human Nav1.7 and Nav1.8 channels have been associated with SFN (Faber et al. 2012a, b; Han et al. 2012, 2014; Hoeijmakers et al. 2012b; Huang et al. 2013). More recently, seven mutations in Nav1.9 channels were identified in patients with SFN (Huang et al. 2014). Functional assessments of two of these mutations showed gain-of-function alterations to Nav1.9 gating properties and caused increased excitability of DRG neurons.
In the present study, we identified a novel mutation, Nav1.9-G699R, in a patient with small fiber neuropathy. This mutation is located in the S4-S5 linker of domain II of Nav1.9 channels. It is known that domain II S4-S5 linker plays an important role in sodium channels activation, because several mutations within this linker in Nav1.4 and Nav1.7 have been reported with pronounced effects on this aspect of channel function (Plassart-Schiess et al. 1998; Bendahhou et al. 2002; Cummins et al. 2004; Han et al. 2006; Hoeijmakers et al. 2012b). However, the effect of the domain II S4-S5 linker on the distinctive gating properties of Nav1.9 is not clear. In the current study, we investigated the effects of the Nav1.9-G699R mutation on the biophysical properties of Nav1.9 channels and studied the effects of expressing this channel on the excitability of DRG neurons.
Materials and Methods
Patient
The patient, a Caucasian man, was evaluated at age 69. The study was approved by an institutional review board at Maastricht University Medical Center, and written informed consent was obtained from the patient before study initiation. Physical examination, skin biopsy, and quantitative sensory testing (QST) were performed as described previously (Faber et al. 2012a). QST assessed thresholds at both left and right thenar eminence and dorsal foot, with temperature threshold testing results using the method of levels. Classification of abnormality was based on Z value statistics. A measurement was considered abnormal when its Z value exceeded 2.5.
Exon Screening
Genomic DNA was extracted from venous blood using the Puregene® genomic DNA isolation kit (Gentra System). Coding exons and flanking intronic sequences were amplified and sequenced as described previously (Faber et al. 2012a). Genomic sequences were compared with the reference Nav1.9 cDNA (NM_014139.2) to identify sequence variation (Dib-Hajj et al. 1999b) using Alamut Mutation-Interpretation Software (Interactive-Biosoftware; Rouen, France).
Plasmids
The GFP-2A-tagged wild-type (WT) construct (pcDNA3-SCN11A) that encodes human Nav1.9 protein was established as previously reported (Huang et al. 2014). The c.2095G>A (p. Gly699Arg) mutation was introduced into the construct using Quickchange® II XL site-directed mutagenesis (Stratagene) and referred to as G699R hereinafter.
Voltage-Clamp: Superior Cervical Ganglion Neuron Isolation and Transfection
All protocols for care and killing of animals were approved by the IACUC at the Veterans Administration Connecticut Healthcare System. Since superior cervical ganglion (SCG) neurons do not express tetrodotoxin-resistent (TTX-R) sodium channels (Rush et al. 2006), transfection of these peripheral neurons with recombinant TTX-R Nav1.9 channels into SCG neurons was utilized for this study. A relatively high level of expression, relatively little rundown of Nav1.9 current facilitated the functional analysis of this channel by voltage-clamp. SCG neurons were isolated from neonatal (birth to 5 days old, gender was not determined) Sprague–Dawley rats and transfected by electroporation as described previously for DRG neurons (Dib-Hajj et al. 2009). Briefly, SCG were harvested, incubated at 37 °C for 20 min in oxygenated complete saline solution (CSS) (in mM: 137 NaCl, 5.3 KCl, 1 MgCl2, 25 sorbitol, 3 CaCl2, and 10 HEPES, adjusted to pH 7.2 with NaOH) containing 1.5 mg/ml Collagenase A (Roche Diagnostics) and 0.6 mM EDTA and then exchanged with an oxygenated, 37°C CSS solution containing 1.5 mg/ml Collagenase D (Roche Diagnostics), 0.6 mM EDTA, and 30 U/ml papain (Worthington Biochemicals) and incubated for another 20 min. SCG were then centrifuged and triturated in DRG media: DMEM/F12 (1:1) with 100 U/ml penicillin, 0.1 mg/ml streptomycin (Invitrogen), and 10% fetal bovine serum (Hyclone), which contained 1.5 mg/ml bovine serum albumin (Sigma-Aldrich) and 1.5 mg/ml trypsin inhibitor (Sigma-Aldrich). After trituration, the cell suspension was transfected with 2.5 µg of GFP-2A-tagged WT or G699R mutant constructs with a Nucleofector IIS device (Lonza), using the Amaxa® Basic Neuron SCN Nucleofector®™ Kit (VSPI-1003) and SCN Basic Neuro Program 6. Transfected neurons were allowed to recover for 5 min at 37°C in calcium-free DMEM media. The cell suspension was then diluted with DRG media containing 1.5 mg/ml bovine serum albumin and 1.5 mg/ml trypsin inhibitor, seeded onto poly-d-lysine/laminin precoated coverslips (BD Biosciences), and incubated at 37°C for 45 min to allow SCG neurons to attach to the coverslips. DRG media containing 50 ng/ml each of mNGF (Alomone Labs, Jerusalem, Israel) and GDNF (Peprotec, Rocky Hill, N.J.) was then added to each well to a final volume of 1.0 ml, and the cells were maintained at 37°C in a 95% air/5% CO2 (vol/vol) incubator for 40–48 h before voltage-clamp recording.
Voltage-Clamp: Recording
Voltage-clamp recordings were obtained at 22 ± 1 °C, within 40–48 h after transfection using an EPC-10 amplifier (HEKA Electronics). Small SCG neurons (<25 μm) with robust green fluorescence and no apparent neurites were selected for voltage-clamp recording. Pipette potential was adjusted to zero before seal formation, and liquid junction potential was not corrected. Capacity transients were canceled, and voltage errors minimized with ~90% series resistance compensation. Voltage-dependent currents were acquired with Patchmaster at 5 min after establishing whole-cell configuration, sampled at 50 kHz, and filtered at 2.9 kHz. The pipette solution contained (in mM) 140 CsF, 10 NaCl, 1 EGTA, and 10 HEPES, pH 7.3 with CsOH (adjusted to 315 mOsmol/l with dextrose). The extracellular bath solution contained (in mM) 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 5 CsCl, 20 tetraethylammonium chloride (TEA·Cl), pH 7.3 with NaOH (327 mOsmol/l). TTX (500 nM), CdCl2 (0.1 mM), and 4-aminopyridine (1 mM) were added in the bath solution to block endogenous TTX-sensitive voltage-gated sodium currents, calcium currents, and potassium currents, respectively.
To generate activation curves, cells were held at −120 mV and stepped to potentials of −100 to +20 mV in 5 mV increments for 100 ms. Peak inward currents obtained from activation protocols were converted to conductance values using the equation, G=I/(V m − E Na ), where G is the conductance, I is the peak inward current, V m is the membrane potential step used to elicit the response, and E Na is the reversal potential for sodium channel. Conductance data were normalized and fit with a Boltzmann equation of the form G=G max /(1+exp[(V 1/2,act − V m )/k)], where V 1/2,act is the midpoint of activation, and k is a slope factor. To generate steady-state fast inactivation curves, cells were stepped to inactivating potentials of −140 to +20 mV for 500 ms followed by a 50 ms step to −40 mV, the voltage where peak current occurs (Huang et al. 2014). The protocol for slow inactivation consisted of a 30-s step to potentials varying from −120 to −30 mV, followed by a 100-ms step to −100 mV to remove fast inactivation and a 50-ms step to −40 mV to elicit a test response. Peak inward currents obtained from steady-state fast inactivation and slow inactivation protocols were normalized by the maximum current amplitude and fit with a Boltzmann equation of the form I = A + (1 − A)/(1+exp[(V m − V 1/2,inact )/k)], where V m represents the inactivating prepulse membrane potential, and V 1/2,inact represents the midpoint of inactivation. Deactivation was estimated from current decay, using a 25-ms short depolarizing pulse to −40 mV followed by a 150-ms repolarizing pulse to potentials ranging from −110 to −60 mV with 5-mV increments. Deactivation kinetics was calculated by fitting the decaying currents with a single exponential function. The response to ramp stimulation was evaluated by a small slow ramp depolarization protocol, which starts from the holding potential of −120 mV and steadily increases to 0 mV over 600 ms for rate of 0.2 mV/ms.
Current-Clamp: DRG Neuron Isolation and Transfection
DRG from 4- to 8-week-old male Sprague–Dawley rats were harvested and dissociated as described previously (Dib-Hajj et al. 2009). Briefly, DRG were harvested, incubated at 37°C for 20 min in oxygenated CSS containing 1.5 mg/ml Collagenase A (Roche Diagnostics) and 0.6 mM EDTA, and then exchanged with an oxygenated, 37°C CSS solution containing 1.5 mg/ml Collagenase D (Roche Diagnostics), 0.6 mM EDTA, and 30 U/ml papain (Worthington Biochemicals) and incubated for another 20 min. A suspension of DRG neurons was then prepared by trituration as described above, 2 ml of DRG media was added to the suspension, and the suspension was filtered using a 70-µm nylon cell strainer (Becton Dickinson). The cell strainer was washed with 2×2 ml of DRG media, the washes were combined with the suspension and then centrifuged at 100 g for 3 min, supernatants were removed, and cells were transfected as described above using the Amaxa® Basic Neuron SCN Nucleofector®™ Kit (VSPI-1003) and SCN Basic Neuro Program 6. After transfection, cells were allowed to recover in calcium-free DMEM, seeded, and incubated for 45 min as described above. DRG media containing 50 ng/ml each of mNGF (Alomone Labs, Jerusalem, Israel) and GDNF (Peprotec, Rocky Hill, N.J.) was then added to each well to a final volume of 1.0 ml, and the cells were maintained at 37°C in a 95% air/5% CO2 (vol/vol) incubator for 40–48 h before current-clamp recording.
Current-Clamp: Recordings
Current-clamp recordings were performed at 22 ± 1°C as previously described (Han et al. 2012) from small (<30-µm diameter) green fluorescent protein-labeled DRG neurons 40–48 h after transfection using EPC-10 amplifier (HEKA). The pipette solution contained (in mM) 140 KCl, 0.5 EGTA, 5 HEPES, and 3 Mg-ATP, pH 7.3 with KOH (adjusted to 315 mOsm with dextrose). The extracellular solution contained (in mM) 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES, pH 7.3 with NaOH (adjusted to 320 mOsm with dextrose). Whole-cell configuration was obtained in voltage-clamp mode before proceeding to current-clamp recording mode. Current threshold was determined by the first action potential elicited by a series of 200-ms depolarizing current injections that increased in 5-pA increments. Voltage threshold was determined by assessing the first-order derivative dV/dt of membrane potential. Cells with stable (<10% variation) resting membrane potentials more negative than −40 mV and overshooting action potentials (>80 mV resting membrane potential to peak) were used for additional data collection. Action potential frequency was determined by quantifying the number of action potentials elicited in response to depolarizing current injections (500-ms).
Data Analysis
Electrophysiological data were analyzed using Fitmaster (HEKA Electronics) and Origin 8.5.1 (Microcal) and presented as mean ± standard error (SE). Statistical significance was determined by unpaired Student’s t tests (current-clamp except firing frequency and spontaneous activity), Mann–Whitney test (firing frequency), or two-proportion z test (comparison of proportion of cells producing spontaneous activity).
Results
Clinical Phenotype and Identification of Nav1.9-G699R Mutation
A 69-year-old man presented with a 5-year history of progressive burning pain of his feet. The patient described the pain as feeling like standing on barbed wire or walking on nails with a tight sensation around his feet. The patient started experiencing pain in his hands 4 years later. The symptoms were aggravated by rising temperature, warm water, lying under a blanket or during and after exercise and were relieved by cold water or cold packs. The patient also reported episodes of redness of the skin of his feet at the end of each day. In addition, the patient noted hypohydrosis, progressive orthostatic dizziness, palpitations, and erectile dysfunction. Family history revealed similar symptoms in his brother (71 years old) for several years; the brother did not consent to provide a DNA sample for analysis. Physical examination showed a slight red discoloration with mildly reduced light touch sense of both soles. No features of large nerve fiber involvement were present. Nerve conduction studies were normal. Quantitative sensory testing showed abnormal temperature thresholds for warmth (right foot 50.0; left foot 50.0 (Z > 2.5, <41.1)) and cold sensation (right foot 3.5; left foot 0.0 (Z > 2.5, >26.1)) in the feet, and abnormal warmth (right thenar 46.4; left thenar 43.9 (Z > 2.5, <33.4)) and cold sensation (right thenar 27.3; left thenar 29.4 (Z > 2.5, >30.1)) in the hands (Table 1). The intraepidermal nerve fiber density in the lower leg (10 cm above the lateral malleolus) was 6.7 per millimeter, which was normal (5th percentile: 2.8/mm). Systemic disorders and non-genetic causes of small fiber neuropathy were ruled out (Lauria et al. 2012). Based on symptoms and abnormal temperature threshold testing, the patient was diagnosed with small fiber neuropathy.
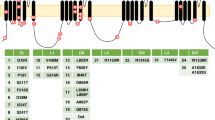
DNA analysis showed no variants in the SCN9A or SCN10A genes encoding Nav1.7 and Nav1.8, respectively, but a new variant (c.2095G>A) was identified in SCN11A encoding Nav1.9. The c.2095G>A variant results in an amino acid substitution of glycine by arginine at position 699 of Nav1.9 (p. Gly699Arg). The glycine at this position in the domain II S4-S5 linker of Nav1.9 is highly conserved in the voltage-gated sodium channel family (Fig. 1). Mutant prediction algorithms suggested that the variant is not tolerated in Nav1.9. Splice-site prediction programs showed no predicted effects on RNA splicing. This variant is not described in literature or in mutation databases, but it is reported in the SNP databases with an A-allele frequency of 0–0.03% (dbSNP database, 0% [n = 0/4552]; 1000 Genomes project/EVS database, 0.03% [n = 4/13006]). The variant was not found previously in our cohort of patients with painful peripheral neuropathy (n = 805 patients), and in a Caucasian control population (n = 234 patients).

Sequence alignment and position of mutation in DII S4-S5 linker in Nav1.9 channel. Schematic shows the 24 transmembrane segments comprising the pore-forming α-subunit of sodium channels and the location of the G699 residue within S4-S5 linker of domain II. The sequence of the DII S4-S5 linker is invariant in the 9 α-subunits of sodium channels from human. The substitution of G699R is highlighted in red type
Functional Assessment of G699R: Voltage-Clamp
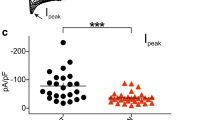
To investigate the effect of the G699R mutation on electrophysiological properties of Nav1.9 channel, SCG neurons were transfected with either Nav1.9 WT or G699R mutant construct followed by voltage-clamp recordings within 40–48 h after transfection. TTX-resistant sodium channels Nav1.8 and Nav1.9 are not detectable in SCG neuron channels (Rush et al. 2006), which facilitate the voltage-clamp analysis of Nav1.9 currents in isolation, by blocking endogenous TTX-S currents with 500 nM TTX in the bath solution. TTX-resistant Nav1.9 currents were recorded from SCG neurons by holding the neurons at −120 mV and stepping to depolarized voltages in 5-mV increments. Figure 2a and b show representative Nav1.9 current family traces recorded from SCG neurons expressing WT and G699R mutant channels, respectively. For both WT and G699R mutant channels, Nav1.9 channel currents display the ultra-slow inactivation, previously reported for Nav1.9 currents recorded from DRG neurons (Cummins et al. 1999; Dib-Hajj et al. 1999a, b; Huang et al. 2014). There is no significant difference of Nav1.9 current densities between WT (52 ± 10 pA/pF, n = 25) and G699R mutant channels (45 ± 6 pA/pF, n = 40, p > 0.05). However, compared with WT channels, activation of G699R mutant channels displays a significant hyperpolarizing shift of 10.1 mV (Fig. 2c). The midpoint of activation for WT and G699R channels is −50.0 ± 1.5 mV (n = 23) and −60.1 ± 1.5 mV (n = 25, P<0.001), respectively. The activation curve slope factor of G699R channel (8.2 ± 0.3 mV, n = 25) is not significantly different from that of WT channel (8.4 ± 0.3 mV, n = 23, p > 0.05). Steady-state fast inactivation also displays change between WT and G699R mutant channels (Fig. 2c). The midpoint of fast inactivation for G699R is depolarized by 6.3 mV (WT: −42.0 ± 1.1 mV, n = 21; G699R: −35.7 ± 1.2 mV, n = 18, P<0.001), and the slope factor of fast inactivation curve of G699R mutant channel (12.8 ± 0.6 mV, n = 18) is also significantly different from that of WT channel (10.3 ± 0.3 mV, n = 21, P<0.001). Additionally, G699R mutant channels also significantly increase the residual current through non-inactivating channels (WT: 13.6% ± 0.4%, n = 21; G699R: 23.6% ± 1.4%, n = 18, P<0.001). The combination of the hyperpolarizing shift in activation and the depolarizing shift in steady-state fast inactivation results in increased overlap between activation and fast inactivation, which predicts a larger window current for G699R mutant channels compared with WT (Fig. 2d).

The G699R mutation of Nav1.9 hyperpolarizes activation and depolarizes fast inactivation. a and b Representative current traces recorded from SCG neurons expressing WT (a) or G699R (b) mutant channels. c Comparison of activation for WT and G699R mutant channels. G699R shifts activation by −10.1 mV (WT: −50.0 ± 1.5 mV, n = 23; G699R: −60.1 ± 1.5 mV, n = 25; p < 0.001). Comparison of steady-state fast inactivation for WT and G699R mutant channels. G699R shifts steady-state fast inactivation by 6.3 mV (WT: −42.0 ± 1.1 mV, n = 21; G699R: −35.7 ± 1.2 mV, n = 18; p < 0.001). d Magnified view of overlap between activation and fast inactivation for WT and G699R mutant channels. Compared with WT, G699R mutant channels produce larger window currents
We also assessed the effects of G699R mutant on the deactivation properties of Nav1.9 channels. Kinetics of deactivation, which reflect the transition from the open to the closed state, were estimated from measurements of current decay at potentials from −110 to −60 mV after briefly activating the channels at −40 mV for 25 ms. Compared with WT, the G699R channels show significantly slower decay rate of deactivation across all deactivation potentials tested (Fig. 3a). However, because of the ultra-slow inactivation of Nav1.9, the trace for tail current does not return back to baseline after a 150-ms repolarizing pulse; this is more obvious for G699R mutant channels (Fig. 3a, inset), possibly due to the bigger window current area for G699R mutant compared with WT channel (Fig. 2d).

The G699R mutation accelerates deactivation and increases ramp currents. a Comparison of deactivation time constants. G699R (n = 15) deactivates significantly more slowly than WT (n = 16), *p < 0.05. Inset Representative currents recorded from two different neurons expressing WT (black) or G699R (red) sodium channels at a repolarization voltage of −70 mV. The WT current peak was scaled to match the G699R current peak for comparing the kinetics of current decay. b Representative ramp currents in response to slow depolarizations (0.2 mV/ms) were normalized to the peak current acquired during activation protocol and plotted as a function of membrane potential for WT (black) and G699R (red) channels. c Steady-state slow inactivation curves for WT and G699R. Steady-state slow inactivation was not altered by the G699R mutation (WT: −73.6 ± 1.3 mV, n = 16; G699R: −76.1 ± 1.1 mV, n = 14; p > 0.05)
It is known that both sodium channel Nav1.7 and Nav1.8 produce ramp currents, and increased ramp currents contribute to the hyperexcitability of DRG neurons (Cummins et al. 1999). Whether Nav1.9 channels also produce ramp currents is less well understood. Expression of Nav1.9 in SCG neurons which do not produce endogenous TTX-R currents permits the evaluation of Nav1.9 ramp currents. In the present study, we used a slow depolarizing stimulus from −120 mV to 0 mV over 600 ms to detect the cell response to the ramp stimulus. Surprisingly, we found that Nav1.9 WT channel produces a large ramp current, which is 75.8 ± 2.2% (n = 19) of the peak transient current. The G699R mutant channel produces a small but significant increase in the normalized ramp current: 83.3 ± 2.7% (n = 20, P<0.05) (Fig. 3b). The voltage at which the peak of the ramp current occurs is significantly shifted by −6.1 mV for the G699R mutant channels (−43.0 ± 1.3 mV, n = 20, P<0.01) compared with WT channels (−36.9 ± 1.2 mV, n = 19).
The effect of G699R mutation on the steady-state slow inactivation of Nav1.9 channel was assessed by inactivating the channel with a prepulse of 30 s from −120 to −30 mV, followed by 100 ms pulses to −100 mV to allow recovery from fast inactivation, then followed by a 50 ms test pulse to −40 mV to detect the available fraction of Nav1.9 channel current. As Fig. 3c shows, the G699R mutation does not significantly shift steady-state slow inactivation compared with WT channels. The midpoint for slow inactivation is −73.6 ± 1.3 mV (n = 16) for WT and −76.1 ± 1.1 mV (n = 14, p > 0.05) for G699R mutant channels. There is also no significant difference for the slope factor of slow inactivation between WT (7.3 ± 0.2 mV, n = 16) and G699R mutant channels (7.7 ± 0.4 mV, n = 14, p > 0.05). G699R mutation produces a small but not statistically significant increase (8.2 ± 1.2%, n = 14) in the residual current through non-inactivating channels of slow inactivation, compared with WT (5.3 ± 0.9%, n = 16, p > 0.05).
Functional Assessment of G699R: Current-Clamp
The effects of the G699R mutation on the excitability of DRG neurons were studied by current-clamp analysis in two groups of DRG neurons, transfected with WT or G699R mutant channels. The input resistance of DRG neurons expressing G699R mutant channels (716 ± 100 MΩ, n = 24) was not significantly different from that of DRG neurons expressing WT channels (778 ± 77 MΩ, n = 30, p > 0.05). The resting membrane potential of DRG neurons expressing the G699R channels was significantly depolarized by 4.3 mV (−50.1 ± 1.2 mV, n = 24), compared with WT channels (−54.4 ± 0.9 mV, n = 30, P<0.01) (Fig. 4a).

The G699R mutation depolarizes resting membrane potential (RMP) and reduces current threshold in DRG neurons. a RMP of DRG neurons expressing WT was depolarized by 4.3 mV compared with that of G699R channels. (WT: −54.4 ± 0.9 mV, n = 30; G699R: −50.1 ± 1.2 mV, n = 24; **p < 0.01) b Current threshold was significantly reduced after expressing G699R channels (WT: 257 ± 21 pA, n = 30; G699R: 174 ± 20 pA, n = 24, **p < 0.01). c Responses of a small DRG neuron transfected with WT channels to the subthreshold (265–270 pA) and the threshold depolarizing current injections (275 pA). d Responses of a small DRG neuron transfected with G699R mutant channels to the subthreshold (130–135 pA) and the threshold depolarizing current injections (140 pA). Arrows with numbers indicate the current amplitude used to elicit the labeled response
Average current threshold for the group of DRG neurons expressing G699R mutant channels (174 ± 20 pA, n = 24) was significantly smaller compared with that of the group of DRG neurons expressing WT channels (257 ± 21 pA, n = 30, P<0.01) (Fig. 4b). Figure 4c shows a small DRG neuron expressing WT channels, which responded to subthreshold current injections (265–270 pA) with graded membrane potential depolarizations. At the current threshold of 275 pA, the first overshooting action potential was generated for this neuron. Representative action potentials recorded from a DRG neuron expressing G699R mutant channels are shown in Fig. 4d. For this neuron, subthreshold graded membrane depolarizations (130–135 pA) were followed by the first overshooting action potential when the injected current was increased to 140 pA, the current threshold for this small DRG neuron. We also compared the voltage threshold (the voltage at which action potential takeoff occurs) between the two group DRG neurons and found no significant difference between the two groups of DRG neurons (WT: −15.8 ± 0.9 mV, n = 30; G699R: −15.4 ± 1.1 mV, n = 24, p > 0.05). There was no significant difference for either action potential amplitude (WT: 106.8 ± 1.8 mV, n = 30; G699R: 101.9 ± 2.0 mV, n = 24, p > 0.05), or half width (WT: 3.74 ± 0.16 ms, n = 30; G699R: 4.07 ± 0.28 ms, n = 24, p > 0.05) between the two groups of DRG neurons.
In order to assess the effects of expressing G699R mutant channels on the repetitive firing properties of DRG neurons, a series of 500-ms current injections from 25 pA to 500 pA in 25-pA increments were applied to DRG neurons. Figure 5a shows the responses of representative DRG neurons expressing either WT or G699R mutant channels to 500-ms current steps at 1X, 1.5X, and 2X current thresholds. The DRG neuron expressing WT channels produced only a single action potential in response to injection of 1X and 1.5X current threshold, and two action potentials to injection of 2X current threshold. Although the DRG neuron expressing G699R mutant channels also generated single action potential in response to 1X current threshold, it produced two action potentials in response to injection of 1.5X current threshold and 5 action potentials in response to injection of 2X current threshold. Figure 5b displays a comparison of the average number of action potential between these two groups of DRG neurons at a spectrum of stimulus intensities. DRG neurons transfected with G699R mutant channels fired at a significantly higher frequency than DRG neurons transfected with WT channels in response to injecting currents from 150 pA to 500 pA (P<0.05, Mann–Whitney test).

The G699R mutation increases excitability in small DRG neurons. a Responses of representative DRG neurons expressing WT (upper) and G699R (lower) channels, respectively, to 500-ms depolarization current steps, which are 1×, 1.5× and 2× (left, center and right traces, respectively) the current threshold for action potential generation. b Comparison of repetitive action potential firing spike number between DRG neurons expressing WT (n = 30) and G699R (n = 24) across a range of 500-ms step current injections from 25 to 500 pA; *p < 0.05
We also observed that among both groups of DRG neurons, some neurons fired spontaneously. G699R caused a nominal increase in the proportion of spontaneously firing neurons, compared with WT, but did not reach statistical significance (WT: 13 out 43 cells, 30.2%; G699R: 19 out 43 cells, 44.2%; p > 0.05, z test).
Discussion
Sodium channel Nav1.9 is preferentially expressed in DRG, trigeminal, and myenteric neurons (Dib-Hajj et al. 1998, 2002; Rugiero et al. 2003). Studies in preclinical animal models have shown impaired somatic inflammatory pain behavior in Nav1.9-null mice (Priest et al. 2005; Amaya et al. 2006; Ostman et al. 2008; Lolignier et al. 2011) and have implicated Nav1.9 in bone cancer pain (Qiu et al. 2012) and visceral pain (Hockley et al. 2014). Recently, gain-of-function mutations in Nav1.9 have been associated with painful neuropathy (Huang et al. 2014) and a syndrome of familial episodic pain (Zhang et al. 2013). Although the mechanism is not well understood, a missense mutation in Nav1.9 has also been reported to be linked to congenital insensitivity to pain (Leipold et al. 2013). Thus, novel mutations in Nav1.9 must be functionally assessed to increase confidence in linking them to pain pathology. In the present paper, we describe the new Nav1.9 mutation G699R in the DII/S4-5 linker, which we identified in a patient with symptoms of painful small fiber neuropathy, with the diagnosis confirmed by quantitative sensory testing. Some of the clinical features in this patient resemble the clinical picture of erythromelalgia, in which mutations within the same part of the Nav1.7 channel produce pain and erythema of the distal limbs, worsened by warmth, and relieved by cold (Drenth and Waxman 2007; Dib-Hajj et al. 2013).
We carried out voltage-clamp assessment of the mutant channels in superior cervical ganglion neurons, which present a platform in which this channel expresses at relatively high levels, with biophysical properties similar to those that have been reported in DRG neurons in other studies. Voltage-clamp analysis shows that G699R causes gain-of-function alterations in Nav1.9 including hyperpolarized activation, depolarized fast inactivation, slowed deactivation, and enhanced response to a slow ramp stimulus. Our current-clamp analysis shows that these pro-excitatory changes in the biophysical properties of Nav1.9 channel increase the excitability of DRG neurons, as manifested by depolarized resting membrane potential, decreased current threshold, and increased firing frequency in response to graded superthreshold stimulation. These data support the conclusion that the G699R mutation contributes to the pathophysiology of painful neuropathy in this individual.
Solving the crystal structures of bacterial voltage-gated sodium channels has enhanced our understanding of molecular mechanisms of sodium channel gating (Payandeh et al. 2011, 2012; Zhang et al. 2012). The NavAb crystal structure demonstrated that during sodium channel activation, the S4-S5 linker moves together with the voltage-sensing domain (VSD) as a molecular unit and that the rotational movements of the VSD and S4-S5 linker pull the S5-S6 helices outward to open the pore (Payandeh et al. 2011). Thus, mutations in the DII/S4-5 linker are expected to impact channel activation. However, the IFM inactivation particle is not located within a membrane-spanning segment that can be accurately modeled based on current crystal structures of bacterial voltage-gated sodium channels. Thus, fast inactivation has not been amenable to analysis by modeling, and the effect of mutations in the DII/S4-5 linker on inactivation is not yet understood at the structural level.
Nav1.9 channels are the least conserved at the primary amino acid level among members of the sodium channel family with human and rodent Nav1.9 sharing only 72% identity, and this divergence in sequence is paralleled by gating properties that are very distinct, compared with the other channels (Dib-Hajj et al. 1998, 1999a, b; Cummins et al. 1999). The G699R mutation substitutes a neutral amino acid glycine with the smallest side group for the positively charged and bulky arginine residue within the DII/S4-S5 linker. As Fig. 1 shows, the amino acid sequence of DII/S4-S5 linker is highly conserved among all nine human voltage-gated sodium channels, and indeed all vertebrate sodium channels, with an invariant glycine in all channels at the corresponding position of G699 in Nav1.9. The high degree of conservation suggests that this linker may play a conserved role in all sodium channels. Mutations in this linker in other sodium channel subtypes have been identified in patients with Dravet syndrome (Nav1.1) (Zuberi et al. 2011), hyperkalemic periodic paralysis (HyperPP) (Nav1.4) (Cannon, 2002), Brugada syndrome (Nav1.5) (Niimura et al. 2004), Lennox-Gastaut syndrome (Nav1.6)(Allen et al. 2013), inherited erythromelalgia (Nav1.7) (Yang et al. 2004; Han et al. 2006; Dib-Hajj et al. 2013), and painful small fiber neuropathy (Nav1.7) (Hoeijmakers et al. 2012b). Among these mutations, two Nav1.4 mutations (L689I and I693T) and four Nav1.7 mutations (I848T, L858H, L858F, and G856D) have been functionally characterized and show a hyperpolarizing shift in voltage-dependent activation (Plassart-Schiess et al. 1998; Bendahhou et al. 2002; Cummins et al. 2004; Han et al. 2006; Hoeijmakers et al. 2012b). In the present study, we found that the Nav1.9-G699R mutation caused a significant hyperpolarizing shift in voltage-dependent activation and this suggests that the DII S4-S5 linker also plays an important role for the activation of the Nav1.9 channel, as in Nav1.4 and Nav1.7 channels. These data support the conclusion, which is based upon the structure of bacterial voltage-gated sodium channels, that the DII/S4-5 linker plays an important role in channel activation and extend it to the Nav1.9 sodium channel and points to a critical role of the conserved G699 residue in this process.
In addition to a hyperpolarizing shift in activation, the G699R mutation causes a 6.3-mV depolarizing shift in fast inactivation. Although it has been previously shown that the sodium channel DIII and DIV S4-S5 linker contribute to steady-state fast inactivation (Smith and Goldin 1997; Lerche et al. 1997; McPhee et al. 1998), a contribution of the DII S4-S5 linker of sodium channels to fast inactivation has not been reported. Mutations in the DII/S4-5 linker of Nav1.4 (L689I and I693T) and Nav1.7 (I848T and L858H) did not alter steady-state fast inactivation (Plassart-Schiess et al. 1998; Bendahhou et al. 2002; Cummins et al. 2004), whereas Nav1.7 mutations L858F and G856D shift steady-state fast inactivation in a depolarized direction (Han et al. 2006; Hoeijmakers et al. 2012b). While the effect on activation of mutations in DII/S4-5 linker is consistent with the pivotal role of this linker in transducing the mechanical force of the movement of the VSD to the pore domain (Payandeh et al. 2011), the effect of a subset of mutations, including Nav1.9-G699R, on fast inactivation suggests that G699 may also play an important role in the fast inactivation of sodium channels, possibly by inducing an allosteric effect that weakens the interaction of the inactivation particle with residues within the DII/S4-5 linker.
Compared with other sodium channels, Nav1.9 displays a dramatically large window current, with the peak around −50 mV (Fig. 2d), within the range of normal resting membrane potential of DRG neurons. The combination of the hyperpolarizing shift in voltage-dependent activation and the depolarizing shift in steady-state fast inactivation results in an increased window current for Nav1.9 G699R mutation compared with WT channels. Increased window current has been shown to contribute to the enhanced ramp current for sodium channel Nav1.3 (Estacion and Waxman, 2013). Here we found that the G699R mutation significantly increased the ramp current of sodium channel Nav1.9, and the coincidence of increased window current for G699R mutant channels suggests that window current may also be an important contributor to the ramp current for Nav1.9 channels. We also showed for the first time that Nav1.9 can generate a very large ramp current (the normalized amplitude is about 75% of transient peak current) in response to slow ramp stimulation. The enhanced window current and ramp current of G699R mutant channels would be expected to increase the excitability of DRG neurons, which was indeed demonstrated by our current-clamp analysis.
In conclusion, this study adds a novel mutation, within a different part of the channel, to the cohort of gain-of-function changes in Nav1.9 that have been linked to painful peripheral neuropathy. This study shows that this new Nav1.9 mutation renders DRG neuron hyperexcitable, providing a basis for the patient’s pain symptoms. Our data show that the DII/S4-5 linker in human Nav1.9 plays a critical role in channel activation, consistent with the mechanism that is based on the crystal structure of bacterial sodium channels.
References
Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E., et al. (2013). De novo mutations in epileptic encephalopathies. Nature, 501(7466), 217–221. doi:10.1038/Nature12439.
Amaya, F., Wang, H., Costigan, M., Allchorne, A. J., Hatcher, J. P., Egerton, J., et al. (2006). The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. Journal of Neuroscience, 26(50), 12852–12860. doi:10.1523/JNEUROSCI.4015-06.2006.
Bendahhou, S., Cummins, T. R., Kula, R. W., Fu, Y. H., & Ptacek, L. J. (2002). Impairment of slow inactivation as a common mechanism for periodic paralysis in DIIS4-S5. Neurology, 58(8), 1266–1272.
Cannon, S. C. (2002). An expanding view for the molecular basis of familial periodic paralysis. Neuromuscular Disorders, 12(6), 533–543. doi:10.1016/S0960-8966(02)00007-X.
Cummins, T. R., Dib-Hajj, S. D., Black, J. A., Akopian, A. N., Wood, J. N., & Waxman, S. G. (1999). A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. Journal of neuroscience, 19(24).
Cummins, T. R., Dib-Hajj, S. D., & Waxman, S. G. (2004). Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. Journal of Neuroscience, 24(38), 8232–8236. doi:10.1523/JNEUROSCI.2695-04.2004.
Dib-Hajj, S., Black, J. A., Cummins, T. R., & Waxman, S. G. (2002). NaN/Nav1.9: a sodium channel with unique properties. Trends in Neurosciences, 25(5), 253–259.
Dib-Hajj, S. D., Choi, J. S., Macala, L. J., Tyrrell, L., Black, J. A., Cummins, T. R., et al. (2009). Transfection of rat or mouse neurons by biolistics or electroporation. Nature Protocols, 4(8), 1118–1126. doi:10.1038/nprot.2009.90.
Dib-Hajj, S. D., Tyrrell, L., Black, J. A., & Waxman, S. G. (1998). NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proceedings of the National Academy of Sciences of the United States of America, 95(15), 8963–8968.
Dib-Hajj, S. D., Tyrrell, L., Cummins, T. R., Black, J. A., Wood, P. M., & Waxman, S. G. (1999a). Two tetrodotoxin-resistant sodium channels in human dorsal root ganglion neurons. FEBS Letters, 462(1–2), 117–120.
Dib-Hajj, S. D., Tyrrell, L., Escayg, A., Wood, P. M., Meisler, M. H., & Waxman, S. G. (1999b). Coding sequence, genomic organization, and conserved chromosomal localization of the mouse gene Scn11a encoding the sodium channel NaN. Genomics, 59(3), 309–318. doi:10.1006/geno.1999.5890.
Dib-Hajj, S. D., Yang, Y., Black, J. A., & Waxman, S. G. (2013). The Na(V)1.7 sodium channel: from molecule to man. Nature Reviews Neuroscience, 14(1), 49–62. doi:10.1038/nrn3404.
Drenth, J. P., & Waxman, S. G. (2007). Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. The Journal of Clinical Investigation, 117(12), 3603–3609. doi:10.1172/JCI33297.
Estacion, M., & Waxman, S. G. (2013). The response of Na(V)1.3 sodium channels to ramp stimuli: multiple components and mechanisms. Journal of Neurophysiology, 109(2), 306–314. doi:10.1152/jn.00438.2012.
Faber, C. G., Hoeijmakers, J. G. J., Ahn, H. S., Cheng, X. Y., Han, C. Y., Choi, J. S., et al. (2012a). Gain of function NaV1.7 mutations in idiopathic small fiber neuropathy. Annals of Neurology, 71(1), 26–39. doi:10.1002/Ana.22485.
Faber, C. G., Lauria, G., Merkies, I. S. J., Cheng, X. Y., Han, C. Y., Ahn, H. S., et al. (2012b). Gain-of-function Na(v)1.8 mutations in painful neuropathy. Proceedings of the National Academy of Sciences of the United States of America, 109(47), 19444–19449. doi:10.1073/pnas.1216080109.
Fjell, J., Hjelmstrom, P., Hormuzdiar, W., Milenkovic, M., Aglieco, F., Tyrrell, L., et al. (2000). Localization of the tetrodotoxin-resistant sodium channel NaN in nociceptors. NeuroReport, 11(1), 199–202. doi:10.1097/00001756-200001170-00039.
Han, C., Hoeijmakers, J. G., Ahn, H. S., Zhao, P., Shah, P., Lauria, G., et al. (2012). Nav1.7-related small fiber neuropathy: impaired slow-inactivation and DRG neuron hyperexcitability. Neurology, 78(21), 1635–1643. doi:10.1212/WNL.0b013e3182574f12.
Han, C., Rush, A. M., Dib-Hajj, S. D., Li, S., Xu, Z., Wang, Y., et al. (2006). Sporadic onset of erythermalgia: a gain-of-function mutation in Nav1.7. Annals of Neurology, 59(3), 553–558. doi:10.1002/ana.20776.
Han, C., Vasylyev, D., Macala, L. J., Gerrits, M. M., Hoeijmakers, J. G., Bekelaar, K. J., et al. (2014). The G1662S NaV1.8 mutation in small fibre neuropathy: impaired inactivation underlying DRG neuron hyperexcitability. Journal of Neurology, Neurosurgery and Psychiatry, 85(5), 499–505. doi:10.1136/jnnp-2013-306095.
Hockley, J. R., Boundouki, G., Cibert-Goton, V., McGuire, C., Yip, P. K., Chan, C., et al. (2014). Multiple roles for NaV1.9 in the activation of visceral afferents by noxious inflammatory, mechanical, and human disease-derived stimuli. Pain, 155(10), 1962–1975, doi:10.1016/j.pain.2014.06.015.
Hoeijmakers, J. G., Faber, C. G., Lauria, G., Merkies, I. S., & Waxman, S. G. (2012a). Small-fibre neuropathies-advances in diagnosis, pathophysiology and management. Nature Reviews Neurology, 8(7), 369–379. doi:10.1038/nrneurol.2012.97.
Hoeijmakers, J. G., Han, C., Merkies, I. S., Macala, L. J., Lauria, G., Gerrits, M. M., et al. (2012b) Small nerve fibres, small hands and small feet: a new syndrome of pain, dysautonomia and acromesomelia in a kindred with a novel NaV1.7 mutation. Brain, 135(Pt 2), 345–358, doi:10.1093/brain/awr349
Huang, J., Han, C., Estacion, M., Vasylyev, D., Hoeijmakers, J. G., Gerrits, M. M., et al. (2014). Gain-of-function mutations in sodium channel Na(V)1.9 in painful neuropathy. Brain, 137, 1627–1642. doi:10.1093/Brain/Awu079.
Huang, J., Yang, Y., Zhao, P., Gerrits, M. M., Hoeijmakers, J. G., Bekelaar, K., et al. (2013). Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. Journal of Neuroscience, 33(35), 14087–14097. doi:10.1523/JNEUROSCI.2710-13.2013.
Lauria, G., Merkies, I. S., & Faber, C. G. (2012). Small fibre neuropathy. Current Opinion in Neurology, 25(5), 542–549. doi:10.1097/WCO.0b013e32835804c5.
Leipold, E., Liebmann, L., Korenke, G. C., Heinrich, T., Giesselmann, S., Baets, J., et al. (2013). A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nature Genetics, 45(11), 1399–1404. doi:10.1038/ng.2767.
Lerche, H., Peter, W., Fleischha, R., Pika-Hartlaub, U., Malina, T., Mitrovic, N., et al. (1997). Role in fast inactivation of the IV/S4-S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. Journal of Physiology, 505(2), 345–352.
Lolignier, S., Amsalem, M., Maingret, F., Padilla, F., Gabriac, M., Chapuy, E., et al. (2011). Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS ONE, 6(8), e23083. doi:10.1371/journal.pone.0023083.
McPhee, J. C., Ragsdale, D. S., Scheuer, T., & Catterall, W. A. (1998). A critical role for the S4-S5 intracellular loop in domain IV of the sodium channel alpha-subunit in fast inactivation. The Journal of biological chemistry, 273(2), 1121–1129.
Niimura, H., Matsunaga, A., Kumagai, K., Ohwaki, K., Ogawa, M., Noguchi, H., et al. (2004). Genetic analysis of Brugada syndrome in western Japan - Two novel mutations. Circulation Journal, 68(8), 740–746. doi:10.1253/Circj.68.740.
Ostman, J. A., Nassar, M. A., Wood, J. N., & Baker, M. D. (2008). GTP up-regulated persistent Na+ current and enhanced nociceptor excitability require NaV1.9. Journal of Physiology, 586(4), 1077–1087. doi:10.1113/jphysiol.2007.147942.
Payandeh, J., El-Din, T. M. G., Scheuer, T., Zheng, N., & Catterall, W. A. (2012). Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature, 486(7401), 135–139. doi:10.1038/Nature11077.
Payandeh, J., Scheuer, T., Zheng, N., & Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature, 475(7356), 353–358. doi:10.1038/Nature10238.
Persson, A. K., Black, J. A., Gasser, A., Cheng, X. Y., Fischer, T. Z., & Waxman, S. G. (2010). Sodium-calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Molecular Pain, 6, doi:10.1186/1744-8069-6-84
Plassart-Schiess, E., Lhuillier, L., George, A. L., Fontaine, B., & Tabti, N. (1998). Functional expression of the Ile693Thr Na+ channel mutation associated with paramyotonia congenita in a human cell line. Journal of Physiology, 507(3), 721–727. doi:10.1111/j.1469-7793.1998.721bs.x.
Priest, B. T., Murphy, B. A., Lindia, J. A., Diaz, C., Abbadie, C., Ritter, A. M., et al. (2005). Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proceedings of the National Academy of Sciences of the United States of America, 102(26), 9382–9387. doi:10.1073/pnas.0501549102.
Qiu, F., Jiang, Y., Zhang, H., Liu, Y., & Mi, W. (2012). Increased expression of tetrodotoxin-resistant sodium channels Nav1.8 and Nav1.9 within dorsal root ganglia in a rat model of bone cancer pain. Neuroscience Letters, 512(2), 61–66. doi:10.1016/j.neulet.2012.01.069.
Rugiero, F., Mistry, M., Sage, D., Black, J. A., Waxman, S. G., Crest, M., et al. (2003). Selective expression of a persistent tetrodotoxin-resistant Na+ current and NaV1.9 subunit in myenteric sensory neurons. Journal of Neuroscience, 23(7), 2715–2725.
Rush, A. M., Dib-Hajj, S. D., Liu, S., Cummins, T. R., Black, J. A., & Waxman, S. G. (2006). A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proceedings of the National Academy of Sciences of the United States of America, 103(21), 8245–8250. doi:10.1073/pnas.0602813103.
Smith, M. R., & Goldin, A. L. (1997). Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophysical Journal, 73(4), 1885–1895.
Waxman, S. G., & Zamponi, G. W. (2014). Regulating excitability of peripheral afferents: emerging ion channel targets. Nature Neuroscience, 17(2), 153–163. doi:10.1038/nn.3602.
Yang, Y., Wang, Y., Li, S., Xu, Z., Li, H., Ma, L., et al. (2004). Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. Journal of Medical Genetics, 41(3), 171–174. doi:10.1136/jmg.2003.012153.
Zhang, X., Ren, W. L., DeCaen, P., Yan, C. Y., Tao, X., Tang, L., et al. (2012). Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature, 486(7401), 130–134. doi:10.1038/Nature11054.
Zhang, X. Y., Wen, J., Yang, W., Wang, C., Gao, L., Zheng, L. H., et al. (2013). Gain-of-function mutations in SCN11A cause familial episodic pain. American Journal of Human Genetics, 93(5), 957–966. doi:10.1016/j.ajhg.2013.09.016.
Zuberi, S. M., Brunklaus, A., Birch, R., Reavey, E., Duncan, J., & Forbes, G. H. (2011). Genotype-phenotype associations in SCN1A-related epilepsies. Neurology, 76(7), 594–600. doi:10.1212/Wnl.0b013e31820c309b.
Acknowledgments
We thank Lawrence J. Macala, Fadia Dib-Hajj, and Palak Shah for technical assistance. We thank Dr. Mark Estacion and Dr. Jianying Huang for valuable comments. This work was supported in part by Grants from the Rehabilitation Research Service and Medical Research Service, Department of Veterans Affairs (SGW and SDH), and Grant #602273 from the European Union Seventh Framework Programme FP7/2007-2013. The Center for Neuroscience & Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University.
Conflict of interest
The authors report no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
On behalf of the PROPANE Study Group.
Rights and permissions
About this article

Cite this article
Han, C., Yang, Y., de Greef, B.T.A. et al. The Domain II S4-S5 Linker in Nav1.9: A Missense Mutation Enhances Activation, Impairs Fast Inactivation, and Produces Human Painful Neuropathy. Neuromol Med 17, 158–169 (2015). https://doi.org/10.1007/s12017-015-8347-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-015-8347-9