
Abstract
With the rise of aging populations, new challenges for health care systems are emerging. Degenerative conditions of the central nervous system share a strikingly great deal of similarities, particularly the production and buildup of malfolded proteins. As a result, stress pathways within the endoplasmic reticulum become activated, triggering widespread neuronal apoptosis. New pharmacological compounds targeting this response are emerging as promising treatment strategies. This review examines the current evidence for protein aggregation in neurodegenerative disease states and discusses future mechanisms of therapeutically targeting the endoplasmic reticulum.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With Alzheimer’s disease alone estimated to affect 24 million people worldwide and predicted to rise fourfold over the next 35 years, the global burden of neurodegenerative diseases (including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis and most recently Huntington’s disease) is only set to rise (Alzheimer’s Association 2014; Fernández-Nogales et al. 2014; Prince et al. 2013).
Despite each disease’s characteristic hallmarks, there are remarkable common pathophysiological features between them. Over the past two decades, it has become well established that all neurodegenerative diseases to some extent exhibit protein folding defects. This therefore implicates the posttranslational environment in the pathogenesis of neurodegeneration and neuronal apoptosis. Hence, this group of conditions is now termed under the umbrella of protein misfolding diseases (Bucciantini et al. 2002). This review will focus on the role of the unfolded protein response (UPR) within Alzheimer’s and Parkinson’s diseases.
The Unfolded Protein Response
Within the eukaryotic cell, the rough endoplasmic reticulum (ER) functions as the primary compartment of protein synthesis, where nascent proteins are folded into their final conformational shape. In order for successful protein folding to occur, there must be adequate supply of adenosine triphosphate (ATP), oxygen and calcium within the luminal domain of the ER. This folding process is governed by the molecular chaperones of the UPR pathway.
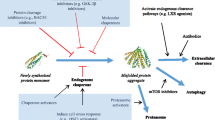
The UPR has demonstrated exceptional potential as a therapeutic target in neurodegenerative disorders (Halliday and Mallucci 2014). The protein folding process is chiefly governed by the ER chaperone, glucose-regulated protein 78 (GRP78/BiP), which functionally regulates ER transmembrane proteins: inositol-requiring enzyme 1 (IRE1), activating transcription factor 6 (ATF-6) and protein kinase R-like endoplasmic reticulum kinase (PERK) (Fig. 1). In the presence certain microenvironment conditions, such as viral infection, inflammation and other cellular stressors (including malfolded proteins) or if prerequisites for protein folding are not met, the aggregation of malfolded proteins occurs, causing GRP78/BiP-mediated activation of the UPR (Fig. 2; Schröder and Kaufman 2005b).

Native binding state of GRP78 and transmembrane stress transducers ATF-6, PERK and IRE1 in the absence of endoplasmic reticulum stress. Note a fraction of GRP78 exists in the cytosolic domain where it can directly inhibit apoptosis

Unfolded protein response signal transduction pathway. When malfolded proteins accumulate in the luminal ER domain, GRP78 dissociates from the ER transmembrane stress transducer proteins and binds to exposed hydrophobic domains on the malfolded protein. This sets a sequence of events in motion. IRE1 phosphorylates and functions to cause XBP1 to become spliced to XBP1S, which upregulates molecular chaperones of the UPR. PERK becomes phosphorylated, which in turn phosphorylates eIF2α. The phosphorylation of eIF2α arrests protein synthesis, preventing MET-tRNA from translating amino acid sequences across the ER membrane with the exception of proteins involved in the UPR. This eIF2α phosphorylation also induces the production of CHOP, a pro-apoptotic protein which induces apoptosis via cytochrome c-mediated activation of caspase 3
The aims of the UPR are threefold:
-
1.
To export potentially cytotoxic malfolded proteins to the proteasome for ubiquitination and atteunate global protein synthesis to prevent further malfolded protein aggregation.
-
2.
If malfolded protein burden persists, to initiate apoptosis.
-
3.
To permit the refolding of misfolded proteins to their intended conformational shape.
Exposed hydrophobic domains of the malfolded proteins permit GRP78/BiP, to dissociate from its bound low-affinity state with IRE1, PERK and ATF-6 and bind with high affinity to the malfolded protein. This dissociation then permits the targeting of malfolded protein by the ER-associated degradation complex (ERAD) for export to the proteasome. This protein targeting further facilitates the phosphorylation of IRE1 and PERK, which go on to mediate downstream targets of the UPR (Quinones et al. 2008).
Key Mediators of the UPR
The UPR mediates neuronal survival and apoptosis by three distinct pathways, as outlined in Fig. 2.
GRP78/BiP
Glucose-regulated protein 78 is a heat-shock-like protein (HSP) belonging to the HSP-70 family, encoded for by the HSPA5 gene (Clarke et al. 2012). In the absence of luminal ER stress, GRP78/BiP remains bound to IRE1, PERK and ATF-6. GRP78/BiP acts as a molecular chaperone, facilitating the correct folding of protein following translation across the ribosomal pores (Lee 2007). In the presence of malfolded protein, GRP78/BiP dissociates from IRE1, PERK and ATF-6, binding with low initial affinity to unfolded proteins through recognition of hydrophobic sequences which, in fully folded proteins, are held within the internal structure, unexposed to the ER lumen.
GRP78/BiP not only acts as a molecular protein folding chaperone, but also has additional functions as a luminal calcium ER binding protein (Li and Lee 2006; Reddy et al. 2003) and maintains cellular calcium homeostasis, which if not maintained may interfere with ER function.
Furthermore, a small percentage of total GRP78/BiP exists as an ER transmembrane protein, much like IRE1, PERK or ATF-6. This confers GRP78/BiP the ability to prevent apoptosis via capsase-7 and Bcl2-associated X protein (BAX) inhibition (Lee 2007) in the cytosol.
GRP78/BiP is a highly conserved protein (Quinones et al. 2008), which is present in all tissues at low basal levels (Lee 2007).
PERK
PERK is an ER transmembrane kinase possessing a luminal stress-sensing domain, which natively remains associated with GRP78/BiP when inactive (Harding et al. 2000).
When GRP78/BiP dissociates in the presence of unfolded proteins, PERK phosphorylates eukaryotic translation initiation factor 2A (EIF2A) and nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (Schröder and Kaufman 2005a, b), which consequently sets in motion a series of cascades resulting in decreased global protein synthesis and altered regulation of the CCAAT/enhancer-binding protein homologous protein (CHOP) and activating transcription factor 4 (ATF-4) pathways.
Inactivation of EIF2A due to PERK phosphorylation prevents the Met transfer RNA (tRNA) ribosome complex from initiating translation, temporarily alleviating the ER from translational activity and attenuating global protein synthesis, which affords the ERAD ability to export unfolded protein to the proteasome for degradation (Harding et al. 2000; Schröder and Kaufman 2005a).
As a result of decreased protein synthesis, levels of cyclin D1, a G1 checkpoint progression protein, are diminished, leading to a characteristic UPR-induced G1 cell cycle arrest (Brewer and Diehl 2000). However, this attenuation is semi-selective, permitting UPR proteins through the EIF2A-dependent blockade and allowing ATF-4 and CHOP production.
Nrf2 is a further target of PERK-mediated phosphorylation, facilitating bZIP translation of cellular antioxidants to maintain cellular and ER redox homeostasis. Nrf2 silencing has been found to lead to an accumulation in reactive oxygen species (ROS) and results in sensitization to ER stressors (Schröder and Kaufman 2005a, b).
IRE1
Inositol-requiring enzyme 1 is an endoplasmic reticulum transmembrane kinase that senses ER luminal stress. The N-terminus of the protein sits within the ER lumen sensing cell stress, while a cytosolic kinase and endoribonuclease feature at the C-terminal facilitate cytosolic and nuclear signal transduction (Tirasophon et al. 1998). When luminal stress is minimal, IRE1 is stored in a monomeric, inactive state through association with molecular ER chaperones. IRE1 becomes activated by luminal ER stress, resulting in dissociation and dimerization of the molecular protein folding chaperone glucose-regulated protein 78 (GRP78/BiP). The luminal N-terminus undergoes transautophosphorylation and homodimerization, thereby activating the cytosolic region of the protein. In eukaryotic organisms, this in turn splices an active fragment from the unspliced, inactive X-box binding protein 1 (XBP1u) mRNA to form the active and spliced XBP1s (Yoshida et al. 2001; Back et al. 2005).
At present, there is debate as to the role of IRE1 as an ER stress sensor as new insights from IRE1 characterization into Saccharomyces cerevisiae emerge. Although it is widely acknowledged that the dissociation of molecular chaperones (such as GRP78/BiP) is associated with IRE1 activation, a mechanistic explanation is currently lacking. Detailed study of the luminal domain of IRE1 has revealed major histocompatibility complex-like structures within IRE1’s quaternary configuration, suggesting a more direct role for the luminal domain of IRE1 in malfolded protein sensing (Credle et al. 2005). These MHC-like domains within IRE1’s luminal domain remain inactive in the absence of misfolded protein, in either a monomeric or an oligomeric configuration. However, in the presence of malfolded protein, client proteins associate with the MHC-like domain and dimerize adjacent IRE1 structures. This oligomerization of the luminal N-terminus activates the IRE1 protein kinase and RNase, which undergo transautophosphorylation rendering them active and able to unconventionally cleave XBP1u. Further evidence for this direct binding model exists for IRE1 with the discovery of two distinct peptide interfaces within the crystal structure of human IRE1 (Korennykh et al. 2010). Here, it is thought that the MHC-like domain of IRE1 resides within the first interface groove, which is proposed to bind malfolded protein (Credle et al. 2005). However, the discovery of a second interface has lent doubt to the role of malfolded proteins as a direct facilitator of luminal IRE1 dimerization (Gardner and Walter 2011). Instead, it is proposed that malfolded protein binding within the groove of the first interface induces conformational change in the luminal IRE1 domain, opening the second interface which thereby initiates IRE1 oligomerization and subsequent IRE1 activation. This direct binding model relegates GRP78/BiP from the original role of activation chaperone and characterizes it as a co-chaperone with the ability to regulate the downstream response of the UPR following IRE1 activation.
X-box binding protein 1 or XBP1 is a bZIP transcription factor that is involved in regulation of the unfolded protein response. It exists in two isoforms: XBP1u and XBP1s; XBP1u dominates in the unstressed cell. Upon initiation of the UPR, the IRE1-activated RNA 2′,3′-cyclic phosphate and 5′-OH ligase (RtcB) then unconventionally cleaves an 26-bp intron from XBP1u mRNA to form the active XBP1s mRNA isoform (Kosmaczewski et al. 2014; Yoshida et al. 2001)). This XBP1s mRNA isoform upregulates transcription of ER molecular chaperones and other stress protein genes. XBP1 is further upregulated by ATF-6 signaling as a result of GRP78/BiP dissociation (Schröder and Kaufman 2005a).
ATF-6
Activating transcription factor-6 is a type II ER transmembrane domain protein that encodes a basic leucine zipper (bZIP) motif in its cytosolic domain (Schröder and Kaufman 2005a, b). Upon dissociation with GRP78/BiP in response to ER lumen stress, ATF-6 translocates to the Golgi complex. Sphingosine-1-phosphate (S1P) then cleaves ATF-6 in the luminal domain and S2P in the N-terminal domain, resulting in the release of a bZIP motif to the nucleus (Schröder and Kaufman 2005a, b) and initiating transcription of genes coding for CHOP, XBP1 and ER stress proteins GRP78/BiP, GRP94 and calreticulin (Okada et al. 2002; Szegezdi et al. 2006).
There are two potential models of how GRP78/BiP functionally regulates ATF-6: the first suggesting the competitive binding of GRP78/BiP from ATF-6 by malfolded proteins and the second model proposing the active dissociation of GRP78/BiP from ATF-6 by signaling caused by malfolding of proteins (Feldman et al. 2005).
The UPR in Disease Pathogenesis
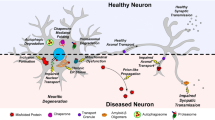
Within neurodegenerative biology, the precise pathophysiology of Alzheimer’s and Parkinson’s diseases remains controversial. It has long been recognized that the presence of malfolded protein aggregation is an early pathophysiological hallmark of both neurodegenerative diseases (Natale et al. 2013).
Activation of the UPR is thought to be an early feature of both Alzheimer’s and Parkinson’s diseases, and upregulation of UPR signaling has been widely observed in ex vivo human disease specimens (Nijholt et al. 2011; Bernales et al. 2012).
The proposed mechanism of ER stress induction differs across the neurodegenerative diseases. In Alzheimer’s disruption of ER calcium homeostasis has been directly implicated in the propagation of misfolded tau and β-amyloid protein, whereas impaired mitochondrial ATP generation is thought to precede the accumulation of α-synuclein in Parkinson’s disease (Brown and Naidoo 2012; Vaughn et al. 2014; Scheper et al. 2011; Gorbatyuk et al. 2012). Although these may both ultimately lead to characteristic UPR activation, these underlying differences have profound effects when considering disease-specific ER therapies (Roussel et al. 2013).
Initially, the UPR appears to offer cytoprotection in response to stressors, through upregulation of GRP78/BiP and selective attenuation of ribosomal translation via inhibition the MET-tRNA complex via EIF2A phosphorylation (Moreno et al. 2012). In rat models of Parkinson’s disease and transgenic Drosophila models of Alzheimer’s disease, substantial UPR-driven cytoprotection has been observed (Loewen and Feany 2010). In the Alzheimer’s model, UPR expression was induced by the accumulation of human tau protein. Reducing the activity of XBP1 using a transgenic loss function allele, significantly increased cytotoxicity of human tau protein. In the Parkinson’s model, overexpression of GRP78/BiP via gene delivery significantly reduced the neurotoxicity of α-synuclein, preventing neuronal loss and maintaining levels of striatal dopamine (Gorbatyuk et al. 2012). These findings are furthermore evidenced in parkinsonian models, where XBP1s modulates the transcription of UPR genes. In these models subsequent XBP1s silencing lead affected death of dopaminergic neurons(Valdés et al. 2014). These findings suggest that initially, the UPR has a protective role within maintaining proteostasis and cellular survival across the central nervous system.
However, there exists a controversy as to whether activation of the UPR does in fact offer neuroprotection during the development of disease, with particular regard to tau pathology. The initial cytoprotection the UPR offers may eventually decline leading to the promotion of tau hyperphosphorylation and overproduction of β-amyloid (Stutzbach et al. 2013).
Disruption in metabolic homeostasis within primary neurons induces the UPR and is widely employed as a means of modeling the effects of the UPR in vivo and in vitro. The phosphorylation of EIF2A has been identified as a key mediator of this response and is associated with an increase in misfolded protein aggregation. β-site APP cleaving enzyme-1 (BACE1), which serves to cleave amyloid precursor protein (APP) to form β-amyloid, is the rate-limiting enzyme in β-amyloid formation (Ferretti et al. 2012; Holsinger et al. 2002). Ex vivo specimens of patients with Alzheimer’s disease demonstrate elevated levels of BACE1 at the posttranscriptional level (Holsinger et al. 2002). Activation of the UPR has been implicated with upregulation of BACE1 activity through phosphorylated EIF2A (O’ Connor et al. 2009). Blocking EIF2A dephosphorylation using salubrinal increases BACE1 levels in the absence of ER stress, thus suggesting BACE1 as a downstream target of EIF2A. O’Connor et al. then demonstrated that inhibition of initial EIF2A phosphorylation activity reduces production of BACE1, emphasizing EIF2A as a potential therapeutic target.
Co-localization of UPR activation and tau phosphorylation by Van der Harg et al. suggests that EIF2A phosphorylation may also mediate the formation of tau aggregates (Van der Harg et al. 2014). Furthermore, phosphorylated Tau has been shown to impair the ERAD complex, thus further exacerbating cellular stress and neuronal apoptosis.
With increasing malfolded protein burden, the UPR transitions from cytoprotection toward activation of downstream, pro-apoptotic pathways. With severe or persistent endoplasmic reticulum stress, the buildup of ATF-4 and upregulation of XBP1s (the spliced active fragment of XBP1) promote the synthesis of CHOP, which through cytochrome c-mediated signaling cleaves caspase 3, leading to apoptosis. The ER transmembrane protein, IRE1, induces apoptosis through the tumor necrosis factor receptor-associated factor 2 (TRAF2)/B cell lymphoma 2 (Bcl2)/c-Jun N-terminal kinase (JNK) signaling pathway (Schröder and Kaufman 2005a; Halliday and Mallucci 2014). Substantial evidence supports that the presence of β-amyloid, tau protein and α-synuclein cause the UPR to activate the pro-apoptotic PERK/ATF-4 and IRE1/TRAF2 pathways inducing widespread ER-mediated neuronal apoptosis (Nakagawa et al. 2000; Bellucci et al. 2011; Abisambra et al. 2013). In both in vivo and in vitro models of Alzheimer’s disease and Parkinson’s disease, silencing of the PERK/ATF-4/CHOP pathway prevents neuronal apoptosis and returns cellular susceptibility to that of controls (Halliday and Mallucci 2014).
The UPR in Disease Progression
Defined patterns of spread have been identified across neurodegenerative disease states, lending support to the concept of prion-like transmission of these malfolded protein aggregates. Evidence suggests that malfolded proteins and other classical pathology demonstrate progression along anatomical planes and neuronal axes (Brundin et al. 2010; Costanzo and Zurzolo 2013).
The molecular basis of this prion-like spread remains elusive; nevertheless, experimental evidence suggests that malfolded proteins may be transported to neighboring cells to facilitate disease transmission. Transmission is thought to be facilitated through 3 pathways:
-
1.
Release of accumulated malfolded protein. A combination of active exocytotic release due to lysosomal export or autophagocystosis and passive release following neuronal death is thought to mediate the process of transmission.
-
2.
Passive or endocytic uptake of malfolded protein via cellular membranes. There is substantial evidence for the ability of neurons to uptake disease-associated proteins such as α-synuclein and amyloids in vitro.
-
3.
Nanotubule formation, permitting active transport of malfolded proteins between neurons.
Once the malfolded protein has been transported to the cytoplasm, it is unclear how this then may go on to influence translation and propagate disease (Brundin et al. 2010).
Regardless of how the mechanism of local transmission occurs, as the main immediate posttranslational cytoprotective pathway against malfolded proteins, UPR dysfunction has been implicated with the propagation of protein misfolding disease and prion-like neurodegeneration.
There is evidence that the UPR may contribute toward the spread of disease outside of the more immediately affected cellular bodies. Microglial activation is seen in response to aggregation of both β-amyloid and α-Synuclein, in addition to a range of other aggregate-forming neurological conditions including human immunodeficiency virus infection (Lee et al. 2010; Nunziante et al. 2011; Luk et al. 2012). Excessive microglial response has been associated with poor prognosis and escalating cognitive decline due to interleukin production within a closed positive feedback loop, which somewhat exacerbates the neurotoxicity of protein aggregates. Activation of the UPR within microglial cells in response to β-amyloid aggregates is associated with increased production of IL-6 and TNF-α, which then in turn leads to further inflammatory response, surmounting to heightened microglia activation (Kakimura et al. 2001).
Furthermore, impairment in proteasomal degradation of ERAD-targeted client proteins has been implicated in the spread of pathologic protein aggregates (Nunziante et al. 2011). Nunziante et al. showed that induction of ER stress and inhibition of the proteasomal degradation pathway resulted in significant enhancement of cellular prion protein (PrPc) trafficking and secretion, which thereby may enhance the formation of extracellular protein aggregates.
Further work is required to precisely characterize the role of the UPR as a mechanism of disease spread and propagation.
Dysfunction of the UPR in Disease
Dysfunction of the UPR and ER export pathways is thought to be key to the pathophysiological changes observed in neuronal tissue. Experimental reports of in vitro tau expression causing significant signal shift of functional ERAD reporters demonstrate that accumulation of tau interferes with the ERAD complex, preventing export to the proteasome and amplifying ER stress, forcing the UPR to remove aberrant protein via autophagy and apoptosis (Nakagawa et al. 2000; Scheper et al. 2011). This is validated by further findings in other Alzheimer’s models, where diminished function of the proteasome β-sub-unit was observed in response to endoplasmic reticulum stress induced in neuronal cells cultured with the UPR inducer tunicamycin (Nijholt et al. 2011).
The mechanisms underpinning UPR dysfunction are unclear. Endoplasmic reticulum chaperone defects and aging are thought to be implicated in driving disease. In both Alzheimer’s disease and Parkinson’s disease, significant disruption of the ER molecular chaperones involved in the UPR appear to be depleted and cannot activate the UPR correctly, despite conditions of ER stress in ex vivo human diseased brain tissue and in vivo models (Lee et al. 2010; Gorbatyuk et al. 2012). A novel GRP78/BiP-inducing compound, BiP inducer X, has been developed and shown promise in vivo, preventing neuronal death from ischemic sources of ER stress (Kudo et al. 2008). However, there have been no studies to date investigating its efficacy in neurodegenerative models, where the efficiency of proteasomal degradation is impaired.
As a consequence of aging, the balance of cytoprotection and pro-apoptotic drive in the UPR signaling pathway shifts to favor the pro-apoptotic functions. The ER is known to undergo structural alterations with age and becomes an increasingly dispersed structure. This is closely mirrored by depletions in the UPR chaperone GRP78/BiP, which therefore reduces the ability of the neuron to sense malfolded protein burden and potentially explaining the increased risk of both Alzheimer’s and Parkinson’s diseases with age (Brown and Naidoo 2012; Lee et al. 2010).
Genetic polymorphisms of UPR mediators may have a role to play in the disruption of ER protein folding. One of the key proponents for this idea is the presence PERK single-nucleotide polymorphisms (SNPs) in autopsy specimens from patients with progressive supranuclear palsy (Höglinger et al. 2011; Stutzbach et al. 2013). However, this area is currently largely unexplored.
Furthermore, microenvironmental factors must be carefully considered as contributors toward the production of misfolded protein states. It is well-known tissue hypoxia and other UPR-inducing states also influence the rapid accumulation of malfolded protein and activation of the UPR (Schröder and Kaufman 2005a). This is well illustrated in experimental ischemic stroke models. Following the occlusion of cerebral arteries of murine models and subsequent reperfusion, both striatal and cortical expression of GRP78/BiP, XBP1 and CHOP were co-localized to areas of focal ischemia (Urban et al. 2009; Rissanen et al. 2006; Morimoto et al. 2007). Interestingly, one of the studies pretreated rats with simvastatin, a HMG-CoA reductase inhibitor, which appeared to attenuate the unfolded protein response within ischemic foci; however, trials of statins in humans with Alzheimer’s disease have failed to provide demonstrable benefit (Sano et al. 2011; Urban et al. 2009). Further studies have additionally identified UPR activation as neuroprotective, with the constitutive expression of Bax Inhibitor-1 (BI-1) in transgenic mice modulating the UPR so as to limit the activity of pathways effecting caspase cleavage and leading to apoptosis (Morimoto et al. 2007; Rissanen et al. 2006).
The UPR as a Therapeutic Target
Due to interest in the UPR from other fields of medicine, namely oncology and organ preconditioning for transplant surgery, pharmacological compounds specifically targeting the UPR in neurodegenerative disease are currently in early stages of development.
Due to the complex nature of the UPR, current controversies in targeting and contrasts of findings are likely to be due to the heterogeneity of current disease models. As modeling and knowledge of neurodegenerative pathogenesis improves so will the ability to screen for compounds to therapeutically target the UPR.
Multiple compounds for targeting the endoplasmic reticulum have been described in the experimental setting and may currently be separated into four distinct classes (Table 1):
-
1.
Heat-shock-protein-inducing compounds.
-
2.
Inhibitors of the pro-apoptotic elements of the UPR.
-
3.
Proteasome-activity promoters.
-
4.
ER environment stabilizers.
As discussed previously, upregulation of GRP78/BiP via novel compounds, such as BiP inducer X, may prove a potent method of disrupting malfolded protein burden on neuronal cells and reversing the decline in the availability of GRP78/BiP with age. Increasing the availability of UPR chaperones increases the capacity of the ER stress response to target and export misfolded client proteins, thereby attenuating global stress and conferring cytoprotection.
Heat-shock binding proteins, or HSBs, include geldanamycin, tanespimycin and retaspimycin, all of which are small molecule inhibitors of HSP-90. This class of HSP-90 inhibitor is currently under investigation primarily in cancers; however, there has been interest in their apparent neuroprotective abilities (Luo et al. 2008). HSP-90 inhibitors are thought to exert this activity in three ways: the direct inhibition of HSP-90, the indirect inhibition of HSP-90-dependent tyrosine kinases (which maintain the neurodegenerative phenotype) and the upregulation of other HSP families (including HSP-70’s and thus GRP78/BiP; Luo et al. 2007; Ochel et al. 2001).
The Bonini group, have most notably produced a substantial body of evidence for the effects of HSP-90 inhibition in neurodegenerative disease. The group has identified that HSP-90 inhibition results in directed HSP-70 family upregulation, leading in turn to suppression of α-synuclein toxicity in Drosophila. Similar work has added weight to this, within both Alzheimer’s and Parkinson’s disease models (Auluck et al. 2002, 2005). In murine–rotenone-based models, administration of the HSP inducer carbenoxolone demonstrated significantly higher striatal dopamine levels and improved motor function versus untreated controls (Thakur and Nehru 2014).
Tanespimycin, a soluble analogue of Geldanamycin, has demonstrated acceptable toxicity profile in patients with both solid tumors and hematological malignancies (Dimopoulos et al. 2011). However, to date there are no registered trials for the investigation of tanespimycin in neurological disease (www.clinicaltrials.gov).
Manipulation of the PERK pathways, in particular the targeting of the downstream targets of phosphorylated EIF2A, can permit the evasion of apoptosis by the neuronal cell under malfolded protein burden. The induction of GADD34 overexpression by novel compounds, a protein which regulates the dephosphorylation of EIF2A, has been shown to prevent neuronal death versus control and lentiviral short hairpin RNA (shRNA) silenced of PrP proteins (Moreno et al. 2012). Hyperphosphorylation of PERK pathways is seen widely in protein misfolding diseases, and targeting EIF2A in this way may prove a novel strategy to prevent CHOP synthesis and mitigate neuronal cell death (Halliday and Mallucci 2014; Moreno et al. 2012).
Increasing the capacity of the ERAD and proteasome for export of misfolded client protein to alleviate luminal ER stress may also pose a novel solution. Of particular interest are compounds with antagonistic activity against arachidonate 5-lipoxygenase (5-LOX). The physiological role of 5-LOX is well characterized within the eicosanoid synthesis pathway, which has a critical role in the propagation of inflammatory processes. Novel curcumin pyrazole derivatives have been identified as having anti-5-LOX activity, which has shown to stimulate misfolded protein clearance in mice models with APP/PS1 transgenic Alzheimer’s mouse models (Valera et al. 2013).
Nevertheless, despite the putative therapeutic potential exhibited by these novel compounds, the immediate commercial availability is somewhat limited, prompting the identification of already licensed compounds which may exert a degree of activity upon the ER.
The tetracycline antibiotic, minocycline, features anti-LOX-5 activity and has been discussed extensively for use in the treatment of neurodegenerative conditions. Minocycline upregulates the anti-apoptotic protein Bcl2 in addition to anti-5-LOX activity, thus enabling suppression of apoptosis and proteasomal promotion and enabling misfolded protein burden to be cleared, in addition to inflammatory mediator suppression (Valera et al. 2013; Zhang and Bazan 2010; Yan 2013; Ferretti et al. 2012). Recent evidence additionally suggests that this may exert an inhibitory effect on BACE1, although it is unclear whether this is via interference with EIF2A phosphorylation (Ferretti et al. 2012). Initial trial results from the NINDS NET PD trial demonstrating non-futility in Parkinson’s disease have paved the way for further studies, including the Phase 2 minocycline in Alzheimer’s disease efficacy trial (MADE), which is due to report results in the latter half of the coming decade (MADE 2013; NINDS NET PD 2006).
Furthermore, experimental targeting of the ryanodine receptor (RyR) with the readily available drug dantrolene has been demonstrated to reduce hippocampal amyloid plaque load and improve cognitive function in murine models (Peng et al. 2012). Dantrolene’s action upon RyR prevents the disruption of intracellular calcium homeostasis, which is seen in Alzheimer’s disease, providing calcium channel stabilization. However, dantrolene as a candidate drug is unlikely to translate effectively to the clinical setting due to its side-effect profile, although it serves to highlight the therapeutic potential of calcium signaling.
In future, one of the more novel solutions may be the clinical use of pharmacological chaperones (pharmacoperones). Pharmacoperones are small lipophilic constructs which selectively bind malfolded protein intermediaries and facilitate folding to the target protein’s final conformational shape (Ulloa-Aguirre and Michael Conn 2012). If targeted to refold pathogenic proteins produced in neurodegenerative disease, this may be a powerful and highly selective means of disease targeting. Much work is required to overcome the technical challenges associated with molecular targeting of protein folding.
The UPR remains an attractive target in neurodegenerative disease for several reasons. The ability to target only cells which are affected by ER stress provides a highly specific means of halting the progression of neurodegenerative diseases at an early stage. Providing the neuron opportunity to recover from transient ER stress, preventing apoptosis and permitting malfolded proteins to be targeted for degradation would disrupt the pathological processes involved in protein misfolding diseases at several levels.
Conclusion
The role of the UPR in the pathogenesis and intercellular propagation of neurodegenerative diseases remains highly complex and poorly understood. It appears that there are significant disruptions within the endoplasmic reticulum, leading to the characteristic accumulation of malfolded protein in neurodegenerative disease.
Nevertheless, the UPR remains a potent target, with the ability to specifically target neurons producing malfolded proteins, preventing both neuronal apoptosis and local disease spread. Both novel and existing licensed compounds have demonstrated promise in the clinical setting and warrant further investigation.
References
Abisambra, J. F., Jinwal, U. K., Blair, L. J., O’Leary, J. C., Li, Q., Brady, S., et al. (2013). Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. Journal of Neuroscience, 33(22), 9498–9507. doi:10.1523/JNEUROSCI.5397-12.2013.Tau.
Alton, E. W. F. W, Boyd, A. C., Cheng, S. H., Cunningham S., Davies, J. C., Gill, D. R., & Griesenbach, U., et al. (2013). A randomised, double-blind, placebo-controlled phase IIB clinical trial of repeated application of gene therapy in patients with cystic fibrosis. Thorax, 68(11), 1075–1077. doi:10.1136/thoraxjnl-2013-203309. http://www.ncbi.nlm.nih.gov/pubmed/23525080.
Alzheimer Association. (2014). Alzheimer’s disease facts and figures.
Auluck, P. K., Chan, H. Y. E., & Trojanowski, J. Q. (2002). Chaperone suppression of {alpha}-synuclein toxicity in a drosophila model for Parkinson’s disease. Science, 295(5556), 865–868.
Auluck, P. K., Meulener, M. C., & Bonini, N. M. (2005). Mechanisms of suppression of {alpha}-synuclein neurotoxicity by Geldanamycin in Drosophila. The Journal of Biological Chemistry, 280(4), 2873–78. doi:10.1074/jbc.M412106200. http://www.ncbi.nlm.nih.gov/pubmed/15556931.
Back, S. H., Schröder, M., Lee, K. Zhang, K. & Kaufman, R. J. (2005). ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods (San Diego, Calif.), 35(4), 395–416. doi:10.1016/j.ymeth.2005.03.001. http://www.ncbi.nlm.nih.gov/pubmed/15804613.
Bellucci, A., Navarria, L., Zaltieri, M., Falarti, E., Bodei, S., Sigala, S., Battistin, L., Spillantini, M., Missale, C. & Spano, P. (2011). Induction of the unfolded protein response by Α-synuclein in experimental models of Parkinson’s Disease. Journal of Neurochemistry, 116(4), 588–605. doi:10.1111/j.1471-4159.2010.07143.x. http://www.ncbi.nlm.nih.gov/pubmed/21166675.
Bernales, S., Soto, M. M. & McCullagh, E. (2012). Unfolded protein stress in the endoplasmic reticulum and mitochondria: A role in neurodegeneration. Frontiers in Aging Neuroscience, 4, 5. doi:10.3389/fnagi.2012.00005. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3336107&tool=pmcentrez&rendertype=abstract.
Brewer, J. W., & Diehl, J. A. (2000). PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proceedings of the National Academy of Sciences of the United States of America, 97(23), 12625–12630. doi:10.1073/pnas.220247197. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=18814&tool=pmcentrez&rendertype=abstract.
Brown, M. K., & Naidoo, N. (2012). The endoplasmic reticulum stress response in aging and age-related diseases. Frontiers in Physiology 3, 263. doi:10.3389/fphys.2012.00263. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3429039&tool=pmcentrez&rendertype=abstract.
Brundin, P., Melki, R., & Kopito, R. (2010). Prion-like transmission of protein aggregates in neurodegenerative diseases. Nature Reviews. Molecular Cell Biology 11. Nature Publishing Group: 301–7. http://dx.doi.org/10.1038/nrm2873.
Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., Taddei, N., Ramponi, G., Dobson, C. M. & Stefani, M. (2002). Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature, 416(6880), 507–511. doi:10.1038/416507a. http://www.ncbi.nlm.nih.gov/pubmed/11932737.
Clarke, R., Cook, K. L., Hu, R., Facey, C. O. B., Tavassoly, I., Schwartz, J. L., & Baumann, W. T. et al. (2012). Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Research, 72(6), 1321–1331. doi:10.1158/0008-5472.CAN-11-3213. http://www.ncbi.nlm.nih.gov/pubmed/22422988.
Connor, T. O., Doherty-Sadleir, K. R., Maus, E., Velliquette, R. A., Cole, S. L., Eimer, W. A., & Hitt, B. et al. (2009). NIH public access 60(6), 988–1009. doi:10.1016/j.neuron.2008.10.047.Phosphorylation.
Costanzo, M., & Zurzolo, C. (2013). The cell biology of prion-like spread of protein aggregates: Mechanisms and implication in neurodegeneration. The Biochemical Journal, 452(1), 1–17. doi:10.1042/BJ20121898. http://www.ncbi.nlm.nih.gov/pubmed/23614720.
Credle, J. J., Finer-Moore, J. S., Papa, F. R., Stroud, R. M. & Walter, P. (2005). On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proceedings of the National Academy of Sciences of the United States of America, 102(52), 18773–84. doi:10.1073/pnas.0509487102. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1316886&tool=pmcentrez&rendertype=abstract.
Dimopoulos, M. -A., Mitsiades, C. S., Anderson, K. C., & Richardson P. G. (2011). Tanespimycin as antitumor therapy. Clinical Lymphoma, Myeloma & Leukemia, 11(1). Elsevier Inc. 17–22. doi:10.3816/CLML.2011.n.002. http://www.ncbi.nlm.nih.gov/pubmed/21454186.
Feldman, D. E., Chauhan, V., & Koong, A. C. (2005). The unfolded protein response: A novel component of the hypoxic stress response in tumors. Mol Cancer Res, 3(11), 597–605. doi:10.1158/1541-7786.mcr-05-0221. http://www.ncbi.nlm.nih.gov/pubmed/16317085.
Fernández-Nogales, M., Cabrera, J. R., Santos-Galindo, M., Hoozemans, J. J. M., Ferrer, I., Rozemuller, A. J. M., Hernández, F., Avila, J. & Lucas, J. J. (2014). Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nature Medicine, 20(8), 881–85. doi:10.1038/nm.3617. http://www.ncbi.nlm.nih.gov/pubmed/25038828.
Ferretti, M. T., Allard, S., Partridge, V., Ducatenzeiler, A., & Cuello, A. C. (2012). Minocycline corrects early, pre-plaque neuroinflammation and inhibits BACE-1 in a transgenic model of Alzheimer’s disease-like amyloid pathology. Journal of Neuroinflammation, 9(1). BioMed Central Ltd: 62. doi:10.1186/1742-2094-9-62. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3352127&tool=pmcentrez&rendertype=abstract.
Gardner, B. M., & Walter, P. (2011). Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science (New York, N.Y.), 333(6051), 1891–1894. doi:10.1126/science.1209126. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3202989&tool=pmcentrez&rendertype=abstract.
Gorbatyuk, M. S., Shabashvili, A., Chen, W., Meyers, C., Sullivan, L. F., Salganik, M., Lin, J. H., Lewin, A. S., Muzyczka, N., & Gorbatyuk, O. S. (2012). Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of Parkinson disease. Molecular Therapy: The Journal of the American Society of Gene Therapy, 20(7). Nature Publishing Group: 1327–1337. doi:10.1038/mt.2012.28. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3392977&tool=pmcentrez&rendertype=abstract.
Halliday, M., & Mallucci, G. R. (2014). Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology, 76 Pt A. Elsevier: 169–174. doi:10.1016/j.neuropharm.2013.08.034. http://www.ncbi.nlm.nih.gov/pubmed/24035917.
Harding, H. P., Zhang, Y., Bertolotti, A., Zeng, H., & Ron, D. (2000). Perk is essential for translational regulation and cell survival during the unfolded protein response. Molecular Cell, 5(5): 897–904. http://www.ncbi.nlm.nih.gov/pubmed/10882126.
Höglinger, G. U., Melhem, N. M., Dickson, D. W., Sleiman, P. M. A., Wang, L. -S., Klei, L., & Rademakers, R. et al. (2011). Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nature Genetics, 43(7). Nature Publishing Group: 699–705. doi:10.1038/ng.859. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3125476&tool=pmcentrez&rendertype=abstract.
Holsinger, D. R. M, McLean, C. A., Beyreuther, K., Masters, C. L., & Evin, G. (2002). Increased expression of the amyloid precursor B-secretase in Alzheimer’s disease. Annals of Neurology, 51(6), 782–786. doi:10.1002/ana.10168. http://www.ncbi.nlm.nih.gov/pubmed/12112082.
Investigators, MADE. (2013). Minocycline in Alzheimer’ S disease efficacy trial: The MADE trial. NIHR Protocol, 1–32.
Investigators, Ninds Net-Pd. (2006). A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology, 66(5). AAN Enterprises: 664–671.
Kakimura, J., Kitamura, Y., Taniguchi, T., Shimohama, S., & Gebicke-Haerter, P. J. (2001). Bip/GRP78-induced production of cytokines and uptake of amyloid-beta(1-42) peptide in microglia. Biochemical and Biophysical Research Communications, 281(1), 6–10. doi:10.1006/bbrc.2001.4299. http://www.ncbi.nlm.nih.gov/pubmed/11178952.
Korennykh, A. V., Egea, P. F., Korostelev, A. A., Finer-moore, J., Shokat, K. M., Stroud, R. M. & Walter, P. (2010). NIH public access, 457(3), 687–693. doi:10.1038/nature07661.The.
Kosmaczewski, S. G., Edwards, T. J., Han, S. M., Eckwahl, M. J., Meyer, B. I., Peach, S., Hesselberth, J. R., Wolin, S. L., & Hammarlund, M. (2014). The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Reports, 15(12), 1278–1285. doi:10.15252/embr.201439531. http://www.ncbi.nlm.nih.gov/pubmed/25366321.
Kudo, T, Kanemoto, S., Hara, H., Morimoto, N., Morihara, T., Kimura, R., Tabira, T., Imaizumi, K., & Takeda, M. (2008). A molecular chaperone inducer protects neurons from ER stress. Cell Death and Differentiation, 15(2), 364–375. doi:10.1038/sj.cdd.4402276. http://www.ncbi.nlm.nih.gov/pubmed/18049481.
Lee, A. S. (2007). GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Research, 67(8), 3496–3499. doi:10.1158/0008-5472.CAN-07-0325. http://www.ncbi.nlm.nih.gov/pubmed/17440054.
Lee, J. H., Won, S. M., Suh, J., Son, S. J., Moon, G. J., Park, U. J., & Gwag, B. J. (2010). Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Experimental & Molecular Medicine, 42(5), 386–94. doi:10.3858/emm.2010.42.5.040. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2877248&tool=pmcentrez&rendertype=abstract.
Lee, E. -J., Woo, M. -S., Moon, P. -G., Baek, M. -C., Choi, I. -Y., Kim, W. -K., Junn, E., & Kim, H. -S. (2010). Alpha-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. Journal of Immunology (Baltimore, Md.: 1950), 185(1), 615–623. doi:10.4049/jimmunol.0903480. http://www.ncbi.nlm.nih.gov/pubmed/20511551.
Li, J., & Lee, A. S. (2006). Stress induction of GRP78/BiP and its role in cancer. Current Molecular Medicine, 6(1), 45–54. http://www.ncbi.nlm.nih.gov/pubmed/16472112.
Loewen, C. A., & Feany, M. B. (2010). The unfolded protein response protects from tau neurotoxicity in vivo. PloS One, 5(9), 1–6. doi:10.1371/journal.pone.0013084. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2947516&tool=pmcentrez&rendertype=abstract.
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., Brien, P. O., Trojanowski, J. Q., & Lee, V. M. (2012). Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science, 338, 949–954.
Luo, W., Dou, F., Rodina, A., Chip, S., Kim, J., Zhao, Q., & Moulick, K., et al. (2007). Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proceedings of the National Academy of Sciences of the United States of America, 104(22): 9511–9516. doi:10.1073/pnas.0701055104. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1890525&tool=pmcentrez&rendertype=abstract.
Luo, W., Rodina, A., & Chiosis, G. (2008). Heat shock protein 90: translation from cancer to Alzheimer’s disease treatment? BMC Neuroscience, 9(Suppl 2), S7. doi:10.1186/1471-2202-9-S2-S7. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2604891&tool=pmcentrez&rendertype=abstract.
McLaughlin, M., & Vandenbroeck, K. (2011). The endoplasmic reticulum protein folding factory and its chaperones: New targets for drug discovery? British Journal of Pharmacology, 162(2), 328–345. doi:10.1111/j.1476-5381.2010.01064.x. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3031055&tool=pmcentrez&rendertype=abstract.
Moreno, J. A., Radford, H., Peretti, D., Steinert, J. R., Verity, N., Martin, M. G., Halliday, M., et al. (2012). Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature, 485(7399). Nature Publishing Group: 507–511. doi:10.1038/nature11058. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3378208&tool=pmcentrez&rendertype=abstract.
Morimoto, N., Oida, Y., Shimazawa, M., Miura, M., Kudo, T., Imaizumi, K., & Hara, H. (2007). Involvement of endoplasmic reticulum stress after middle cerebral artery occlusion in mice. Neuroscience, 147(4). IBRO: 957–967. doi:10.1016/j.neuroscience.2007.04.017. http://www.ncbi.nlm.nih.gov/pubmed/17590517.
Nakagawa, T., Zhu, H., Morishima, N., & Li, E. (2000). Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. 403, 8–13.
Natale, G., Pompili, E., Biagioni, F., Paparelli, S., Lenzi, P., & Fornai, F. (2013). Histochemical approaches to assess cell-to-cell transmission of misfolded proteins in neurodegenerative diseases. European Journal of Histochemistry: EJH, 57(1), e5. doi:10.4081/ejh.2013.e5. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3683612&tool=pmcentrez&rendertype=abstract.
Nijholt, D. A. T., de Graaf, T. R., van Haastert, E. S., Osório Oliveira, A., Berkers, C. R., Zwart, R., Ovaa, H., Baas, F., Hoozemans, J. J. M., & Scheper, W. (2011). Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: Implications for Alzheimer’s disease. Cell Death and Differentiation, 18(6), 1071–1081. doi:10.1038/cdd.2010.176. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3131935&tool=pmcentrez&rendertype=abstract.
Nunziante, M., Ackermann, K., Dietrich, K., Wolf, H., Gädtke, L., Gilch, S., Vorberg, I., Groschup, M., & Schätzl, H. M. (2011). Proteasomal dysfunction and endoplasmic reticulum stress enhance trafficking of prion protein aggregates through the secretory pathway and increase accumulation of pathologic prion protein. The Journal of Biological Chemistry, 286(39). 9650 Rockville Pike, Bethesda, MD 20814, U.S.A.: American Society for Biochemistry and Molecular Biology: 33942–33953. doi:10.1074/jbc.M111.272617. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3190803/.
Ochel, H. -J., Eichhorn, K., & Gademann, G. (2001). Geldanamycin: The prototype of a class of antitumor drugs targeting the heat shock protein 90 family of molecular chaperones. Cell Stress & Chaperones, 6(2), 105. doi:10.1379/1466-1268(2001)006<0105:GTPOAC>2.0.CO;2. http://cest.allenpress.com/perlserv/?request=get-abstract&doi=10.1379%2F1466-1268(2001)006%3C0105%3AGTPOAC%3E2.0.CO%3B2.
Okada, T., Yoshida, H., Akazawa, R., Negishi, M., & Mori, K. (2002). Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. The Biochemical Journal, 366(Pt 2), 585–594. doi:10.1042/BJ20020391. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1222788&tool=pmcentrez&rendertype=abstract.
Peng, J., Liang, G., Inan, S., Joseph, D. J., Meng, Q., Peng, Y., et al. (2012). Dantrolene ameliorates cognitive decline and neuropathology in alzheimer triple transgenic mice. Neuroscience Letters, 516(2), 274–279. doi:10.1016/j.neulet.2012.04.008.Dantrolene.
Prince, M., Bryce, R., Albanese, E., Wimo, A., Ribeiro, W., & Ferri, C. P. (2013). The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 9(1). Elsevier Ltd: 63–75.e2. doi:10.1016/j.jalz.2012.11.007. http://www.ncbi.nlm.nih.gov/pubmed/23305823.
Quinones, Q. J., de Ridder, G. G., & Pizzo, S. V. (2008). GRP78: A chaperone with diverse roles beyond the endoplasmic reticulum. Histology and Histopathology, 23(11), 1409–1416. http://www.ncbi.nlm.nih.gov/pubmed/18785123.
Reddy, R. K, Mao, C., Baumeister, P., Austin, R. C., Kaufman, R. J., & Lee, A. S. (2003). Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. The Journal of Biological Chemistry, 278(23), 20915–20924. doi:10.1074/jbc.M212328200. http://www.ncbi.nlm.nih.gov/pubmed/12665508.
Rissanen, A., Sivenius, J., & Jolkkonen, J. (2006). Prolonged bihemispheric alterations in unfolded protein response related gene expression after experimental stroke. Brain Research, 1087(1), 60–66. doi:10.1016/j.brainres.2006.02.095. http://www.ncbi.nlm.nih.gov/pubmed/16684512.
Roussel, B. D., Kruppa, A. J., Miranda, E., Crowther, D. C., Lomas, D. A., & Marciniak, S. J. (2013) Endoplasmic reticulum dysfunction in neurological disease. The Lancet Neurology, 12(1). Elsevier Ltd: 105–118. doi:10.1016/S1474-4422(12)70238-7. http://www.ncbi.nlm.nih.gov/pubmed/23237905.
Sano, M., Thomas, R. G., & Van Dyck, C. H. (2011). Placebo-controlled trial of simvastatin to treat Alzheimer sisease.
Scheper, W., Nijholt, D. A. T., & Hoozemans, J. J. M. (2011). The unfolded protein response and proteostasis in Alzheimer disease: Preferential activation of autophagy by endoplasmic reticulum stress. Autophagy, 7(8), 910–911. doi:10.4161/auto.7.8.15761. http://www.landesbioscience.com/journals/autophagy/article/15761/.
Schröder, M., & Kaufman, R. J. (2005a). The mammalian unfolded protein response. Annual Review of Biochemistry, 74, 739–789. doi:10.1146/annurev.biochem.73.011303.074134. http://www.ncbi.nlm.nih.gov/pubmed/15952902.
Schröder, M., & Kaufman, R. J. (2005b). ER stress and the unfolded protein response. Mutation Research, 569(1–2), 29–63. doi:10.1016/j.mrfmmm.2004.06.056. http://www.ncbi.nlm.nih.gov/pubmed/15603751.
Stutzbach, L. D., Xie, S. X., Naj, A. C., Albin, R., Gilman, S., Lee, V. M. Y., Trojanowski, J. Q., Devlin, B., & Schellenberg, G. D. (2013). The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear Palsy and Alzheimer’s disease. Acta Neuropathologica Communications, 1(1), 31. doi:10.1186/2051-5960-1-31. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3893579&tool=pmcentrez&rendertype=abstract.
Szegezdi, E., Logue, S. E., Gorman, A. M., & Samali, A. (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Reports, 7(9), 880–885. doi:10.1038/sj.embor.7400779. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1559676&tool=pmcentrez&rendertype=abstract.
Thakur, P., & Nehru, B. (2014). Long-term heat shock proteins (HSPs) induction by carbenoxolone improves hallmark features of Parkinson’s disease in a rotenone-based model. Neuropharmacology, 79. Elsevier Ltd: 190–200. doi:10.1016/j.neuropharm.2013.11.016. http://www.ncbi.nlm.nih.gov/pubmed/24296154.
Tirasophon, W., Welihinda, A. A., & Kaufman, R. J. (1998). A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes & Development, 12(12), 1812–1824. doi:10.1101/gad.12.12.1812. http://www.genesdev.org/cgi/doi/10.1101/gad.12.12.1812.
Ulloa-Aguirre, A., & Conn, P. M. (2012). Pharmacoperones: A new therapeutic approach for diseases caused by misfolded G protein-coupled receptors. Recent Patents on Endocrine, Metabolic & Immune Drug Discovery, 5(1), 13–24.
Urban, P., Pavlíková, M., Sivonová, M., Kaplán, P., Tatarková, Z., Kaminska, B., & Lehotský, J. (2009). Molecular analysis of endoplasmic reticulum stress response after global forebrain ischemia/reperfusion in rats: Effect of neuroprotectant simvastatin. Cellular and Molecular Neurobiology 29 (2): 181–92. doi:10.1007/s10571-008-9309-7. http://www.ncbi.nlm.nih.gov/pubmed/18807172.
Valdés, P., Mercado, G., Vidal, R. L., Molina, C., Parsons, G., Court, F. A., Martinez, A., et al. (2014). Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proceedings of the National Academy of Sciences of the United States of America, 111(18), 6804–6809. doi:10.1073/pnas.1321845111. http://www.ncbi.nlm.nih.gov/pubmed/24753614.
Valera, E., Dargusch, R., Maher, P. A., & Schubert, D. (2013). Modulation of 5-lipoxygenase in proteotoxicity and Alzheimer’s disease. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 33(25), 10512–10525. doi:10.1523/JNEUROSCI.5183-12.2013. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3685841&tool=pmcentrez&rendertype=abstract.
Van der Harg, J. M., Nölle, A., Zwart, R., Boerema, A. S., van Haastert, E. S., Strijkstra, A. M., Hoozemans, J. J., & Scheper, W. (2014). The unfolded protein response mediates reversible tau phosphorylation induced by metabolic stress. Cell Death & Disease, 5, e1393. doi:10.1038/cddis.2014.354. http://www.ncbi.nlm.nih.gov/pubmed/25165879.
Vaughn, L. S., Snee, B., & Patel, R. C. (2014). Inhibition of PKR protects against tunicamycin-induced apoptosis in neuroblastoma cells. Gene, 536(1). Elsevier B.V. 90–96. doi:10.1016/j.gene.2013.11.074. http://www.ncbi.nlm.nih.gov/pubmed/24334130.
Yan, Y. (2013). Minocycline alleviates beta-amyloid protein and tau pathology via restraining neuroinflammation induced by diabetic metabolic disorder, 1089–1095.
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., & Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell, 107(7), 881–891. http://www.ncbi.nlm.nih.gov/pubmed/11779464.
Zhang, C., & Bazan, N. G. (2010). Lipid-mediated cell signaling protects against. doi:10.3945/jn.109.114884.858.
Conflict of interest
The author has no conflicts of interest to declare. This work was unfunded.
Ethical standard
This manuscript does not contain clinical studies, patient or animal data.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Drake, T.M. Unfolding the Promise of Translational Targeting in Neurodegenerative Disease. Neuromol Med 17, 147–157 (2015). https://doi.org/10.1007/s12017-015-8346-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-015-8346-x