
Abstract
Pemphigus is a group of rare, potentially devastating autoimmune diseases of the skin and mucous membranes with high morbidity and potentially lethal outcome. The major clinical variant, pemphigus vulgaris (PV) is caused by a loss of intercellular adhesion of epidermal keratinocytes which is induced by IgG autoantibodies against components of desmosomes. Specifically, IgG against the desmosomal adhesion proteins, desmoglein 3 (Dsg3) and desmoglein 1 (Dsg1), preferentially target their ectodomains which are presumably critical for the transinteraction and signalling function of these adhesion molecules. There is a close immunogenetic association of PV with the human leukocyte antigen (HLA) class II alleles, HLA-DRB1*04:02 and HLA-DQB1*05:03. These have been shown to be critical for the presentation of immunodominant peptides to autoreactive CD4+ T helper cells. The importance of autoaggressive T-B cell interaction in the induction of pathogenic IgG autoantibodies which directly cause epidermal loss of adhesion has been demonstrated both clinically (by the use of the anti-CD20 monoclonal antibody rituximab) and experimentally (in PV mouse models). The strong association of clinically active pemphigus with autoantibodies of the IgG4 and IgE subclasses strongly suggests that T helper 2 cells are critical regulators of the immune pathogenesis of pemphigus. Novel therapeutic approaches target autoreactive T and B cells to specifically interfere with the T cell-dependent activation of B cells leading to the generation of autoantibody-producing plasma cells. Our improved understanding of the autoantibody-driven effector phase of pemphigus has led to the introduction of novel therapies that target pathogenic autoantibodies such as immunoadsorption and drugs that block pathogenic autoantibody-induced cell signalling events.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pemphigus encompasses a group of life-threatening autoimmune bullous diseases characterized by flaccid blisters and erosions of the mucous membranes and skin [1] (Table 1). The underlying loss of epidermal cell-adhesion is caused by IgG autoantibodies against the desmosomal adhesion proteins, desmoglein 3 (Dsg3) and/or Dsg1, on epidermal keratinocytes. In general, anti-Dsg IgG serum concentrations relate to the clinical activity of pemphigus [1,2,3]. The presence of anti-Dsg3 IgG and anti-Dsg1 IgG is in general well-correlated to a distinct clinical phenotype, i.e., mucosal and cutaneous involvement, respectively [4]. Pemphigus vulgaris (PV), the major clinical variant, is rare and is linked to distinct human leukocyte antigen (HLA) class II alleles, namely HLA-DRB1*04:02 and HLA-DQB1*05:03 [5,6,7,8,9,10]. Based on its well-characterized immune pathogenesis with precisely identified autoantigens showing a tissue-specific expression pattern, PV can be considered as a paradigm of an organ-specific autoimmune disorder. Clinically, PV poses a therapeutic challenge for the clinician and is associated with high mortality [1, 11,12,13]. The therapeutic mainstay of pemphigus are systemic glucocorticoids which cause considerable co-morbidities upon chronic use [1, 14]. Immunosuppressants have a glucocorticoid-sparing effect but are not morbostatic per se [14]. Thus, there is urgent need to treat pemphigus more specifically and to reduce side effects of treatment. The B cell targeting mAb, rituximab has shown great promise in inducing clinical remissions [15,16,17]. Still, long-term clinical remissions require repeated immunosuppressive treatment with rituximab. We will describe here therapeutic options which are based on our improved understanding of the immune pathogenesis of pemphigus.
Pemphigus Vulgaris (PV)
PV is the most common clinical pemphigus variant with an incidence of 0.1–0.5/105 persons without a sex preference. PV affects preferentially adults with a peak at the 4th–6th decade while children and elderly are rarely affected [1]. Juvenile PV in young adults below the 3rd decade is extremely rare and shows a clinical course with preferential involvement of the oral mucosa as in adulthood. Prior to the advent of systemic corticosteroids and immunosuppressants, the prognosis of pemphigus was fatal within 5 years and reached a mortality of 50% already after 2 years as the consequence of secondary complications including infections and malnutrition [14]. Upon treatment with corticosteroids, the mortality of PV has decreased to 10% although there is still a significant morbidity due to chronic immunosuppressive therapy such as infections.
The etiology of PV is largely unknown. Among the factors discussed are environmental agents, infections, drugs and tumors. A major confounding factor is the significant association of PV with distinct human leukocyte antigen (HLA) class II alleles. Specifically, an increased prevalence of PV in distinct ethnic groups such as Jews, Iraqi, Indians and Iranians is well-known. This is explained by the high prevalence of distinct HLA class II alleles in these ethnic groups which are associated with PV. In addition to HLA-DRB1*0402 in Jewish people [6] and HLA-DQB1*0503 in non-Jewish populations [5, 7], PV is also associated with HLA-G and the transporter associated processing (TAP) gene [18].
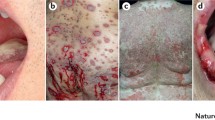
In most instances, PV initially manifests as painful erosions of the oral mucosa (70%) or the genital mucosa (20%), rarely also with erosions of the conjunctivae which are extremely painful and tend to progress [1, 11]. Other affected sites are lips, palate, cheeks and gingiva. When larynx and pharynx are also involved, acute hoarseness is a clinical hallmark. However, intact bullae are hard to find at the oral mucosa rather than painful erosions with remnants of blisters in the periphery. Affected patients are severely impaired with eating, swallowing and talking. Accompanying symptoms are sialorrhoea, bloody saliva and fetor ex ore. Esophageal involvement is rare. The majority of the patients develop flaccid blisters on the trunk, preferentially on the seborrheic skin areas which transform into slowly healing crusty erosions with a tendency to progression (Fig. 1a–c). The initial (mucosal) lesions of PV are often misdiagnosed as herpetic infection, aphthosis, oral candidosis, erosive lichen planus or gingivitis without major therapeutic success (Fig. 1d–f). For this reason, a diagnostic biopsy and immune serological diagnostics to rule out pemphigus are essential.

Pemphigus vulgaris (PV). Extensive, partly confluent erosions in the seborrheic areas of the face and upper thorax (a), trunk (b) and shoulders (c). Sharply demarcated erosions of the palate (d) and upper jaw (e) and flaccid blister at the mucosal surface of the lower lip (f)
Pemphigus Foliaceus (PF)
PF is much rarer than PV and occurs as a sporadic form (Cazenave type) and a related endemic form which mainly occurs in Brazil (pemphigus braziliensis or fogo selvagem) and North Africa (Tunisia). In general, epidemic PF is associated with skin lesions only which may preferentially occur at the seborrheic skin areas, i.e., scalp, face, chest and shoulders. Initially, PF presents with flaccid bullae and later on, with erythematous hyperkeratotic plaques and scaly crusts at the seborrheic body sites (Fig. 2). Eventually, if left untreated, the lesions may progress and lead to erythroderma accompanied by pruritus and burning of the skin. As in PV, there is no sex predilection. Localized PF variants, which mainly affect the seborrheic areas of the face and scalp, are pemphigus seborrhoicus and pemphigus erythematosus which is associated with antinuclear antibodies. These PF variants do occur in children but are rare as PF is less frequently found in children than PV [19].

Pemphigus foliaceus (PF). Scaly erosions and plaques at the seborrheic areas of the trunk (a), scaly superficial erosions clearly visible at higher magnification (b)
Brazilian PF (“fogo selvagem”) is an endemic variant of PF which occurs in the rural areas of Limao Verde. Clinically, it resembles sporadic PF but is more frequent in children and young adults [19]. In the endemic regions, there is a greater prevalence of anti-Dsg1 IgG autoantibodies also in clinically healthy individuals. A local rural factor is considered as the initiator of the autoimmune response against Dsg1 such as the saliva of endemic sand flies [20].
Paraneoplastic Pemphigus (PNP)
PNP is a pemphigus variant which is part of an autoimmune syndrome associated with neoplasms of various origins. Clinically, PNP is usually characterized by painful, extensive mucosal erosions and lichenoid papules of the extremities (Fig. 3). Characteristic is the pronounced mucositis and variable skin lesions ranging from erythema multiforme, lichenoid papules, extensive erythemas with epidermolysis which may transform into toxic-epidermal necrolysis. PNP is mostly associated with lymphoproliferative disorders such as chronic lymphocytic leukemia, plasmocytoma, Castleman’s disease and B cell lymphoma. Concomitant thymoma is also relatively common [21]. Unlike other pemphigus disorders, loss of adhesion of the lung epithelium (obliterating bronchiolitis), which is linked to the presence of anti-plakin autoantibodies, can be fatal.

Paraneoplastic pemphigus. Extensive erosions of the conjunctivae and oral mucosa and lips (a) and polymorphic erythemas of the palmae (b)
PNP should be a consideration if mucosal lesions show a clear progression despite initiation of immunosuppressive treatment with systemic glucocorticoids and/or adjuvant immunosuppressants. In the case of PNP, perilesional biopsies reveal a mixed epidermal keratinocyte surface staining of tissue-bound IgG and/or complement and IgG and/or C3 deposits at the dermal-epidermal basement membrane zone. In addition, PNP sera do not only bind the surface of epithelial cells of monkey esophagus but also to plakin-rich urinary bladder epithelium which is not targeted by PV sera.
PNP is primarily associated with IgG autoantibodies against several members of the plakin family such as the desmosomal plaque proteins desmoplakin I, II, envoplakin, periplakin and against hemidesmosomal adhesion molecules such as plektin and BP230 [22,23,24,25]. Recently, we identified a PNP patient with IgG reactivity against BP180 [26]. The 170 kD autoantigen, which is recognized by the majority of PNP sera and thus of high diagnostic value, has been recently identified as the protease inhibitor, alpha-2 macroglobulin-like 1 (A2ML1). The role of A2ML1-specific IgG autoantibodies in the pathogenesis of PNP is yet unclear [29].
Once the diagnosis of PNP is established by immunoserological measures, a thorough work-up should be performed to identify or exclude associated internal malignancies, in particular lymphatic disorders as mentioned above. The overall prognosis of PNP is closely linked to effective treatment of the underlying malignancy.
Rare Pemphigus Variants
A rare (< 5%) variant of PV is pemphigus vegetans which presents with hypertrophic vegetating plaques preferentially at the intertriginous body areas, i.e., inguinal, anogenital and neck region. The chronic vegetating lesions are consequence of the incomplete and persisting wound healing of chronic erosions. The more aggressive Neumann type presents with centrifugally developing purulent hypertrophic, papillomatous, partly verruciform plaques of the intertriginous body folds and lips, more rarely on the trunk and extremities with a fetid odor. The less aggressive Hallopeau type is associated with vegetating plaques and concomitant pustules. Pemphigus herpetiformis presents with herpetiform flaccid cutaneous bullae, mucosal involvement is rare [1, 11].
IgA Pemphigus
IgA pemphigus is characterized clinically rather by pustules than by flaccid blisters. Characteristic are annular pustular lesions which are more strongly pronounced at the periphery (Fig. 4). Mucosal involvement is rare. IgA pemphigus may be associated with gammopathies and hematological disorders. Direct immunofluorescence microscopy (DIF) of perilesional skin shows IgA deposits on the surface of epidermal keratinocytes in a netlike pattern. By indirect IF (IIF) (ca. 50%), IgA pemphigus sera show anti-epithelial IgA antibodies which bind to the surface of epithelial cells of monkey esophagus in a netlike pattern. Two variants of IgA pemphigus can be distinguished: intraepidermal neutrophilic disease is commonly associated with IgA antibodies against Dsg1 or Dsg3, and a subcorneal pustular dermatosis which is associated with IgA against desmocollin 1 (Dsc1) [28,29,30].

IgA pemphigus. Erythematous plaques with peripheral scaly pearl-like erosions of the trunk (a)
Diagnostic Work-Up
Pemphigus presents with a broad spectrum of clinical symptoms. The mainstay of diagnostics consists of histopathology (loss of epidermal adhesion), DIF and IIF which are complemented by the serological detection of IgG autoantibodides against distinct autoantigens by ELISA and immunoblot analysis [1, 11].
By histopathology, pemphigus is characterized by an intraepidermal loss of cellular adhesion. PV shows acantholysis as a consequence of suprabasal loss of epidermal keratinocyte adhesion (Fig. 5a). Single or grouped acantholytic cells can be retrieved by smears from blisters or erosions (Tzanck test). Acantholysis is also present in hair follicles and the ducts of sebaceous glands. Characteristic is eosinophilic spongiosis, i.e. the influx of eosinophils which is associated with loss of epidermal cell-cell adhesion. In PF, acantholysis is more superficial, i.e., subcorneal at the level of the stratum spinosum. Pemphigus vegetans shows extensive epidermal acanthosis and papillomatosis, acantholytic areas and a marked neutrophilic and eosinophil-rich infiltrate with intraepidermal pustules. IgA pemphigus is also associated with the epidermal influx of neutrophils. PNP shows, in addition to acantholysis, a band-like lichenoid dermal infiltrate.

Diagnostics in pemphigus. Suprabasilar loss of keratinocyte adhesion (a) by histopathology of lesional skin, IgG deposits on the surface of epidermal keratinocytes with a netlike epidermal staining pattern by direct immunofluorescence microscopy (DIF) (b), netlike staining pattern of PV sera monkey esophagus (c) and rat urinary bladder (d) by indirect IF
Critical for the diagnosis of pemphigus is the detection of tissue-bound and serum IgG or IgA autoantibodies by DIF and IIF. For proper DIF diagnostics, biopsies are taken from perilesional skin as tissue inflammation may lead to rapid degradation of tissue-bound autoantibodies. Tissues samples are stored in Michel’s medium or saline solution [14]. DIF show deposits of IgG and/or C3 on the surface of epidermal keratinocytes with a netlike intraepidermal staining pattern (Fig. 5b).
IIF of patients’ sera is performed on different substrates including monkey esophagus (Fig. 5c), human skin and rat or rabbit urinary bladder epithelium (Fig. 5d). Upon incubation of the pemphigus sera with these substrates, IIF shows a netlike cell surface staining with IgG and IgA deposits on the surface of epithelial cells. The sensitivity of IIF in detecting desmosomal autoantibodies is in the range of 80–90%. Monkey esophagus is the main substrate for the detection of Dsg autoantibodies, while rat or monkey urinary bladder serve as a substrate for the detection of plakin-specific IgG. Cell surface deposits of serum IgG on monkey esophagus epithelial cells by IIF are also found in individuals with IgG against certain blood group factors which may cross-react. Antibodies against desmoplakin with the identical staining pattern on monkey esophagus are occasionally seen in patients with severe erythema multiforme.
The identification of the autoantigens of pemphigus has significantly improved the serological diagnosis and classification of this group of immunobullous skin disorders. The major autoantigens of pemphigus, Dsg1 and Dsg3, are available in recombinant form and are widely used to identify and monitor IgG autoantibodies by ELISA. PV with exclusive involvement of the mucous membranes is associated with IgG against Dsg3, while the mucocutaneous variant of PV is associated with both, anti-Dsg1 and anti-Dsg3 IgG. In contrast, PF is characterized by IgG against Dsg1 only. Mucosal involvement in PF is extremely rare. In PV, in addition to anti-Dsg IgG, autoantibodies against other desmosomal adhesion molecules, such as Dsc and plakins, as well as non-related proteins such as acetyl choline receptors and pemphaxin, have been reported [31,32,33]. The pathogenic significance of the latter autoantibodies has not yet been proven. As the concentrations of serum IgG autoantibodies against Dsg1 and Dsg3 generally correlate with the clinical activity, Dsg ELISA is also considered as a good serological marker of disease activity in pemphigus. Anti-Dsg autoantibodies are mainly of the IgG4 and IgE subtypes and are presumably regulated by autoreactive T helper 2 cells.
Immunoblot and immune precipitation are additional immune serological assays which are of secondary importance for routine diagnostics. However, immunoblot assays with recombinant autoantigenic proteins are available in specialized laboratories to complement the commercially available Dsg ELISA in cases of atypical pemphigus or PNP. In atypical pemphigus, IgG autoantibodies against Dsg1 and Dsg3 are not detectable in the patients’ sera. In these cases, the Dsc, particularly Dsc3 represent the major autoantigen(s) [34, 35]. The pathogenicity of anti-Dsc3 IgG has been shown ex vivo with IgG purified from patients’ sera. Anti-Dsc3 IgG has been also identified in PNP, pemphigus vegetans and pemphigus herpetiformis [35].
Dsg-Specific IgG Autoantibodies
A critical role of Dsg-specific autoantibodies in the pemphigus pathogenesis was confirmed by clinical observations and various in vitro and in vivo studies. The emergence of Dsg-specific autoantibodies in pemphigus patients and a general correlation of autoantibody titers with disease activity indicate a tight connection between Dsg-specific autoantibodies and disease development and progression [36,37,38,39,40]. Furthermore, due to diaplacentar transfer of maternal autoantibodies, newborns of mothers suffering from pemphigus also develop intraepidermal blisters [41, 42]. In animal models, passive transfer of purified patient IgG or Dsg-specific IgG and the transfer of Dsg3-specific lymphocytes lead to a pemphigus-like phenotype [43,44,45,46,47,48,49]. In vitro, Dsg-specific autoantibodies lead to keratinocyte dissociation induced by the loss of desmosomal integrity [50,51,52,53,54,55,56].
Although anti-Dsg IgG serum concentrations generally correlate with disease activity, some patients also exhibit anti-Dsg3 IgG in clinical remission, strongly suggesting the existence of non-pathogenic Dsg-specific IgG [57]. Further investigations revealed that these autoantibodies, considered as non-pathogenic, were not able to induce keratinocyte dissociation in vitro [58, 59]. Thus, the autoantibody profile of pemphigus patients consists of pathogenic and non-pathogenic Dsg-specific antibodies [60, 61].
In PV, their pathogenic potential is presumably dependent on recognition of distinct Dsg epitopes [45, 60,61,62,63,64,65,66,67]. Pathogenic IgG autoantibodies preferentially target the NH2-terminal portion of Dsg3 whereas non-pathogenic autoantibodies are thought to recognize epitopes of the membrane proximal COOH-terminus of the Dsg ectodomains. Amagai et al. [45] identified IgG reactive with the NH2-terminal EC1-EC2 subdomains of Dsg3 which induced intraepidermal blisters in neonatal mice, while IgG targeting the COOH-terminal EC3-EC5 subdomains of Dsg3 failed to induce intraepidermal blistering. Utilizing Dsg3-specific mAb from an active mouse model of PV, the group of Amagai confirmed this pathogenicity concept [61]. Along this line, based on domain swapped recombinant Dsg proteins, they showed that most of the Dsg-specific IgG autoantibodies from PV patients recognize epitopes inside the EC1-EC2 subdomains of Dsg3 [68,69,70]. Furthermore, it was shown that in active PV and PF, the Dsg-specific IgG autoantibody profile mainly targets the NH2-terminus of the Dsg ectodomain [68, 69]. Within the NH2 terminal domains, major conformational B cell epitopes are mapped to aa26-aa87 of Dsg1 (EC1) and aa25-aa88 of Dsg3 (EC1) [71].
Autoantibody binding largely depends on the degree of maturation of the Dsg. Prior to cleavage in the Golgi apparatus and integration into the plasma membrane, cadherins, like Dsg1 and Dsg3, are synthesized as a premature form in the endoplasmic reticulum. In PF, sera recognize the mature (mat) form of Dsg1 while non-pathogenic mAb presumably target either the mature or the premature (pre) form of Dsg1 [65, 66]. This is in line with previous findings in PV where pathogenic anti-Dsg3 mAb preferentially bound to matDsg3 [72]. Interestingly, for pathogenic mAb specific for the matDsg3 and preDsg3 the binding was blocked by most of PV sera whereas for matDsg3 specific mAb only, like the murine anti-Dsg3 mAb, AK23, the binding capacity was mostly unaffected [64]. This indicates that despite similar epitope recognition, these mAb exhibit different binding properties.
Autoantibody Isotypes
IgG4 presumably represents the major pathogenic antibody isotype in the pemphigus pathogenesis [63, 64, 67, 73,74,75,76]. As an example, the switch from non-pathogenic IgG1 to pathogenic IgG4 has been described in fogo selvagem (FS), the endemic Brazilian subtype of PF. Specifically, 55% of healthy individuals in that rural area exhibit anti-Dsg1 (EC5) IgG1. Upon transition into clinically apparent PF, patients experience a subclass and epitope switch to EC1/EC2-reactive IgG4 [77,78,79]. Recently, a pathogenic anti-Dsg3 IgG1 antibody was isolated from a PV patient which strongly suggests that an interplay of different pathogenic IgG subclasses eventually leads to loss of desmosomal integrity. As monovalent (Fab), bivalent (F(ab)’)2) or recombinant single-chain variable (scFV) autoantibody fragments of Dsg-specific IgG antibodies are able to induce dissociation of keratinocytes in vitro and in vivo, the role of the Fc portion of pathogenic pemphigus autoantibodies is presumably of minor importance [47, 60, 65, 80].
Dsg1/Dsg3 Compensation Theory
Differential expression of the autoantigens of PF (Dsg1) and PV (Dsg3) are the basis for the so-called Dsg1/Dsg3 compensation theory which explain why skin and mucosal membranes are differentially affected by anti-Dsg IgG autoantibodies in both pemphigus variants [4, 81]. Dsg1 is mainly expressed in the upper epidermis while Dsg3 is predominantly expressed in the lower, i.e. suprabasal layers of the epidermis. Thus, PV sera with Dsg3-specific IgG do not induce suprabasal loss of adhesion in human skin as Dsg1 is sufficiently expressed throughout the epidermis of the skin to compensate epidermal adhesion in the absence of Dsg3 adhesive capacity. On the other hand, anti-Dsg3 IgG causes loss of epidermal adhesion in the mucous membranes which express only low amounts of Dsg1, insufficient to compensate impaired Dsg3 adhesion. In the case of PV with both anti-Dsg1 and anti-Dsg3 IgG, loss of epidermal adhesion occurs both at the skin and mucous membranes [82].
Mode of Action of Pathogenic Autoantibodies
There is increasing evidence that both, direct inhibition of Dsg transinteraction via steric hindrance and altered outside-in-signaling by pemphigus autoantibodies leads to loss of desmosomal integrity and blister formation [83] (Fig. 6). The concept of steric hindrance is supported by data which show that PF and PV sera mainly contain IgG autoantibodies reactive with the NH2-terminal EC1 subdomains of Dsg1 and Dsg3, respectively [69, 84] where they interfere the trans-interaction of Dsg [52, 55]. In contrast to IgG autoantibodies which target other epitopes of Dsg1 and Dsg3, respectively, the serum concentrations of these IgG autoantibodies correlate with disease activity [52, 55, 85]. Beside the impact on steric hindrance, PV-IgG and the Dsg3 (EC1)-specific mAb, AK23, induce internalization and depletion of Dsg3 from the cell membrane [51, 86, 87]. It was further shown that the Dsg3-depleting activity of anti-Dsg3 mAb correlate with their pathogenicity. Thus, pemphigus is also known as a desmosome-remodeling disease [82]. The depletion process of Dsg leading to acantholysis can be divided in two steps: first, probably mainly induced by a direct interference of trans-interaction of Dsg, desmosomes lose adhesive properties and become more susceptible for subsequent depletion processes. The second step is induced by signal transduction events which lead to Dsg endocytosis and depletion resulting in loss of demosomal integrity and adhesion [82]. Völlner et al. showed that Dsg interact with flotillins, components of lipid rafts in the cell membrane outside of desmosomes [88]. Antibodies against Dsg interfere with Dsg-flotillin interaction leading to the internalization of cell membrane-associated Dsg. Depletion of extradesmosomal Dsg interferes with proper desmosomal function which largely depends on the integration of extra-desmosomal Dsg.

Mode of action of pathogenic autoantibodies. Autoantibodies inhibit desmosomal adhesion via steric hindrance (a), altered cellular signaling (b), depletion of extradesmosomal Dsg (c) and not yet fully understood mechanisms including inhibition of the protease inhibitor, A2ML1, binding to acetyl choline receptors on epidermal keratinocytes, induction of inflammasome-dependent caspase 8 and subsequent pro-inflammatory cytokines (d)
Autoantibodies Induce Cell Signaling Events
Dsg-specific IgG autoantibodies alter major cellular signaling events involving p38 MAPK, protein kinase C (PKC), c-Jun N-terminal kinases (JNK), RhoA and caspases 3, 6, 8 and 9 [55, 78, 89,90,91,92]. One of the best characterized anti-Dsg IgG-induced signaling cascades is the p38MAPK pathway. Specifically, p38MAPK directly influences intermediate filament organization and thereby maintenance of the desmosomal structure. Drug-induced inhibition of p38MAPK prevents blister formation in mice treated with anti-Dsg3 and anti-Dsg1 IgG, respectively [93, 94]. Moreover, the activated phosphorylated form of p38 was detected in PF and PV lesions [93]. Furthermore, IgG from PV sera induced p38MAPK activation occurs in vivo and in vitro after 15–30 min [78, 89, 92]. PKC also regulates desmosomal functions by induction of Dsg3 depletion from the desmosome. In vitro and in vivo, an anti-Dsg1/Dsg3-IgG induced activation of PKC and its downstream pathway was already observed after 30 s [95]. Moreover, blockade of PKC inhibits blister formation by PV IgG in vivo and in vitro [55, 90]. The process of acantholysis and cell death share similar signaling complexes and both were triggered by binding of PV IgG [91]. There are some data describing signs of keratinocyte apoptosis upon binding of PV-IgG binding which is tightly connected to detachment processes and blister formation [96,97,98,99,100,101]. Along this line, inhibition of p38MAPK leads to a blockade of both, acantholysis and apoptosis and caspase inhibitors are able to prevent acantholysis in vivo and in vitro [94].
Non-Dsg IgG Autoantibodies
In addition to anti-Dsg IgG autoantibodies, various IgG autoantibodies that target unrelated autoantigens were detected in the different pemphigus variants [91]. IgG reactive with Dsc3 have been identified in PNP and in atypical pemphigus cases which do not show IgG reactivity with the Dsg. Anti-Dsc3 IgG induce loss of keratinocyte adhesion similar to anti-Dsg1/Dsg3 IgG [34, 35, 102]. Anti-mitochondrial antibodies found in PV patients lead to the activation of JNK and p38MAPK in keratinocytes. Depletion of these antibodies from PV sera abolished loss of keratinocyte adhesion in vivo and in vitro [103]. Nguyen et al. detected anti-keratinocyte α-acetylcholine receptor (AChR) IgG autoantibodies in 85% of PV patients and blocking of AChR lead to desmosomal disassembly [91]. Other targeted molecules like Dsc1, desmoplakin 1 and 2, Dsg4 (which cross-reacts with Dsg1), pemphaxin, E-cadherin and PMP-22/gas [102, 104,105,106,107,108,109,110] were also described in PV. So far, the role of most non-Dsg IgG autoantibodies is still unclear. Some of these autoantibodies may act synergistically with anti-Dsg IgG and thus contribute to pemphigus pathogenesis. Some other may be only phenomena of epitope spreading induced by a general loss of tolerance upon progression of pemphigus. The recent identification of A2ML1 as a major targeted autoantigen in PNP has change our view that the pemphigus pathogenesis largely or exclusively depends on IgG recognition of adhesion molecules of the skin [27]. As A2ML1 is a protease inhibitor, its inhibited function could lead to an aggravation of the inflammatory response which is initially induced by anti-epithelial and anti-hemidemosomal IgG autoantibodies.
Autoantibody-Independent Factors
Sera of PV patients show mainly an increase of Th2 cytokines like IL-6, IL-4 and IL-10 [111,112,113] and rather suppressed Th1 cytokines like IL-2 and IFN-γ [113]. Of note, the Th17 cell-related cytokine IL-17a and the follicular T helper cell (Tfh) associated cytokines IL-21 and IL-27 are also elevated in PV sera [114]. In addition, IL-17a was also detected in PV skin lesions [114,115,116]. In the blister fluids of PV skin lesions, complement activation, cytotoxic proteases and high concentrations of IL-4 and IL-10 were detected [77, 111, 113, 117]. Furthermore, TNFα RNA is widely expressed in PV skin lesions and TNFα serum concentrations largely correlate with disease activity and IgG autoantibody titers [112, 118,119,120]. Fas ligand (FasL) detected in PV sera was described to induce apoptosis in keratinocytes by caspase 8 activation. Inhibition of FasL protein leads to an inhibition of PV IgG-induced apoptosis of epidermal keratinocytes in vitro and in vivo strongly suggesting that FasL plays a critical role in PV pathogenesis [101, 121]. Other groups are in favor of the concept that apoptosis of epidermal keratinocytes is rather a late, secondary event in the immune pathogenesis of pemphigus [122]. Additionally, the combination of TNFα, FasL and PV-IgG synergistically induced loss of keratinocyte adhesion in vitro. At present, the aforementioned antibody-independent factors may contribute to development of pemphigus but their distinct role in disease pathogenesis is still unclear.
Mouse Models of Pemphigus
Intensive research over the past decades using different animal models of pemphigus led to enormous insights into the underlying autoimmune mechanisms including loss of tolerance, activation of autoreactive T and B cells and production of pathogenic IgG antibodies (Fig. 7). Beginning with the passive transfer model that uses transfer of pemphigus IgG into neonatal mice to induce a blistering phenotype in vivo, mouse model development proceeded with the generation of an active disease model by transferring Dsg3−/− splenocytes into Rag-2−/− immunodeficient mouse. Recently developed mouse models use humanization of the autoantigen (Dsg3) or disease associated HLA molecules (HLA-DRB1*04:02) to model the autoimmune response in pemphigus more closely to the human situation. However, to date a wider understanding of the pathogenesis of pemphigus is hindered by a lack of a spontaneous mouse model which does not require disease induction by antibody transfer or active immunization. Furthermore, despite the fact that the mouse models presented here show key features of PV, it is still a matter of debate whether these models can reproduce pemphigus in its all complexity. For instance, current models focus on autoimmunity against Dsg3 as the main auto-antigen of PV. However, several other desmosomal and also non-desmosomal autoantigens have been identified in pemphigus patients [123] and IgG autoantibodies against non-desmosomal targets were shown to be pathogenic in vivo [32, 33]. These findings support the concept that pemphigus results not only from monopathogenic autoimmunity against Dsg1 and Dsg3 but rather from multipathogenic mechanisms with synergistic and cumulative effects of various autoantibodies targeting different autoantigens of epidermal keratinocytes [2, 32].

Mouse models of pemphigus. Passive transfer of pemphigus IgG into neonatal mice (a), active mouse model of PV by transfer of T and B cells from Dsg3-immunized Dsg3−/− donor mice to Dsg3+/+ Rag-2−/− immunodeficient recipient mice (b), immunization of mice transgenic for human CD4 and the PV-associated HLA-DRB1*04:02 allele with human Dsg3 (c), injection of IgG from pemphigus sera into mice transgenic for human Dsg3 (d)
Passive Transfer Mouse Model of PV
To reproduce IgG autoantibody-dependent loss of epidermal adhesion in vivo, Anhalt et al. developed a neonatal mouse model which was induced by passive transfer of IgG fractions from sera of PV patients with active disease (Fig. 7a) [47, 124]. Utilizing the passive transfer model, it was shown that binding of PV autoantibodies to their target antigens on epidermal keratinocytes results in cutaneous blister formation in vivo. Specifically, IgG fractions from PV sera were intraperitoneally injected into neonatal Balb/c mice at different doses (1.5–16.0 mg/g body weight) resulting in the induction of cutaneous blisters and erosions with histologic and DIF features reminiscent of pemphigus [47]. The model allowed further investigations on the mechanisms of autoantibody-induced acantholysis in pemphigus. In a time-course study, the ultrastructural changes after IgG autoantibody transfer were closely examined using electron microscopy. Early detachment of epidermis was observed as a widening of the intercellular space between keratinocytes as early as 1 h after IgG transfer leading to complete cell detachment within 6 h [125]. Passive transfer of PV IgG into C5-deficient neonatal mice led the same extensive blistering indicating that complement activation is not essential for IgG-induced acantholysis in PV [126]. Similarly, IgG purified from PF sera induced blisters with the characteristic subcorneal loss of epidermal adhesion when passively transferred into neonatal Balb/c mice [48, 127,128,129]. In summary, the passive transfer model using Balb/c neonates reproduces clinical features of pemphigus disease in mice and provided first evidence that PV IgG alone are pathogenic in vivo and facilitated the dissection of the blister formation in pemphigus in vivo.
Active Disease Mouse Model of Pemphigus
With the identification of Dsg1 and Dsg3 as major target autoantigens of pemphigus [81, 127, 130], an active disease model of pemphigus was developed to study the autoimmune mechanisms leading to the generation of pathogenic Dsg3-specific autoantibodies and the evolving clinical phenotype (Fig. 7b).The major limitation in the development of an active mouse model using forced immunization with mouse Dsg3 was to break immunological self-tolerance. Repeated immunizations and several adjuvants were tried to break tolerance. To overcome the limitation of self-tolerance against mouse Dsg3, the active disease model of PV used Dsg3−/− mice that lacked immunological tolerance against naturally expressed Dsg3 [131]. To circumvent this problem, Dsg3−/− mice were immunized with recombinant mouse Dsg3 and isolated splenocytes, containing Dsg3-reactive T and B cells, were subsequently transferred into immunodeficient Rag2−/− Dsg3+/+ recipients to induce a Dsg3-specific autoimmune response in vivo [46, 132]. The recipient mice showed oral erosions around the snout with suprabasal acantholysis induced by in vivo production of Dsg3-specific IgG [133, 134].
Dsg3-specific autoantibodies binding to native mouse Dsg3 with different pathogenic properties were generated in this in vivo model. Anti-Dsg3 IgG which possessed pathogenic properties bound to the NH2-terminal EC1 subdomain of Dsg3 while non-pathogenic IgG antibodies were mapped to the mid portion or COOH-terminus of the Dsg3 ectodomain [135]. Among those, the IgG antibody AK23 shows high pathogenic activity by binding to the adhesive interface of Dsg3 [135, 136] presumably leading to steric hindrance of desmosomal trans-interaction [137].
In addition, Dsg3-reactive T cell clones were generated and further characterized in this PV mouse model. After transfer of Dsg3-reactive T cells into Rag2−/− Dsg3+/+ recipients, a subset of T cell clones was able to prime naïve B cells to produce pathogenic anti-Dsg3 IgG [138]. This effect could be suppressed by adding soluble IL-4Rα pointing towards IL-4 as a critical mediator driving autoantibodies production in this mouse model [138]. By titration of T cells it was demonstrated that one potent Dsg3-reactive T cell clone is sufficient to induce anti-Dsg3 IgG production and, eventually, also a clinical phenotype [139]. Anti-Dsg3 IgG production was greatly reduced by treatment of the mice with anti-CD154 which blocked CD40/CD154 interaction [140]. Peripheral T cell tolerance to Dsg3 could be restored by Treg cells as CD4+CD25+ Treg cells suppressed IgG antibody production and Treg cell depletion augmented anti-Dsg3 IgG production [141]. Further modification of the model by transferring naїve splenocytes from non-immunized Dsg3−/− mice into Rag2−/− Dsg3+/+ recipient mice showed that priming of T cells and B cells with Dsg3 was not essential prior to transfer into the Dsg3-competent immunodeficient mice [142]. However, anti-Dsg3 IgG serum concentrations were lower and showed reduced pathogenicity when compared to Dsg3-specific IgG obtained after active immunization [142, 143].
Humanized Dsg3-Transgenic Mice
Mouse models using human Dsg3-specific IgG antibodies have been hampered as anti-human Dsg3 IgG is not able to induce a clinical phenotype in adult mice [144, 145], suggesting that certain pathogenic epitopes are not conserved between mouse and human Dsg3 although they share a homology of 85.6% [146]. To further characterize the pathogenicity of anti-human Dsg3 IgG in vivo, a humanized transgenic Dsg3 mouse model was generated by introducing human Dsg3 in Dsg3−/− mice [131, 147]. In this model, passive transfer of anti-Dsg3 IgG from PV patients induced mucosal erosions and suprabasilar acantholysis and bound preferentially to human Dsg3 when compared with mouse Dsg3 (Fig. 7c) [131, 147].
Humanized HLA-Transgenic Mouse Model of PV
The majority of PV patients show a genetic predisposition towards certain HLA class II alleles [7, 148, 149], i.e., HLA-DRB1*0402 in the Jewish and HLA-DQB1*0503 in the non-Jewish population [6, 150]. It has been shown that specific charges within the active binding site of the HLA class II binding pocket confer selective binding of a limited set of immunodominant Dsg3-peptides to HLA-DRB1*0402 [151, 152]. This mouse model thus reproduces T cell recognition of human Dsg3 in PV (Fig. 7d). Proof for this HLA class II–Dsg3 peptide algorithm is given by the observation that Dsg3-reactive T cells from PV patients recognize immunodominant epitopes of Dsg3 in association with the PV-associated HLA class II alleles, HLA-DRB1*0402 and HLA-DQB1*0503, both of which show similar peptide binding motifs [153].
After immunization with recombinant human Dsg3, the HLA-transgenic mice develop a robust anti-Dsg3 IgG response which induces loss of cell adhesion of cultured human keratinocytes and ex vivo epidermal acantholysis in human skin biopsies [154]. Anti-Dsg3 IgG production was completely diminished upon blocking of T cell–B cell interaction with anti-CD40L or depletion of CD4+ T cells showing that T cell dependent B cell activation is critical for the induction of pathogenic anti-Dsg3 IgG. Furthermore, recognition of Dsg3 was highly specific for the PV-associated HLA class II alleles used in this model as Dsg3 immunization with mice transgenic for HLA-DRB1*04:01 (associated with rheumatoid arthritis (RA)) did not lead to the formation of pathogenic anti-Dsg3 IgG [154]. Moreover, immunization of the HLA-transgenic mice with a set of immunodominant Dsg3-peptides which share a positively charged anchor motif for HLA-DRB1*04:02 and show strong binding to HLA-DRB1*04:02, induced a conformational anti-Dsg3 IgG response while immunization with HLA-non-binding Dsg3-peptides did not induce pathogenic Dsg3-specific IgG antibodies [154].
Using the same mouse model, the role of Treg cells in shaping the T-cell mediated Dsg3-specific immune response was recently investigated. Induction of Treg cells by the superagonistic anti-CD28 antibody D665 led to a strongly reduced production of anti-Dsg3 IgG [155]. The HLA-transgenic mouse model thus reproduces the specificities of autoreactive T- and B-cell responses against human Dsg3 which are seen in PV patients. The model is suitable for investigating the activation and interaction of Dsg3-specific, HLA class II-restricted T cells with autoreactive B cells in the autoimmune response in PV and will serve as a preclinical model to test specific immune interventions in PV.
Treatment of Pemphigus
Although a broad spectrum of therapeutic interventions has been described for pemphigus, a generally accepted treatment strategy remains still unclear. As demonstrated by current meta-analyses on treatment options in pemphigus, both the lack of adequately powered high-quality randomized clinical trials and the use of various outcome measures lead to inconclusive evidence for standard of care recommendations in pemphigus [156].
In the initial treatment of pemphigus, a combination of high-dose systemic corticosteroids (1.0–1.5 mg/kg/d prednisolone) with an adjuvant steroid-sparing immunosuppressant, mostly azathioprine (1.5–2.5 mg/kg/d according to thiopurine methyltransferase activity) or mycophenolate mofetil (2 g/d)/ mycophenolic acid (1440 mg/d), is considered as the mainstay of systemic treatment in pemphigus [14]. A steroid-sparing effect in pemphigus has been shown for different immunosuppressive drugs, such as azathioprine [157], mycophenolate mofetil [158] or cyclophosphamide [159]. Azathioprine, mycophenolate mofetil, or methotrexate (15–25 mg/week) are the immunosuppressive standards. Although highly effective, cyclophosphamide given per os or as a bolus is not recommended in the first intention as there are too many adverse side-effects. It is recommended that patients receiving corticosteroids or immunosuppressive therapy be vaccinated against seasonal flu, H1N1, and pneumococcae prior to immunosuppressive therapy. Certain immunosuppressants contraindicate the use of live vaccines [14].
In patients who are refractory to the initial treatment or have contraindications to high-dose corticosteroids, second-line options targeting IgG autoantibodies or autoreactive B cells in pemphigus are recommended. High-dose intravenous immunoglobulins (IVIg) (2 g/kg/cycle) have been shown to rapidly reduce the titers of serum autoantibodies in pemphigus patients mostly accompanied by clinical remission [160].
The therapeutic removal of circulating IgG by immunoadsorption leads to a very rapid and dramatic reduction of autoantibodies in the serum of pemphigus patients leading to a fast clinical response [161]. A recent German multicenter trial has studied the efficacy of adjuvant immunoadsorption in inducing clinical remission in PV and PF compared to immunosuppressive treatment only. A total of 72 pemphigus patients were equally allocated to both treatment arms and their clinical response was monitored during a 12 month observation period. First results suggest that pemphigus patients with extensive skin involvement seem to profit more from adjuvant immunoadsorption (Eming et al., in preparation).
The B cell depleting monoclonal anti-CD20 antibody rituximab has shown efficacy especially in refractory pemphigus patients [162]. Based on the recent clinical trial by Pascal Joly and the French Study Group of Bullous Diseases (Groupe Bulle) the efficacy of rituximab in the treatment of pemphigus has been raised on a high level of evidence [163]. Joly and colleagues compared first-line use of rituximab (1000 mg on days 0 and 14 and 500 mg at months 12 and 18) combined with short-term prednisone versus prednisone alone in a cohort of 90 newly diagnosed, untreated pemphigus patients. The rituximab treated patients received prednisone either at an initial dose of 0.5 mg/kg per day which was tapered for 3 months (moderate disease) or 1.0 mg/kg per day which was tapered for 6 months (severe disease). Patients in the prednisone group were given 1.0 mg/kg per day (moderate disease) and 1.5 mg/kg per day (severe disease), tapered over a period of 12 or 18 months. 41 of 46 patients (89%) in the rituximab group were in complete clinical remission of therapy at month 24, whereas 34% (15 of 44 patients) in the group receiving only prednisone reached this primary endpoint of the study. The cumulative prednisone dose as well as the severe adverse events were significantly lower the group of patients assigned to rituximab plus prednisone compared to those patients receiving prednisone alone.
Outlook: Pathogenesis-Based Therapeutic Approaches
The pathogenesis of pemphigus is relatively well known, compared to other, especially systemic, autoimmune disorders. The pathogenicity of Dsg-specific IgG autoantibodies has been clearly demonstrated in various in vitro and in vivo systems [164, 165]. Considering pemphigus as a paradigm of an autoantibody-mediated autoimmune disease, the role of Dsg3-specific CD4+ T cells in this disease has been in the focus of our group and others [153, 166]. There is increasing evidence that loss of tolerance to Dsg on the CD4+ T cell level resulting in autoreactive T cells is a crucial event in the initiation and probably in the perpetuation of the autoantibodies response in pemphigus [139, 140, 152, 167, 168]. Since the original identification of autoantibodies in the sera of pemphigus patients by Beutner and Jordan [169], and the characterization of Dsg3 as the major autoantigen in PV [127] the pathogenic relevance of Dsg3-specific autoantibodies and the autoantibodies producing cells are taking center stage in pemphigus research. Thus, it seems obvious that both the immune cells that are relevant in the autoimmune cascade of pemphigus and the mechanisms finally leading to the loss of epidermal keratinocyte adhesion upon binding of Dsg-reactive autoantibodies have been identified as promising drug targets in pemphigus. The overall goal of developing targeted pathogenesis-driven therapies is to avoid the well-known side effects of systemic immunosuppression and ultimately to induce long-term remission in pemphigus (Table 2).
Targeting Formation of Pathogenic Autoantibodies
The successful use of the B cell depleting mAb, rituximab (Rtx) in pemphigus has been previously described in several studies. However, a recent study by the French groupe bulle demonstrated that during a median follow-up period of 79 month 81% of PV patients who previously received Rtx treatment experienced a disease relapse [170]. In several Rtx studies, in most PV patients the clinical remission was paralleled by a decline in serum Dsg3-reactive IgG autoantibodies, suggesting that autoreactive plasma cells (PC) in PV are short-lived and that long-lived CD20 negative PC do not seem to play a relevant role in the autoantibodies response in pemphigus [171]. Moreover, Ohyama et al. [70] demonstrated in a longitudinal serological study that epitope spreading in PV seems to be a rare event, since B cell epitopes in Dsg3 did not significantly change in acute disease and clinical remission, respectively [70].
These results have been underlined by a study on the B cell receptor (BCR) repertoire in PV patients. Hammers et al. [172] investigated the BCR repertoire in Dsg3-specific B cell clones in PV patients during different stages of disease and the authors could show that a defined cohort of autoreactive B cell clones persists over time, probably causing a clinical relapse after a preceding period of remission [172]. Thus, long-lived PC residing in bone marrow niches and inflamed tissue are not likely to be promising therapeutic targets in PV. However, in other autoimmune disorders, such as systemic lupus erythematosus (SLE), these autoantibodies secreting PC have been shown to be effective targets. A cohort of refractory systemic lupus SLE patients received the proteasome inhibitor bortezomib and demonstrated a significant decrease in disease activity remaining stable for 6 months [173]. Previously, several in vivo studies using mouse models of SLE indicated that targeting the proteasome and type-I interferon activity by inhibitors such as bortezomib and carfilzomib, results in a reduction of clinical disease activity [173,174,175]. It remains to be elucidated whether this novel approach which is being investigated in various autoimmune diseases at the moment will be relevant for future therapies in PV [176].
In addition to the recently published phase III trial using rituximab as first line therapy in pemphigus by Joly and co-workers [163], other B cell directed strategies are being investigated in clinical trials at the moment. Ofatumumab (Arzerra, HuMax-CD20) is a fully human anti-CD20 IgG1 antibody which has been approved by the FDA for therapy of B-CLL in April 2010. In a multicentre, randomized, double-blind, placebo-controlled phase III study that started in August 2013, efficacy, tolerability and safety of subcutaneous injection of ofatumumab in PV patients have been investigated. In this trial, 20 mg of the antibody is administered subcutaneously once every 4 weeks through week 56 (NCT01920477/OPV116910). A long-term extension of this trial has been performed by GlaxoSmithKline; the primary objective is to provide continued treatment with subcutaneous ofatumumab for eligible study patients who completed the aforementioned trial in order to obtain further long-term safety and tolerability information in PV patients receiving ofatumumab every 4 weeks (NCT02613910/OPV117059). Both studies evaluating ofatumumab in PV have been terminated by Novartis.
In the same line of thought, Ellebrecht and colleagues [177], published a case report treating a patient with refractory PV with the second-generation humanized anti-CD20 antibody veltuzumab (Immunomedics). The anti-CD20 antibody was administered as two 320 mg (188 mg/m2) subcutaneous doses 2 weeks apart resulting in complete clinical remission and immunosuppressive therapy was terminated 22 weeks after veltuzumab application in this patient. A clinical relapse occurred 2 years after velutuzumab therapy and the patients received a second treatment cycle using the same dosage regimen that induced again complete clinical remission lasting for 9 months. The patient was followed for 35 months and did not show any serious side effects of B cell depletion. The clinical response to veltuzumab correlated with anti-Dsg3-IgG serum levels [177]. A clinically relevant advantage of veltuzumab over Rtx is the ability of subcutaneous administration in low doses, thus facilitating its clinical application at reduced health care costs. Another humanized anti-CD20 antibody, ocrelizumab (Ocrevus, Roche), has been recently approved by the FDA for treatment of primary progressive and relapsing-remitting type of multiple sclerosis (MS). This humanized mAb is being evaluated in phase III trials in other autoantibody-mediated disorders such as RA and SLE as well. Other second- and third-generation anti-CD20 antibodies with enhanced B cell depleting activity and improved binding affinities to B cells compared to Rtx might be promising tools in PV in the future, once their clinical efficacy in pemphigus has been shown [178].
A different approach of B cell targeting aims at inactivating or blocking factors that are important for B cell survival and differentiation into PC, respectively. Belimumab (Benlysta, GSK) a monoclonal human IgG1 antibody that binds to soluble B lymphocyte stimulator, (BLyS) or B cell activating factor belonging to the TNF family (BAFF), among other effects, impairs B cell differentiation to PC. It is approved as adjuvant immunosuppressive /immunomodulatory therapy in SLE patients who are refractory to basic immunosuppression alone [179]. Atacicept is a fully human recombinant fusion protein that blocks not only BLyS but also the proliferation-inducing ligand (APRIL), another B-cell activating factor. In order to evaluate its efficacy and safety, its biological and clinical activity has been studied in animal models and in patients with RA, MS and SLE, respectively. So far, atacicept seems to provide an added value in treating autoantibody-mediated autoimmune disorders such as SLE but further investigations are necessary to establish its efficacy and safety in these conditions [180]. The results from clinical trials in the above mentioned autoimmune diseases are encouraging and this strategy might be of interest in PV as an add-on opportunity in patients demonstrating incomplete clinical responses to B cell depleting therapies, such as Rtx. Safety, tolerability and efficacy of a monoclonal anti-BAFF-receptor antibody (VAY736) is being investigated in a randomized, partial-blind, placebo-controlled multicentre trial (NCT01930175).
Recently, a novel antigen-specific therapeutic approach by Ellebrecht, Payne and colleagues from the University of Pennsylvania has raised a lot of attention in the pemphigus field. This group developed Dsg3-specific T cells that are directed to specifically target Dsg3-reactive B cells in PV patients by using the chimeric autoantibody receptor (CAAR) technology [181]. Chimeric autoantibody receptor (CAAR)-T cells have been shown to be effective in cancer therapy, such as B cell leukemia showing remarkable clinical responses including long-lasting remission [182,183,184]. To generate Dsg3 CAAR T cells, Ellebrecht et al. fused fragments of the Dsg3 extracellular domain to CD137/CD3 signalling domains in primary human T cells [181]. The aim is that Dsg3 CAAR T cells recognize the Dsg3-specific BCR on autoreactive B cells in PV patients and kill these cells in an HLA-independent but antigen-specific manner. The authors provided proof of the ability of Dsg3 CAAR T cells to kill B cells expressing a Dsg3-reactive BCR first in vitro followed by in vivo experiments using Dsg3 B cell hybridomas in mice. In each system, Dsg3-reactive B cell hybridomas were specifically killed by CAAR T cells and mice showed decreased titers of Dsg3-specific IgG, thus preventing the manifestation of a clinical phenotype in these animals [181]. An important point in this study was to show that circulating Dsg3-reactive IgG autoantibodies did not interfere with the ability of Dsg3 CAAR T cells to kill autoreactive B cells by binding to the Dsg3 antigen expressed on the CAAR T cell. Ellebrecht et al. demonstrated that CAAR T cells retained their killing activity against the B cell hybridomas even in the presence of Dsg3 reactive IgG. Finally, the authors showed that Dsg3 CAAR T cells did not bind to Dsg3-expressing epidermal keratinocytes, thus excluding relevant off-target effects of Dsg3 CAAR T cells. This approach appears promising because Dsg3 CAAR T cells seem to act very specifically on B cells expressing an autoreactive Dsg3-specific BCR. It may also overcome the limitation of current B cell directed therapies by leading to long-lasting elimination of Dsg3-specific B cell clones in PV patients. However, more preclinical efficacy data need to be obtained before CAAR T cell technology can be translated to the patients.
Targeting CD4+ Autoreactive T Cells
In PV, several lines of evidence from preclinical animal studies as well as ex vivo experiments using human peripheral mononuclear cells (PBMC) suggest that loss of tolerance to Dsg3 at the level of CD4+ T cells is mandatory for the production of Dsg3-specific IgG autoantibodies [46, 114, 153, 185]. The major immunoglobulin isotype present in the pool of Dsg3-specific autoantibodies in PV is IgG4 in acute disease and IgG1 in chronic active and in some patients in remitting disease [186,187,188]. Our group showed in a retrospective serological study that in addition to Dsg3-reactive IgG4, IgE autoantibodies were present in acute phases of PV as well [189]. The serological data speaks for a crucial role of Dsg3-reactive Th2 cells providing B cell help for the secretion of autoantibodies in PV [153]. However, other CD4+ T cell subsets, such as Th17 and especially Tfh cells, have been characterized in PV patients as well [114, 190, 191]. Thus, it is an intriguing approach to try to restore Dsg3-specific T cell tolerance in PV in order to interfere with the autoimmune cascade at an early time point. In general, efforts to induce antigen-specific tolerance in autoimmune disorders by administration of the autoantigen have been made and the studies demonstrated miscellaneous results depending on the system used. In animal models of autoimmunity, allograft transplantation, and allergy, mainly in the model of experimental autoimmune encephalomyelitis (EAE), it has been extensively shown that the administration of immunodominant peptides under tolerogenic conditions induces a robust and long lasting tolerance to the respective auto-/alloantigen [192, 193]. Cell-based approaches have been successfully applied in the Th1/Th17-dominated autoimmune models of EAE, a mouse model of human MS, and the non-obese diabetic (NOD) model of type 1 diabetes [194, 195]. In these animal models, a single i.v.-injection of immunodominant peptides that are chemically linked to syngeneic splenocytes by using ethylene carbodiimide (ECDI) is sufficient to prevent the onset of the autoimmune disease. The immunological mode of action in these animal models has been investigated in numerous studies and the induction of antigen-specific tolerance has been mainly attributed to the induction of tolerogenic antigen presenting cells (APC) in the recipient animals, such as splenic macrophages, B cells and dendritic cells (DC) that give rise to Treg cells [195]. Based on these preclinical studies, Lutterotti, Martin and co-workers performed a first-in-man trial to assess the feasibility, safety and tolerability of a tolerization approach in MS patients [196]. Seven MS patients who were off-treatment for standard therapies were included in this open-label, single-center, dose-escalation study. The study participants received a single i.v.-infusion of seven myelin peptides cross-linked to autologous PBMC. It was required that the patients demonstrated T cell reactivity against at least one of the myelin peptides used in the trial. This trial showed that the administration of antigen-coupled PBMC was feasible, safe and well tolerated by the patients. Preliminary ex vivo studies revealed decreased proliferation of autoreactive T cells in peripheral blood in those patients who had received a higher dose of peptide-coupled PBMC [196]. Future phase II studies are warranted to further explore this promising therapeutic approach. Resulting from basic immunological research, studies in preclinical models and translational approaches, different strategies to induce antigen-specific tolerance in autoimmune diseases proceed from proof of concept studies to clinical trials. In addition to the above mentioned investigations, other cell-based approaches using red blood cells as well as particle-based (e.g. nanoparticles as carriers of antigens) methods are evaluated for clinical use in the near future [197,198,199].
Another field of novel therapeutic strategies in treating autoimmune diseases is based on the hypothesis that the balance of Treg cells and potentially self-reactive T effector cells is disturbed in the context of autoimmunity suggesting a therapeutic potential of Treg cells. In PV, studies by our group characterized Dsg3-specific type 1 regulatory (Tr1) cells in peripheral blood of PV patients and HLA-matched healthy controls [200]. Ex vivo these Dsg3-reactive Tr1 cells suppressed the proliferation of Dsg3-specific effector T cell clones in an IL-10- and TGF-dependent manner [200]. Interestingly, the frequencies of these Tr1 cells was significantly decreased in peripheral blood of PV patients compared to HLA-matched healthy controls, supporting the above mentioned hypothesis of an imbalance of Treg and T effector cells in autoimmune diseases [200].
In this context, low-dose IL-2 therapy in autoimmune and inflammatory diseases has gained interest based on recent proof-of-concept clinical trials in vasculitis, alopecia areata and SLE [201]. In HCV-induced vasculitis [202], in GVHD [203] and recently in SLE [204] low-dose IL-2 application has been shown to specifically and safely activate and expand Treg cells in humans. Moreover, in these clinical trials, IL-2 improved the inflammatory and autoimmune conditions, respectively. An ongoing open-label phase II clinical trial investigates the stimulatory effect of low-dose IL-2 administration on Treg cells in 11 different autoimmune diseases, such as SLE, RA, inflammatory bowel diseases, vasculitis, etc. The TRANSREG trial aims to select those autoimmune/inflammatory diseases in which further therapeutic development, using low-dose IL-2, will be performed in more detail (NCT01988506).
Finally, in other autoantibody-mediated autoimmune diseases, such as SLE, there is increasing evidence that Tfh cells play a critical role in autoantibodies formation [205] and therefore this CD4+ T cell subset is considered as a potential therapeutic candidate in the context of autoimmunity. In PV, our group recently identified increased frequencies of circulating CD4+CXCR5+IL-21+ Tfh cells in peripheral blood of PV patients compared to healthy individuals [114]. Moreover, PV patients with chronic active disease demonstrated elevated IL-21 serum levels, which were dramatically decreased upon systemic immunosuppression. For the first time, we were able to detect Dsg3-reactive IL-21-secreting cells using ELISpot-assay [114]. Several therapeutic substances targeting Tfh activity have been identified already and have been shown to exert immunological as well as clinical effects, mostly in animal models of SLE [205]. The therapeutic tools undergoing preclinical and clinical testing include the humanized anti-IL-6-receptor antibody tocilizumab, an anti-IL-21-receptor antibody (ATR-07) and JAK inhibitors, such as tofacitinib and ruxolitinib, which are investigated in RA and alopecia areata as well [206, 207]. In PV, the role of IL-21 secreting Tfh cells in the pathogenesis needs to be defined more precisely, prior to specifically targeting this cell subset.
Targeting Dsg-Specific Autoantibodies
PV is one of the rare autoimmune disorders in which the major autoantigen is clearly defined and the pathogenic relevance of serum autoantigen-specific autoantibodies has been extensively demonstrated in vitro and in vivo. The transfer of PV autoantibodies is sufficient to induce a blistering phenotype with suprabasilar acantholysis in recipient organisms, as demonstrated using the passive mouse model of PV [47]. Various therapeutic strategies to interfere with the action of PV autoantibodies or to deplete these immunoglobulins have been in the focus of pemphigus research. Immunoadsorption (IA), the therapeutic removal of immunoglobulins and circulating immune complexes from plasma, has been proven to be a useful treatment option in several auto- immune disorders or in renal transplant recipients [208]. Plasmapheresis removes plasma proteins non-specifically including clotting factors, hormones and albumin, thus requiring the subsequent substitution of fresh frozen plasma or albumin. In contrast, IA represents a more specific approach by selectively eliminating immunoglobulins and circulating immune complexes (CIC). A recent German multicenter, prospective, randomized, parallel-group trial has studied the efficacy and safety of adjuvant IA in inducing clinical remission in PV and PF compared to immunosuppressive treatment, consisting of prednisolone and azathioprine or mycophenolate only. Seventy-two pemphigus patients were equally allocated to both treatment arms and their clinical response and immunological parameters were monitored during a 12-month observation period. First results suggest that patients with extensive skin involvement profit from adjuvant IA and the adjuvant treatment showed a steroid-sparing effect (Eming, Zillikens, Hertl, Schmidt, in preparation). In this study, total plasma IgG was removed from the patients by using protein A adsorber columns. A potential future development of IA treatment in PV is the use of antigen-specific adsorbers that mainly extract pathogenic Dsg-specific IgG autoantibodies. Recently, Langenhan and colleagues demonstrated that antigen-specific IA requires the entire ectodomains of Dsg1 and Dsg3, respectively [209]. Future clinical trials using Dsg-coated adsorber columns in IA are needed to demonstrate a potential clinical advantage over non-specific IA.
Targeting the Pathogenic Effects of IgG Autoantibodies
The exact mechanisms leading to loss of epidermal keratinocyte adhesion upon binding of autoantibodies to their antigenic target structures is not completely understood, yet [210]. Several research groups have identified different signaling pathways in human keratinocytes that are activated upon binding of IgG autoantibodies from PV patients [211]. Recently, Walter and co-workers demonstrated that different signalling patterns contribute to loss of keratinocyte adhesion depending on the autoantibodies reactivities, i.e. anti-Dsg1-and/or anti-Dsg3-reactivity [212]. As p38MAPK activation is one of the signaling events that has been shown to be mandatory for complete loss of keratinocyte adhesion, p38MAPK signaling appears to be a promising therapeutic target. Egu et al. showed that in vitro the pharmacologic inhibition of p38MAPK ameliorated interdesmosomal widening and loss of keratin insertion and finally prevented PV IgG-induced blister formation in human skin biopsies [213].
These recent in vitro findings are in line with previous results by Berkowitz et al. demonstrating that p38MAPK inhibitors were capable of preventing acantholysis and skin blistering in an in vivo mouse model of PV [94]. However, pharmacologic inhibition of p38MAPK activation in different inflammatory disorders, such as RA, Crohn’s disease or psoriasis, has raised significant safety concerns in clinical trials [214]. In PV, a phase II multicenter, open-label trial applying an allosteric p38MAPK inhibitor (KC-706) had to be terminated due to severe adverse events in the study patients [215]. Thus, the future clinical use of p38MAPK inhibitors might be limited by their safety profile. The relevance of apoptosis in loss of keratinocyte adhesion in PV is controversially discussed in the field [121, 122]. However, using the passive transfer model of PV, Pacheco-Tovar and colleagues demonstrated that in a preventive setting the irreversible caspase-3 inhibitor Ac-DEVD-CMK prevented blister formation upon injection of pemphigus IgG in vivo [216]. This study confirmed previous results by Li et al. using PF IgG in the neonatal mouse model of pemphigus who showed that inhibition of caspase-3/7 as well as a broad-spectrum caspase inhibitor (Bok-D-fmk) protected the recipient mice from blister formation [217]. Anyway, clinical trials that investigate the efficacy and safety of caspase inhibitors in PV are still expected.
Conclusions
Based on its well-understood immune pathogenesis, pemphigus can be considered as a model disease of an antibody-mediated autoimmune disorder of the skin. Our increasing understanding of the immunological networks which regulate B cell activation and autoantibody production have led to novel therapeutic regimens which target distinct pathways in the pemphigus pathogenesis. Moreover, the identification of the major autoantigens of pemphigus has led to the development for more refined and specific diagnostic measures which help to better define clinical subtypes with different outcomes. The therapeutic achievements of ongoing research in pemphigus may be largely applicable to not yet fully defined autoimmune disorders of the skin and beyond.
References
Kneisel A, Hertl M (2011) Autoimmune bullous skin diseases. Part 2: diagnosis and therapy. J Dtsch Dermatol Ges 9(11):927–947. https://doi.org/10.1111/j.1610-0387.2011.07809.x
Di Zenzo G, Borradori L, Muller EJ (2016) The pathogenesis of pemphigus: controversy vs complexity. Exp Dermatol. https://doi.org/10.1111/exd.13176
Di Zenzo G, Amber KT, Sayar BS, Muller EJ, Borradori L (2016) Immune response in pemphigus and beyond: progresses and emerging concepts. Semin Immunopathol 38(1):57–74. https://doi.org/10.1007/s00281-015-0541-1
Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR (1999) Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest 103(4):461–468. https://doi.org/10.1172/JCI5252
Sinha AA, Brautbar C, Szafer F, Friedmann A, Tzfoni E, Todd JA, Steinman L, McDevitt HO (1988) A newly characterized HLA DQ beta allele associated with pemphigus vulgaris. Science 239(4843):1026–1029
Ahmed AR, Yunis EJ, Khatri K, Wagner R, Notani G, Awdeh Z, Alper CA (1990) Major histocompatibility complex haplotype studies in Ashkenazi Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci U S A 87(19):7658–7662
Ahmed AR, Wagner R, Khatri K, Notani G, Awdeh Z, Alper CA, Yunis EJ (1991) Major histocompatibility complex haplotypes and class II genes in non-Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci U S A 88(11):5056–5060
Tron F, Gilbert D, Joly P, Mouquet H, Drouot L, Ayed MB, Sellami M, Masmoudi H, Makni S (2006) Immunogenetics of pemphigus: an update. Autoimmunity 39(7):531–539
Shams S, Amirzargar AA, Yousefi M, Rezaei N, Solgi G, Khosravi F, Ansaripour B, Moradi B, Nikbin B (2009) HLA class II (DRB, DQA1 and DQB1) allele and haplotype frequencies in the patients with pemphigus vulgaris. J Clin Immunol 29(2):175–179
Yan L, Wang JM, Zeng K (2012) Association between HLA-DRB1 polymorphisms and pemphigus vulgaris: a meta-analysis. Br J Dermatol 167(4):768–777
Kneisel A, Hertl M (2011) Autoimmune bullous skin diseases. Part 1: clinical manifestations. J Dtsch Dermatol Ges 9(10):844–856; quiz 857. https://doi.org/10.1111/j.1610-0387.2011.07793.x
Martin LK, Werth V, Villanueva E, Segall J, Murrell DF (2009) Interventions for pemphigus vulgaris and pemphigus foliaceus. Cochrane Database Syst Rev 1:CD006263
Otten JV, Hashimoto T, Hertl M, Payne AS, Sitaru C (2014) Molecular diagnosis in autoimmune skin blistering conditions. Curr Mol Med 14(1):69–95
Hertl M, Jedlickova H, Karpati S, Marinovic B, Uzun S, Yayli S, Mimouni D, Borradori L, Feliciani C, Ioannides D, Joly P, Kowalewski C, Zambruno G, Zillikens D, Jonkman MF (2015) Pemphigus. S2 guideline for diagnosis and treatment--guided by the European dermatology forum (EDF) in cooperation with the European academy of dermatology and venereology (EADV). J Eur Acad Dermatol Venereol 29(3):405–414. https://doi.org/10.1111/jdv.12772
Ahmed AR, Kaveri S, Spigelman Z (2015) Long-term remissions in recalcitrant pemphigus vulgaris. N Engl J Med 373(27):2693–2694. https://doi.org/10.1056/NEJMc1508234
Ahmed AR, Spigelman Z, Cavacini LA, Posner MR (2006) Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med 355(17):1772–1779. https://doi.org/10.1056/NEJMoa062930
Joly P, Mouquet H, Roujeau JC, D'Incan M, Gilbert D, Jacquot S, Gougeon ML, Bedane C, Muller R, Dreno B, Doutre MS, Delaporte E, Pauwels C, Franck N, Caux F, Picard C, Tancrede-Bohin E, Bernard P, Tron F, Hertl M, Musette P (2007) A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med 357(6):545–552. https://doi.org/10.1056/NEJMoa067752
Slomov E, Loewenthal R, Korostishevsky M, Goldberg I, Brenner S, Gazit E (2005) Pemphigus vulgaris is associated with the transporter associated with antigen processing (TAP) system. Hum Immunol 66(12):1213–1222. https://doi.org/10.1016/j.humimm.2005.11.004
Kanwar AJ, Kaur S (1991) Pemphigus in children. Int J Dermatol 30(5):343–346
Qian Y, Jeong JS, Maldonado M, Valenzuela JG, Gomes R, Teixeira C, Evangelista F, Qaqish B, Aoki V, Hans G Jr, Rivitti EA, Eaton D, Diaz LA (2012) Cutting edge: Brazilian pemphigus foliaceus anti-desmoglein 1 autoantibodies cross-react with sand fly salivary LJM11 antigen. J Immunol 189(4):1535–1539. https://doi.org/10.4049/jimmunol.1200842
Ohzono A, Sogame R, Li X, Teye K, Tsuchisaka A, Numata S, Koga H, Kawakami T, Tsuruta D, Ishii N, Hashimoto T (2015) Clinical and immunological findings in 104 cases of paraneoplastic pemphigus. Br J Dermatol 173(6):1447–1452. https://doi.org/10.1111/bjd.14162
Amagai M, Nishikawa T, Nousari HC, Anhalt GJ, Hashimoto T (1998) Antibodies against desmoglein 3 (pemphigus vulgaris antigen) are present in sera from patients with paraneoplastic pemphigus and cause acantholysis in vivo in neonatal mice. J Clin Invest 102(4):775–782. https://doi.org/10.1172/jci3647
Brandt O, Rafei D, Podstawa E, Niedermeier A, Jonkman MF, Terra JB, Hein R, Hertl M, Pas HH, Muller R (2012) Differential IgG recognition of desmoglein 3 by paraneoplastic pemphigus and pemphigus vulgaris sera. J Invest Dermatol 132(6):1738–1741. https://doi.org/10.1038/jid.2012.1
Hashimoto T, Amagai M, Watanabe K, Chorzelski TP, Bhogal BS, Black MM, Stevens HP, Boorsma DM, Korman NJ, Gamou S et al (1995) Characterization of paraneoplastic pemphigus autoantigens by immunoblot analysis. J Invest Dermatol 104(5):829–834
Kazerounian S, Mahoney MG, Uitto J, Aho S (2000) Envoplakin and periplakin, the paraneoplastic pemphigus antigens, are also recognized by pemphigus foliaceus autoantibodies. J Invest Dermatol 115(3):505–507. https://doi.org/10.1046/j.1523-1747.2000.00088-2.x
Maier L, Udvardi A, Hertl M, Eming R, Schmidt E, Zillikens D, Volc-Platzer B (2017) Paraneoplastic pemphigus with anti-BP180 autoantibodies and Castleman disease. Br J Dermatol 176(3):824–826. https://doi.org/10.1111/bjd.14877
Schepens I, Jaunin F, Begre N, Laderach U, Marcus K, Hashimoto T, Favre B, Borradori L (2010) The protease inhibitor alpha-2-macroglobulin-like-1 is the p170 antigen recognized by paraneoplastic pemphigus autoantibodies in human. PLoS One 5(8):e12250. https://doi.org/10.1371/journal.pone.0012250
Hashimoto T, Kiyokawa C, Mori O, Miyasato M, Chidgey MA, Garrod DR, Kobayashi Y, Komori K, Ishii K, Amagai M, Nishikawa T (1997) Human desmocollin 1 (Dsc1) is an autoantigen for the subcorneal pustular dermatosis type of IgA pemphigus. J Invest Dermatol 109(2):127–131
Karpati S, Amagai M, Liu WL, Dmochowski M, Hashimoto T, Horvath A (2000) Identification of desmoglein 1 as autoantigen in a patient with intraepidermal neutrophilic IgA dermatosis type of IgA pemphigus. Exp Dermatol 9(3):224–228
Yasuda H, Kobayashi H, Hashimoto T, Itoh K, Yamane M, Nakamura J (2000) Subcorneal pustular dermatosis type of IgA pemphigus: demonstration of autoantibodies to desmocollin-1 and clinical review. Br J Dermatol 143(1):144–148
Ahmed AR, Carrozzo M, Caux F, Cirillo N, Dmochowski M, Alonso AE, Gniadecki R, Hertl M, López-Zabalza MJ, Lotti R, Pincelli C, Pittelkow M, Schmidt E, Sinha AA, Sprecher E, Grando SA (2016) Monopathogenic vs multipathogenic explanations of pemphigus pathophysiology. Exp Dermatol 25(11):839–846. https://doi.org/10.1111/exd.13106
Nguyen VT, Ndoye A, Shultz LD, Pittelkow MR, Grando SA (2000) Antibodies against keratinocyte antigens other than desmogleins 1 and 3 can induce pemphigus vulgaris-like lesions. J Clin Invest 106(12):1467–1479. https://doi.org/10.1172/jci10305
Vu TN, Lee TX, Ndoye A, Shultz LD, Pittelkow MR, Dahl MV, Lynch PJ, Grando SA (1998) The pathophysiological significance of nondesmoglein targets of pemphigus autoimmunity. Development of antibodies against keratinocyte cholinergic receptors in patients with pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol 134(8):971–980
Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, Leiferman KM, Ishii N, Hashimoto T, Amagai M, Zone JJ, Payne AS (2010) Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am J Pathol 177(6):2724–2730. https://doi.org/10.2353/ajpath.2010.100483
Rafei D, Muller R, Ishii N, Llamazares M, Hashimoto T, Hertl M, Eming R (2011) IgG autoantibodies against desmocollin 3 in pemphigus sera induce loss of keratinocyte adhesion. Am J Pathol 178(2):718–723. https://doi.org/10.1016/j.ajpath.2010.10.016
Abasq C, Mouquet H, Gilbert D, Tron F, Grassi V, Musette P, Joly P (2009) ELISA testing of anti-desmoglein 1 and 3 antibodies in the management of pemphigus. Arch Dermatol 145(5):529–535. https://doi.org/10.1001/archdermatol.2009.9
Amagai M, Komai A, Hashimoto T, Shirakata Y, Hashimoto K, Yamada T, Kitajima Y, Ohya K, Iwanami H, Nishikawa T (1999) Usefulness of enzyme-linked immunosorbent assay using recombinant desmogleins 1 and 3 for serodiagnosis of pemphigus. Br J Dermatol 140(2):351–357
Cheng SW, Kobayashi M, Kinoshita-Kuroda K, Tanikawa A, Amagai M, Nishikawa T (2002) Monitoring disease activity in pemphigus with enzyme-linked immunosorbent assay using recombinant desmogleins 1 and 3. Br J Dermatol 147(2):261–265
Daneshpazhooh M, Chams-Davatchi C, Khamesipour A, Mansoori P, Taheri A, Firooz A, Mortazavi H, Esmaili N, Dowlati Y (2007) Desmoglein 1 and 3 enzyme-linked immunosorbent assay in Iranian patients with pemphigus vulgaris: correlation with phenotype, severity, and disease activity. J Eur Acad Dermatol Venereol 21(10):1319–1324. https://doi.org/10.1111/j.1468-3083.2007.02254.x
Schmidt E, Dahnrich C, Rosemann A, Probst C, Komorowski L, Saschenbrecker S, Schlumberger W, Stocker W, Hashimoto T, Brocker EB, Recke A, Rose C, Zillikens D (2010) Novel ELISA systems for antibodies to desmoglein 1 and 3: correlation of disease activity with serum autoantibody levels in individual pemphigus patients. Exp Dermatol 19(5):458–463. https://doi.org/10.1111/j.1600-0625.2010.01069.x
Ruach M, Ohel G, Rahav D, Samueloff A (1995) Pemphigus vulgaris and pregnancy. Obstet Gynecol Surv 50(10):755–760
Walker DC, Kolar KA, Hebert AA, Jordon RE (1995) Neonatal pemphigus foliaceus. Arch Dermatol 131(11):1308–1311
Amagai M, Hashimoto T, Green KJ, Shimizu N, Nishikawa T (1995) Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. J Invest Dermatol 104(6):895–901
Amagai M, Hashimoto T, Shimizu N, Nishikawa T (1994) Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J Clin Invest 94(1):59–67. https://doi.org/10.1172/JCI117349
Amagai M, Karpati S, Prussick R, Klaus-Kovtun V, Stanley JR (1992) Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J Clin Invest 90(3):919–926. https://doi.org/10.1172/JCI115968
Amagai M, Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, Nishikawa T (2000) Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. J Clin Invest 105(5):625–631. https://doi.org/10.1172/JCI8748
Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA (1982) Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med 306(20):1189–1196. https://doi.org/10.1056/NEJM198205203062001
Roscoe JT, Diaz L, Sampaio SA, Castro RM, Labib RS, Takahashi Y, Patel H, Anhalt GJ (1985) Brazilian pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Invest Dermatol 85(6):538–541
Schulze K, Galichet A, Sayar BS, Scothern A, Howald D, Zymann H, Siffert M, Zenhausern D, Bolli R, Koch PJ, Garrod D, Suter MM, Muller EJ (2012) An adult passive transfer mouse model to study desmoglein 3 signaling in pemphigus vulgaris. J Invest Dermatol 132(2):346–355. https://doi.org/10.1038/jid.2011.299
Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, Muller E (2001) A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol 153(4):823–834
Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, Kowalczyk AP (2006) Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem 281(11):7623–7634. https://doi.org/10.1074/jbc.M512447200
Heupel WM, Zillikens D, Drenckhahn D, Waschke J (2008) Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol 181(3):1825–1834
Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, Hanakawa Y, Amagai M, Kowalczyk AP (2011) Desmosome disassembly in response to pemphigus vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Invest Dermatol 131(3):706–718. https://doi.org/10.1038/jid.2010.389
Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, Amagai M, Kowalczyk AP (2012) Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS One 7(12):e50696. https://doi.org/10.1371/journal.pone.0050696
Spindler V, Endlich A, Hartlieb E, Vielmuth F, Schmidt E, Waschke J (2011) The extent of desmoglein 3 depletion in pemphigus vulgaris is dependent on ca(2+)-induced differentiation: a role in suprabasal epidermal skin splitting? Am J Pathol 179(4):1905–1916. https://doi.org/10.1016/j.ajpath.2011.06.043
Spindler V, Rotzer V, Dehner C, Kempf B, Gliem M, Radeva M, Hartlieb E, Harms GS, Schmidt E, Waschke J (2013) Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest 123(2):800–811. https://doi.org/10.1172/JCI60139
Belloni-Fortina A, Faggion D, Pigozzi B, Peserico A, Bordignon M, Baldo V, Alaibac M (2009) Detection of autoantibodies against recombinant desmoglein 1 and 3 molecules in patients with pemphigus vulgaris: correlation with disease extent at the time of diagnosis and during follow-up. Clin Dev Immunol 2009:187864. https://doi.org/10.1155/2009/187864
Kamiya K, Aoyama Y, Shirafuji Y, Hamada T, Morizane S, Fujii K, Hisata K, Iwatsuki K (2012) Detection of antibodies against the non-calcium-dependent epitopes of desmoglein 3 in pemphigus vulgaris and their pathogenic significance. Br J Dermatol 167(2):252–261. https://doi.org/10.1111/j.1365-2133.2012.10929.x
Kamiya K, Aoyama Y, Shirafuji Y, Hamada T, Morizane S, Fujii K, Iwatsuki K (2013) A higher correlation of the antibody activities against the calcium-dependent epitopes of desmoglein 3 quantified by ethylenediaminetetraacetic acid-treated enzyme-linked immunosorbent assay with clinical disease activities of pemphigus vulgaris. J Dermatol Sci 70(3):190–195. https://doi.org/10.1016/j.jdermsci.2013.02.011
Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, Tsunoda K, Amagai M, Stanley JR, Siegel DL (2005) Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest 115(4):888–899. https://doi.org/10.1172/JCI24185
Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M (2003) Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol 170(4):2170–2178
Bhol KC, Ahmed AR (2002) Production of non-pathogenic human monoclonal antibodies to desmoglein 3 from pemphigus vulgaris patient. Autoimmunity 35(2):87–91
Cho MJ, Lo AS, Mao X, Nagler AR, Ellebrecht CT, Mukherjee EM, Hammers CM, Choi EJ, Sharma PM, Uduman M, Li H, Rux AH, Farber SA, Rubin CB, Kleinstein SH, Sachais BS, Posner MR, Cavacini LA, Payne AS (2014) Shared VH1-46 gene usage by pemphigus vulgaris autoantibodies indicates common humoral immune responses among patients. Nat Commun 5:4167. https://doi.org/10.1038/ncomms5167
Di Zenzo G, Di Lullo G, Corti D, Calabresi V, Sinistro A, Vanzetta F, Didona B, Cianchini G, Hertl M, Eming R, Amagai M, Ohyama B, Hashimoto T, Sloostra J, Sallusto F, Zambruno G, Lanzavecchia A (2012) Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest 122(10):3781–3790. https://doi.org/10.1172/JCI64413
Ishii K, Lin C, Siegel DL, Stanley JR (2008) Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol 128(4):939–948. https://doi.org/10.1038/sj.jid.5701132
Yamagami J, Kacir S, Ishii K, Payne AS, Siegel DL, Stanley JR (2009) Antibodies to the desmoglein 1 precursor proprotein but not to the mature cell surface protein cloned from individuals without pemphigus. J Immunol 183(9):5615–5621. https://doi.org/10.4049/jimmunol.0901691
Yeh SW, Cavacini LA, Bhol KC, Lin MS, Kumar M, Duval M, Posner MR, Ahmed AR (2006) Pathogenic human monoclonal antibody against desmoglein 3. Clin Immunol 120(1):68–75. https://doi.org/10.1016/j.clim.2006.03.006
Chan PT, Ohyama B, Nishifuji K, Yoshida K, Ishii K, Hashimoto T, Amagai M (2010) Immune response towards the amino-terminus of desmoglein 1 prevails across different activity stages in nonendemic pemphigus foliaceus. Br J Dermatol 162(6):1242–1250. https://doi.org/10.1111/j.1365-2133.2010.09696.x
Futei Y, Amagai M, Sekiguchi M, Nishifuji K, Fujii Y, Nishikawa T (2000) Use of domain-swapped molecules for conformational epitope mapping of desmoglein 3 in pemphigus vulgaris. J Invest Dermatol 115(5):829–834. https://doi.org/10.1046/j.1523-1747.2000.00137.x
Ohyama B, Nishifuji K, Chan PT, Kawaguchi A, Yamashita T, Ishii N, Hamada T, Dainichi T, Koga H, Tsuruta D, Amagai M, Hashimoto T (2012) Epitope spreading is rarely found in pemphigus vulgaris by large-scale longitudinal study using desmoglein 2-based swapped molecules. J Invest Dermatol 132(4):1158–1168. https://doi.org/10.1038/jid.2011.448
Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M (2001) Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J Immunol 167(9):5439–5448
Sharma PM, Choi EJ, Kuroda K, Hachiya T, Ishii K, Payne AS (2009) Pathogenic anti-desmoglein MAbs show variable ELISA activity because of preferential binding of mature versus proprotein isoforms of desmoglein 3. J Invest Dermatol 129(9):2309–2312. https://doi.org/10.1038/jid.2009.41
Allen EM, Giudice GJ, Diaz LA (1993) Subclass reactivity of pemphigus foliaceus autoantibodies with recombinant human desmoglein. J Invest Dermatol 100(5):685–691
Dmochowski M, Hashimoto T, Nishikawa T (1992) The analysis of IgG subclasses of anti-intercellular antibodies in pemphigus by an immunoblot technique. Arch Dermatol Res 284(5):309–311
Jones CC, Hamilton RG, Jordon RE (1988) Subclass distribution of human IgG autoantibodies in pemphigus. J Clin Immunol 8(1):43–49
Wilson CL, Wojnarowska F, Dean D, Pasricha JS (1993) IgG subclasses in pemphigus in Indian and UK populations. Clin Exp Dermatol 18(3):226–230
Aoki V, Rivitti EA, Diaz LA, Cooperative Group on Fogo Selvagem R (2015) Update on fogo selvagem, an endemic form of pemphigus foliaceus. J Dermatol 42(1):18–26. https://doi.org/10.1111/1346-8138.12675
Lee HE, Berkowitz P, Jolly PS, Diaz LA, Chua MP, Rubenstein DS (2009) Biphasic activation of p38MAPK suggests that apoptosis is a downstream event in pemphigus acantholysis. J Biol Chem 284(18):12524–12532. https://doi.org/10.1074/jbc.M808204200
Warren SJ, Arteaga LA, Rivitti EA, Aoki V, Hans-Filho G, Qaqish BF, Lin MS, Giudice GJ, Diaz LA (2003) The role of subclass switching in the pathogenesis of endemic pemphigus foliaceus. J Invest Dermatol 120(1):104–108. https://doi.org/10.1046/j.1523-1747.2003.12017.x
de Bruin A, Caldelari R, Williamson L, Suter MM, Hunziker T, Wyder M, Muller EJ (2007) Plakoglobin-dependent disruption of the desmosomal plaque in pemphigus vulgaris. Exp Dermatol 16(6):468–475. https://doi.org/10.1111/j.1600-0625.2007.00557.x
Amagai M, Stanley JR (2012) Desmoglein as a target in skin disease and beyond. J Invest Dermatol 132(3 Pt 2):776–784. https://doi.org/10.1038/jid.2011.390
Kitajima Y (2014) 150(th) anniversary series: desmosomes and autoimmune disease, perspective of dynamic desmosome remodeling and its impairments in pemphigus. Cell Commun Adhes 21(6):269–280. https://doi.org/10.3109/15419061.2014.943397
Waschke J, Spindler V (2014) Desmosomes and extradesmosomal adhesive signaling contacts in pemphigus. Med Res Rev 34(6):1127–1145. https://doi.org/10.1002/med.21310
Hacker-Foegen MK, Janson M, Amagai M, Fairley JA, Lin MS (2003) Pathogenicity and epitope characteristics of anti-desmoglein-1 from pemphigus foliaceus patients expressing only IgG1 autoantibodies. J Invest Dermatol 121(6):1373–1378. https://doi.org/10.1111/j.1523-1747.2003.12608.x
Muller R, Svoboda V, Wenzel E, Gebert S, Hunzelmann N, Muller HH, Hertl M (2006) IgG reactivity against non-conformational NH-terminal epitopes of the desmoglein 3 ectodomain relates to clinical activity and phenotype of pemphigus vulgaris. Exp Dermatol 15(8):606–614. https://doi.org/10.1111/j.1600-0625.2006.00451.x
Aoyama Y, Nagai M, Kitajima Y (2010) Binding of pemphigus vulgaris IgG to antigens in desmosome core domains excludes immune complexes rather than directly splitting desmosomes. Br J Dermatol 162(5):1049–1055. https://doi.org/10.1111/j.1365-2133.2010.09672.x
Aoyama Y, Owada MK, Kitajima Y (1999) A pathogenic autoantibody, pemphigus vulgaris-IgG, induces phosphorylation of desmoglein 3, and its dissociation from plakoglobin in cultured keratinocytes. Eur J Immunol 29 (7):2233–2240. doi: https://doi.org/10.1002/(SICI)1521-4141(199907)29:07&%2360;2233::AID-IMMU2233&%2362;3.0.CO;2-4
Vollner F, Ali J, Kurrle N, Exner Y, Eming R, Hertl M, Banning A, Tikkanen R (2016) Loss of flotillin expression results in weakened desmosomal adhesion and pemphigus vulgaris-like localisation of desmoglein-3 in human keratinocytes. Sci Rep 6:28820. https://doi.org/10.1038/srep28820
Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, Rubenstein DS (2005) Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem 280(25):23778–23784. https://doi.org/10.1074/jbc.M501365200
Cirillo N, Lanza A, Prime SS (2010) Induction of hyper-adhesion attenuates autoimmune-induced keratinocyte cell-cell detachment and processing of adhesion molecules via mechanisms that involve PKC. Exp Cell Res 316(4):580–592. https://doi.org/10.1016/j.yexcr.2009.10.005
Grando SA (2012) Pemphigus autoimmunity: hypotheses and realities. Autoimmunity 45(1):7–35. https://doi.org/10.3109/08916934.2011.606444
Kawasaki Y, Aoyama Y, Tsunoda K, Amagai M, Kitajima Y (2006) Pathogenic monoclonal antibody against desmoglein 3 augments desmoglein 3 and p38 MAPK phosphorylation in human squamous carcinoma cell line. Autoimmunity 39(7):587–590. https://doi.org/10.1080/08916930600971943
Berkowitz P, Diaz LA, Hall RP, Rubenstein DS (2008) Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Invest Dermatol 128(3):738–740. https://doi.org/10.1038/sj.jid.5701080
Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS (2006) p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A 103(34):12855–12860. https://doi.org/10.1073/pnas.0602973103
Kitajima Y (2013) New insights into desmosome regulation and pemphigus blistering as a desmosome-remodeling disease. Kaohsiung J Med Sci 29(1):1–13. https://doi.org/10.1016/j.kjms.2012.08.001
Arredondo J, Chernyavsky AI, Karaouni A, Grando SA (2005) Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in pemphigus. Am J Pathol 167(6):1531–1544. https://doi.org/10.1016/S0002-9440(10)61239-4
Frusic-Zlotkin M, Pergamentz R, Michel B, David M, Mimouni D, Bregegere F, Milner Y (2005) The interaction of pemphigus autoimmunoglobulins with epidermal cells: activation of the fas apoptotic pathway and the use of caspase activity for pathogenicity tests of pemphigus patients. Ann N Y Acad Sci 1050:371–379. https://doi.org/10.1196/annals.1313.040
Gniadecki R, Jemec GB, Thomsen BM, Hansen M (1998) Relationship between keratinocyte adhesion and death: anoikis in acantholytic diseases. Arch Dermatol Res 290(10):528–532
Pacheco-Tovar MG, Avalos-Diaz E, Vega-Memije E, Bollain-y-Goytia JJ, Lopez-Robles E, Hojyo-Tomoka MT, Dominguez-Soto L, Herrera-Esparza R (2009) The final destiny of acantholytic cells in pemphigus is Fas mediated. J Eur Acad Dermatol Venereol 23(6):697–701. https://doi.org/10.1111/j.1468-3083.2009.03162.x
Pelacho B, Natal C, Espana A, Sanchez-Carpintero I, Iraburu MJ, Lopez-Zabalza MJ (2004) Pemphigus vulgaris autoantibodies induce apoptosis in HaCaT keratinocytes. FEBS Lett 566(1–3):6–10. https://doi.org/10.1016/j.febslet.2004.03.107
Puviani M, Marconi A, Cozzani E, Pincelli C (2003) Fas ligand in pemphigus sera induces keratinocyte apoptosis through the activation of caspase-8. J Invest Dermatol 120(1):164–167. https://doi.org/10.1046/j.1523-1747.2003.12014.x
Kalantari-Dehaghi M, Anhalt GJ, Camilleri MJ, Chernyavsky AI, Chun S, Felgner PL, Jasinskas A, Leiferman KM, Liang L, Marchenko S, Nakajima-Sasaki R, Pittelkow MR, Zone JJ, Grando SA (2013) Pemphigus vulgaris autoantibody profiling by proteomic technique. PLoS One 8(3):e57587. https://doi.org/10.1371/journal.pone.0057587
Marchenko S, Chernyavsky AI, Arredondo J, Gindi V, Grando SA (2010) Antimitochondrial autoantibodies in pemphigus vulgaris: a missing link in disease pathophysiology. J Biol Chem 285(6):3695–3704. https://doi.org/10.1074/jbc.M109.081570
Evangelista F, Dasher DA, Diaz LA, Prisayanh PS, Li N (2008) E-cadherin is an additional immunological target for pemphigus autoantibodies. J Invest Dermatol 128(7):1710–1718. https://doi.org/10.1038/sj.jid.5701260
Kljuic A, Bazzi H, Sundberg JP, Martinez-Mir A, O'Shaughnessy R, Mahoney MG, Levy M, Montagutelli X, Ahmad W, Aita VM, Gordon D, Uitto J, Whiting D, Ott J, Fischer S, Gilliam TC, Jahoda CA, Morris RJ, Panteleyev AA, Nguyen VT, Christiano AM (2003) Desmoglein 4 in hair follicle differentiation and epidermal adhesion: evidence from inherited hypotrichosis and acquired pemphigus vulgaris. Cell 113(2):249–260
Nagasaka T, Nishifuji K, Ota T, Whittock NV, Amagai M (2004) Defining the pathogenic involvement of desmoglein 4 in pemphigus and staphylococcal scalded skin syndrome. J Clin Invest 114(10):1484–1492. https://doi.org/10.1172/JCI20480
Oliveira ME, Culton DA, Prisayanh P, Qaqish BF, Diaz LA (2013) E-cadherin autoantibody profile in patients with pemphigus vulgaris. Br J Dermatol 169(4):812–818. https://doi.org/10.1111/bjd.12455
Dmochowski M, Hashimoto T, Garrod DR, Nishikawa T (1993) Desmocollins I and II are recognized by certain sera from patients with various types of pemphigus, particularly Brazilian pemphigus foliaceus. J Invest Dermatol 100(4):380–384
Kim SC, Chung YL, Kim J, Cho NJ, Amagai M (2001) Pemphigus vulgaris with autoantibodies to desmoplakin. Br J Dermatol 145(5):838–840
Nguyen VT, Ndoye A, Grando SA (2000) Pemphigus vulgaris antibody identifies pemphaxin. A novel keratinocyte annexin-like molecule binding acetylcholine. J Biol Chem 275(38):29466–29476. https://doi.org/10.1074/jbc.M003174200
Bhol KC, Rojas AI, Khan IU, Ahmed AR (2000) Presence of interleukin 10 in the serum and blister fluid of patients with pemphigus vulgaris and pemphigoid. Cytokine 12(7):1076–1083. https://doi.org/10.1006/cyto.1999.0642
D'Auria L, Bonifati C, Mussi A, D'Agosto G, De Simone C, Giacalone B, Ferraro C, Ameglio F (1997) Cytokines in the sera of patients with pemphigus vulgaris: interleukin-6 and tumour necrosis factor-alpha levels are significantly increased as compared to healthy subjects and correlate with disease activity. Eur Cytokine Netw 8(4):383–387
Satyam A, Khandpur S, Sharma VK, Sharma A (2009) Involvement of T(H)1/T(H)2 cytokines in the pathogenesis of autoimmune skin disease-pemphigus vulgaris. Immunol Investig 38(6):498–509
Hennerici T, Pollmann R, Schmidt T, Seipelt M, Tackenberg B, Mobs C, Ghoreschi K, Hertl M, Eming R (2016) Increased frequency of T follicular helper cells and elevated Interleukin-27 plasma levels in patients with pemphigus. PLoS One 11(2):e0148919. https://doi.org/10.1371/journal.pone.0148919
Timoteo RP, da Silva MV, Miguel CB, Silva DA, Catarino JD, Rodrigues Junior V, Sales-Campos H, Freire Oliveira CJ (2017) Th1/Th17-related cytokines and chemokines and their implications in the pathogenesis of pemphigus vulgaris. Mediat Inflamm 2017:7151285. https://doi.org/10.1155/2017/7151285
Zebrowska A, Wozniacka A, Juczynska K, Ociepa K, Waszczykowska E, Szymczak I, Pawliczak R (2017) Correlation between IL36alpha and IL17 and activity of the disease in selected autoimmune blistering diseases. Mediat Inflamm 2017:8980534. https://doi.org/10.1155/2017/8980534
Cho MJ, Ellebrecht CT, Payne AS (2015) The dual nature of interleukin-10 in pemphigus vulgaris. Cytokine 73(2):335–341. https://doi.org/10.1016/j.cyto.2014.11.002
Ameglio F, D'Auria L, Cordiali-Fei P, Trento E, D'Agosto G, Mastroianni A, Giannetti A, Giacalone B (1999) Anti-intercellular substance antibody log titres are correlated with serum concentrations of interleukin-6, interleukin-15 and tumor necrosis factor-alpha in patients with pemphigus vulgaris relationships with peripheral blood neutrophil counts, disease severity and duration and patients' age. J Biol Regul Homeost Agents 13(4):220–224
Feliciani C, Toto P, Amerio P, Pour SM, Coscione G, Shivji G, Wang B, Sauder DN (2000) In vitro and in vivo expression of interleukin-1alpha and tumor necrosis factor-alpha mRNA in pemphigus vulgaris: interleukin-1alpha and tumor necrosis factor-alpha are involved in acantholysis. J Invest Dermatol 114(1):71–77. https://doi.org/10.1046/j.1523-1747.2000.00835.x
Lopez-Robles E, Avalos-Diaz E, Vega-Memije E, Hojyo-Tomoka T, Villalobos R, Fraire S, Domiguez-Soto L, Herrera-Esparza R (2001) TNFalpha and IL-6 are mediators in the blistering process of pemphigus. Int J Dermatol 40(3):185–188
Lotti R, Marconi A, Pincelli C (2012) Apoptotic pathways in the pathogenesis of pemphigus: targets for new therapies. Curr Pharm Biotechnol 13(10):1877–1881
Janse IC, van der Wier G, Jonkman MF, Pas HH, Diercks GF (2014) No evidence of apoptotic cells in pemphigus acantholysis. J Invest Dermatol 134(7):2039–2041. https://doi.org/10.1038/jid.2014.60
Kurzen H, Brenner S (2006) Significance of autoimmunity to non-desmoglein targets in pemphigus. Autoimmunity 39(7):549–556. https://doi.org/10.1080/08916930600971554
Anhalt GJ, Labib RS, Patel H, Diaz LA (1983) Animal models. Clin Dermatol 1(2):98–105
Takahashi Y, Patel HP, Labib RS, Diaz LA, Anhalt GJ (1985) Experimentally induced pemphigus vulgaris in neonatal BALB/c mice: a time-course study of clinical, immunologic, ultrastructural, and cytochemical changes. J Invest Dermatol 84(1):41–46
Anhalt GJ, Till GO, Diaz LA, Labib RS, Patel HP, Eaglstein NF (1986) Defining the role of complement in experimental pemphigus vulgaris in mice. J Immunol (Baltimore, Md : 1950) 137(9):2835–2840
Amagai M, Klaus-Kovtun V, Stanley JR (1991) Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 67(5):869–877. https://doi.org/10.1016/0092-8674(91)90360-b
Rock B, Labib RS, Diaz LA (1990) Monovalent Fab' immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Invest 85(1):296–299. https://doi.org/10.1172/jci114426
Rock B, Martins CR, Theofilopoulos AN, Balderas RS, Anhalt GJ, Labib RS, Futamura S, Rivitti EA, Diaz LA (1989) The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem). N Engl J Med 320(22):1463–1469. https://doi.org/10.1056/nejm198906013202206
Koulu L, Kusumi A, Steinberg MS, Klaus-Kovtun V, Stanley JR (1984) Human autoantibodies against a desmosomal core protein in pemphigus foliaceus. J Exp Med 160(5):1509–1518
Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, Murphy GF, Whitaker-Menezes D, Stanley JR (1997) Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol 137(5):1091–1102
Amagai M (2008) Pemphigus vulgaris and its active disease mouse model. Curr Dir Autoimmun 10:167–181. https://doi.org/10.1159/000131453
Ohyama M, Amagai M, Tsunoda K, Ota T, Koyasu S, J-i H, Umezawa A, Nishikawa T (2002) Immunologic and histopathologic characterization of an active disease mouse model for pemphigus vulgaris. J Invest Dermatol 118(1):199–204. https://doi.org/10.1046/j.0022-202x.2001.01643.x
Shimizu A, Ishiko A, Ota T, Tsunoda K, Koyasu S, Amagai M, Nishikawa T (2002) Ultrastructural changes in mice actively producing antibodies to desmoglein 3 parallel those in patients with pemphigus vulgaris. Arch Dermatol Res 294(7):318–323. https://doi.org/10.1007/s00403-002-0341-z
Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M (2003) Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol (Baltimore, Md : 1950) 170(4):2170–2178
Shimizu A, Ishiko A, Ota T, Saito H, Oka H, Tsunoda K, Amagai M, Nishikawa T (2005) In vivo ultrastructural localization of the desmoglein 3 adhesive interface to the desmosome mid-line. J Invest Dermatol 124(5):984–989. https://doi.org/10.1111/j.0022-202X.2005.23706.x
Shimizu A, Ishiko A, Ota T, Tsunoda K, Amagai M, Nishikawa T (2004) IgG binds to desmoglein 3 in desmosomes and causes a desmosomal split without keratin retraction in a pemphigus mouse model. J Invest Dermatol 122(5):1145–1153. https://doi.org/10.1111/j.0022-202X.2004.22426.x
Takahashi H, Amagai M, Nishikawa T, Fujii Y, Kawakami Y, Kuwana M (2008) Novel system evaluating in vivo pathogenicity of desmoglein 3-reactive T cell clones using murine pemphigus vulgaris. J Immunol (Baltimore, Md : 1950) 181(2):1526–1535
Takahashi H, Kuwana M, Amagai M (2009) A single helper T cell clone is sufficient to commit polyclonal naive B cells to produce pathogenic IgG in experimental pemphigus vulgaris. J Immunol (Baltimore, Md : 1950) 182(3):1740–1745
Aoki-Ota M, Kinoshita M, Ota T, Tsunoda K, Iwasaki T, Tanaka S, Koyasu S, Nishikawa T, Amagai M (2006) Tolerance induction by the blockade of CD40/CD154 interaction in pemphigus vulgaris mouse model. J Invest Dermatol 126(1):105–113. https://doi.org/10.1038/sj.jid.5700016
YOKOYAMA T, Matsuda S, Takae Y, Wada N, Nishikawa T, Amagai M, Koyasu S (2011) Antigen-independent development of Foxp3+ regulatory T cells suppressing autoantibody production in experimental pemphigus vulgaris. Int Immunol 23(6):365–373. https://doi.org/10.1093/intimm/dxr020
Aoki-Ota M, Tsunoda K, Ota T, Iwasaki T, Koyasu S, Amagai M, Nishikawa T (2004) A mouse model of pemphigus vulgaris by adoptive transfer of naive splenocytes from desmoglein 3 knockout mice. Br J Dermatol 151(2):346–354. https://doi.org/10.1111/j.1365-2133.2004.06056.x
Kawasaki H, Tsunoda K, Hata T, Ishii K, Yamada T, Amagai M (2006) Synergistic pathogenic effects of combined mouse monoclonal anti-desmoglein 3 IgG antibodies on pemphigus vulgaris blister formation. J Invest Dermatol 126(12):2621–2630. https://doi.org/10.1038/sj.jid.5700450
Fan JL, Memar O, McCormick DJ, Prabhakar BS (1999) BALB/c mice produce blister-causing antibodies upon immunization with a recombinant human desmoglein 3. J Immunol (Baltimore, Md : 1950) 163(11):6228–6235
Kaithamana S, Fan J-L, Memar O, Li K, Uitto J, Seetharamaiah GS, Prabhakar BS (2003) Relevance of differential immunogenicity of human and mouse recombinant desmoglein-3 for the induction of acantholytic autoantibodies in mice. Clin Exp Immunol 132(1):16–23
Ishikawa H, Li K, Tamai K, Sawamura D, Uitto J (2000) Cloning of the mouse desmoglein 3 gene (Dsg3): interspecies conservation within the cadherin superfamily. Exp Dermatol 9(4):229–239
CULTON DA, McCray SK, Park M, Roberts JC, Li N, Zedek DC, Anhalt GJ, Cowley DO, Liu Z, Diaz LA (2015) Mucosal pemphigus vulgaris anti-Dsg3 IgG is pathogenic to the oral mucosa of humanized Dsg3 mice. J Invest Dermatol 135(6):1590–1597. https://doi.org/10.1038/jid.2015.54
Loiseau P, Lecleach L, Prost C, Lepage V, Busson M, Bastuji-Garin S, Roujeau JC, Charron D (2000) HLA class II polymorphism contributes to specify desmoglein derived peptides in pemphigus vulgaris and pemphigus foliaceus. J Autoimmun 15(1):67–73. https://doi.org/10.1006/jaut.2000.0388
Svecova D, Parnicka Z, Pastyrikova L, Urbancek S, Luha J, Buc M (2015) HLA DRB1* and DQB1* alleles are associated with disease severity in patients with pemphigus vulgaris. Int J Dermatol 54(2):168–173. https://doi.org/10.1111/ijd.12418
Lee E, Lendas KA, Chow S, Pirani Y, Gordon D, Dionisio R, Nguyen D, Spizuoco A, Fotino M, Zhang Y, Sinha AA (2006) Disease relevant HLA class II alleles isolated by genotypic, haplotypic, and sequence analysis in north American Caucasians with pemphigus vulgaris. Hum Immunol 67(1–2):125–139. https://doi.org/10.1016/j.humimm.2005.09.003
Wucherpfennig KW, Strominger JL (1995) Selective binding of self peptides to disease-associated major histocompatibility complex (MHC) molecules: a mechanism for MHC-linked susceptibility to human autoimmune diseases. J Exp Med 181(5):1597–1601
Wucherpfennig KW, Yu B, Bhol K, Monos DS, Argyris E, Karr RW, Ahmed AR, Strominger JL (1995) Structural basis for major histocompatibility complex (MHC)-linked susceptibility to autoimmunity: charged residues of a single MHC binding pocket confer selective presentation of self-peptides in pemphigus vulgaris. Proc Natl Acad Sci U S A 92(25):11935–11939
Hertl M, Eming R, Veldman C (2006) T cell control in autoimmune bullous skin disorders. J Clin Invest 116(5):1159–1166. https://doi.org/10.1172/jci28547
Eming R, Hennerici T, Bäcklund J, Feliciani C, Visconti KC, Willenborg S, Wohde J, Holmdahl R, Sønderstrup G, Hertl M (2014) Pathogenic IgG antibodies against desmoglein 3 in pemphigus vulgaris are regulated by HLA-DRB1*04:02-restricted T cells. J Immunol (Baltimore, Md : 1950) 193(9):4391–4399. https://doi.org/10.4049/jimmunol.1401081
Schmidt T, Willenborg S, Hünig T, Deeg CA, Sonderstrup G, Hertl M, Eming R (2016) Induction of T regulatory cells by the superagonistic anti-CD28 antibody D665 leads to decreased pathogenic IgG autoantibodies against desmoglein 3 in a HLA-transgenic mouse model of pemphigus vulgaris. Exp Dermatol 25(4):293–298. https://doi.org/10.1111/exd.12919
Zhao CY, Murrell DF (2015) Pemphigus vulgaris: an evidence-based treatment update. Drugs 75(3):271–284. https://doi.org/10.1007/s40265-015-0353-6
Beissert S, Werfel T, Frieling U, Bohm M, Sticherling M, Stadler R, Zillikens D, Rzany B, Hunzelmann N, Meurer M, Gollnick H, Ruzicka T, Pillekamp H, Junghans V, Luger TA (2006) A comparison of oral methylprednisolone plus azathioprine or mycophenolate mofetil for the treatment of pemphigus. Arch Dermatol 142(11):1447–1454. https://doi.org/10.1001/archderm.142.11.1447
Beissert S, Mimouni D, Kanwar AJ, Solomons N, Kalia V, Anhalt GJ (2010) Treating pemphigus vulgaris with prednisone and mycophenolate mofetil: a multicenter, randomized, placebo-controlled trial. J Invest Dermatol 130(8):2041–2048. https://doi.org/10.1038/jid.2010.91
Chams-Davatchi C, Mortazavizadeh A, Daneshpazhooh M, Davatchi F, Balighi K, Esmaili N, Akhyani M, Hallaji Z, Seirafi H, Mortazavi H (2013) Randomized double blind trial of prednisolone and azathioprine, vs. prednisolone and placebo, in the treatment of pemphigus vulgaris. J Eur Acad Dermatol Venereol 27(10):1285–1292. https://doi.org/10.1111/j.1468-3083.2012.04717.x
Enk AH, Hadaschik EN, Eming R, Fierlbeck G, French LE, Girolomoni G, Hertl M, Jolles S, Karpati S, Steinbrink K, Stingl G, Volc-Platzer B, Zillikens D (2016) European guidelines (S1) on the use of high-dose intravenous immunoglobulin in dermatology. J Eur Acad Dermatol Venereol 30(10):1657–1669. https://doi.org/10.1111/jdv.13725
Eming R, Hertl M (2006) Immunoadsorption in pemphigus. Autoimmunity 39(7):609–616. https://doi.org/10.1080/08916930600972040
Kasperkiewicz M, Eming R, Behzad M, Hunzelmann N, Meurer M, Schulze-Koops H, von Wussow P, Hertl M, Zillikens D, Freivogel K, Dorner T, Schmidt E (2012) Efficacy and safety of rituximab in pemphigus: experience of the German registry of autoimmune diseases. J Dtsch Dermatol Ges 10(10):727–732. https://doi.org/10.1111/j.1610-0387.2012.07931.x
Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, Caillot F, Golinski ML, Labeille B, Picard-Dahan C, Paul C, Richard MA, Bouaziz JD, Duvert-Lehembre S, Bernard P, Caux F, Alexandre M, Ingen-Housz-Oro S, Vabres P, Delaporte E, Quereux G, Dupuy A, Debarbieux S, Avenel-Audran M, D'Incan M, Bedane C, Beneton N, Jullien D, Dupin N, Misery L, Machet L, Beylot-Barry M, Dereure O, Sassolas B, Vermeulin T, Benichou J, Musette P, French study group on autoimmune bullous skin d (2017) First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet 389 (10083):2031–2040. doi:https://doi.org/10.1016/S0140-6736(17)30070-3
Hammers CM, Stanley JR (2016) Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol 11:175–197. https://doi.org/10.1146/annurev-pathol-012615-044313
Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, Amagai M (2017) Pemphigus. Nat Rev Dis Primers 3:17026. https://doi.org/10.1038/nrdp.2017.26
Amber KT, Staropoli P, Shiman MI, Elgart GW, Hertl M (2013) Autoreactive T cells in the immune pathogenesis of pemphigus vulgaris. Exp Dermatol 22(11):699–704. https://doi.org/10.1111/exd.12229
Hertl M, Karr RW, Amagai M, Katz SI (1998) Heterogeneous MHC II restriction pattern of autoreactive desmoglein 3 specific T cell responses in pemphigus vulgaris patients and normals. J Invest Dermatol 110(4):388–392. https://doi.org/10.1046/j.1523-1747.1998.00156.x
Veldman CM, Gebhard KL, Uter W, Wassmuth R, Grotzinger J, Schultz E, Hertl M (2004) T cell recognition of desmoglein 3 peptides in patients with pemphigus vulgaris and healthy individuals. J Immunol 172(6):3883–3892
Beutner EH, Jordon RE (1964) Demonstration of skin antibodies in sera of pemphigus vulgaris patients by indirect immunofluorescent staining. Proc Soc Exp Biol Med 117:505–510
Colliou N, Picard D, Caillot F, Calbo S, Le Corre S, Lim A, Lemercier B, Le Mauff B, Maho-Vaillant M, Jacquot S, Bedane C, Bernard P, Caux F, Prost C, Delaporte E, Doutre MS, Dreno B, Franck N, Ingen-Housz-Oro S, Chosidow O, Pauwels C, Picard C, Roujeau JC, Sigal M, Tancrede-Bohin E, Templier I, Eming R, Hertl M, D'Incan M, Joly P, Musette P (2013) Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci Transl Med 5 (175):175ra130. doi:https://doi.org/10.1126/scitranslmed.3005166
Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M (2008) Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol 128(12):2850–2858. https://doi.org/10.1038/jid.2008.172
Hammers CM, Chen J, Lin C, Kacir S, Siegel DL, Payne AS, Stanley JR (2015) Persistence of anti-desmoglein 3 IgG(+) B-cell clones in pemphigus patients over years. J Invest Dermatol 135(3):742–749. https://doi.org/10.1038/jid.2014.291
Alexander T, Sarfert R, Klotsche J, Kuhl AA, Rubbert-Roth A, Lorenz HM, Rech J, Hoyer BF, Cheng Q, Waka A, Taddeo A, Wiesener M, Schett G, Burmester GR, Radbruch A, Hiepe F, Voll RE (2015) The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis 74(7):1474–1478. https://doi.org/10.1136/annrheumdis-2014-206016
Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T, Barnard J, Nevarez S, Goldman BI, Kirk CJ, Looney RJ, Anolik JH (2012) Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum 64(2):493–503. https://doi.org/10.1002/art.33333
Taddeo A, Khodadadi L, Voigt C, Mumtaz IM, Cheng Q, Moser K, Alexander T, Manz RA, Radbruch A, Hiepe F, Hoyer BF (2015) Long-lived plasma cells are early and constantly generated in New Zealand black/New Zealand white F1 mice and their therapeutic depletion requires a combined targeting of autoreactive plasma cells and their precursors. Arthritis Res Ther 17:39. https://doi.org/10.1186/s13075-015-0551-3
Hiepe F, Radbruch A (2016) Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat Rev Nephrol 12(4):232–240. https://doi.org/10.1038/nrneph.2016.20
Ellebrecht CT, Choi EJ, Allman DM, Tsai DE, Wegener WA, Goldenberg DM, Payne AS (2014) Subcutaneous veltuzumab, a humanized anti-CD20 antibody, in the treatment of refractory pemphigus vulgaris. JAMA Dermatol 150(12):1331–1335. https://doi.org/10.1001/jamadermatol.2014.1939
Huang A, Madan RK, Levitt J (2016) Future therapies for pemphigus vulgaris: rituximab and beyond. J Am Acad Dermatol 74(4):746–753. https://doi.org/10.1016/j.jaad.2015.11.008
Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, Ruiz-Irastorza G, Hughes G (2016) Systemic lupus erythematosus. Nat Rev Dis Primers 2:16039. https://doi.org/10.1038/nrdp.2016.39
Stohl W (2017) Inhibition of B cell activating factor (BAFF) in the management of systemic lupus erythematosus (SLE). Expert Rev Clin Immunol 13(6):623–633. https://doi.org/10.1080/1744666X.2017.1291343
Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, Milone MC, Payne AS (2016) Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353(6295):179–184. https://doi.org/10.1126/science.aaf6756
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, Raffeld M, Feldman S, Lu L, Li YF, Ngo LT, Goy A, Feldman T, Spaner DE, Wang ML, Chen CC, Kranick SM, Nath A, Nathan DA, Morton KE, Toomey MA, Rosenberg SA (2015) Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 33(6):540–549. https://doi.org/10.1200/JCO.2014.56.2025
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL (2015) T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385(9967):517–528. https://doi.org/10.1016/S0140-6736(14)61403-3
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371(16):1507–1517. https://doi.org/10.1056/NEJMoa1407222
Eming R, Hennerici T, Backlund J, Feliciani C, Visconti KC, Willenborg S, Wohde J, Holmdahl R, Sonderstrup G, Hertl M (2014) Pathogenic IgG antibodies against desmoglein 3 in pemphigus vulgaris are regulated by HLA-DRB1*04:02-restricted T cells. J Immunol 193(9):4391–4399. https://doi.org/10.4049/jimmunol.1401081
Bhol K, Natarajan K, Nagarwalla N, Mohimen A, Aoki V, Ahmed AR (1995) Correlation of peptide specificity and IgG subclass with pathogenic and nonpathogenic autoantibodies in pemphigus vulgaris: a model for autoimmunity. Proc Natl Acad Sci U S A 92(11):5239–5243
Lo AS, Mao X, Mukherjee EM, Ellebrecht CT, Yu X, Posner MR, Payne AS, Cavacini LA (2016) Pathogenicity and epitope characteristics do not differ in IgG subclass-switched anti-desmoglein 3 IgG1 and IgG4 autoantibodies in pemphigus vulgaris. PLoS One 11(6):e0156800. https://doi.org/10.1371/journal.pone.0156800
Spaeth S, Riechers R, Borradori L, Zillikens D, Budinger L, Hertl M (2001) IgG, IgA and IgE autoantibodies against the ectodomain of desmoglein 3 in active pemphigus vulgaris. Br J Dermatol 144(6):1183–1188
Nagel A, Lang A, Engel D, Podstawa E, Hunzelmann N, de Pita O, Borradori L, Uter W, Hertl M (2010) Clinical activity of pemphigus vulgaris relates to IgE autoantibodies against desmoglein 3. Clin Immunol 134(3):320–330. https://doi.org/10.1016/j.clim.2009.11.006
Xu RC, Zhu HQ, Li WP, Zhao XQ, Yuan HJ, Zheng J, Pan M (2013) The imbalance of Th17 and regulatory T cells in pemphigus patients. Eur J Dermatol 23(6):795–802. https://doi.org/10.1684/ejd.2013.2177
Asothai R, Anand V, Das D, Antil PS, Khandpur S, Sharma VK, Sharma A (2015) Distinctive Treg associated CCR4-CCL22 expression profile with altered frequency of Th17/Treg cell in the immunopathogenesis of pemphigus vulgaris. Immunobiology 220(10):1129–1135. https://doi.org/10.1016/j.imbio.2015.06.008
Miller SD, Turley DM, Podojil JR (2007) Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol 7(9):665–677. https://doi.org/10.1038/nri2153
Pearson RM, Casey LM, Hughes KR, Miller SD, Shea LD (2017) In vivo reprogramming of immune cells: technologies for induction of antigen-specific tolerance. Adv Drug Deliv Rev. https://doi.org/10.1016/j.addr.2017.04.005
Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD (2012) Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9-23 epitope and involves functional epitope spreading. J Autoimmun 39(4):347–353. https://doi.org/10.1016/j.jaut.2012.04.005
Turley DM, Miller SD (2007) Peripheral tolerance induction using ethylenecarbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J Immunol 178(4):2212–2220
Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C, Schippling S, Miller SD, Sospedra M, Martin R (2013) Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med 5 (188):188ra175. doi:https://doi.org/10.1126/scitranslmed.3006168
Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD, Miller SD (2012) Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol 30(12):1217–1224. https://doi.org/10.1038/nbt.2434
Pearson RM, Casey LM, Hughes KR, Wang LZ, North MG, Getts DR, Miller SD, Shea LD (2017) Controlled delivery of single or multiple antigens in tolerogenic nanoparticles using peptide-polymer bioconjugates. Mol Ther 25(7):1655–1664. https://doi.org/10.1016/j.ymthe.2017.04.015
Serra P, Santamaria P (2015) Nanoparticle-based autoimmune disease therapy. Clin Immunol 160(1):3–13. https://doi.org/10.1016/j.clim.2015.02.003
Veldman C, Pahl A, Beissert S, Hansen W, Buer J, Dieckmann D, Schuler G, Hertl M (2006) Inhibition of the transcription factor Foxp3 converts desmoglein 3-specific type 1 regulatory T cells into Th2-like cells. J Immunol 176(5):3215–3222
Klatzmann D, Abbas AK (2015) The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 15(5):283–294. https://doi.org/10.1038/nri3823
Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, Sene D, Cacoub P, Klatzmann D (2011) Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med 365(22):2067–2077. https://doi.org/10.1056/NEJMoa1105143
Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP 3rd, Armand P, Cutler C, Ho VT, Treister NS, Bienfang DC, Prasad S, Tzachanis D, Joyce RM, Avigan DE, Antin JH, Ritz J, Soiffer RJ (2011) Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med 365(22):2055–2066. https://doi.org/10.1056/NEJMoa1108188
He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, Jin Y, Gan Y, Hu X, Jia R, Xu C, Hou Z, Leong YA, Zhu L, Feng J, An Y, Jia Y, Li C, Liu X, Ye H, Ren L, Li R, Yao H, Li Y, Chen S, Zhang X, Su Y, Guo J, Shen N, Morand EF, Yu D, Li Z (2016) Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med 22(9):991–993. https://doi.org/10.1038/nm.4148
Sawaf M, Dumortier H, Monneaux F (2016) Follicular helper T cells in systemic lupus erythematosus: why should they be considered as interesting therapeutic targets? J Immunol Res 2016:5767106. https://doi.org/10.1155/2016/5767106
Elwood F, Witter DJ, Piesvaux J, Kraybill B, Bays N, Alpert C, Goldenblatt P, Qu Y, Ivanovska I, Lee HH, Chiu CS, Tang H, Scott ME, Deshmukh SV, Zielstorff M, Byford A, Chakravarthy K, Dorosh L, Rivkin A, Klappenbach J, Pan BS, Kariv I, Dinsmore C, Slipetz D, Dandliker PJ (2017) Evaluation of JAK3 biology in autoimmune disease using a highly selective, irreversible JAK3 inhibitor. J Pharmacol Exp Ther 361(2):229–244. https://doi.org/10.1124/jpet.116.239723
Winthrop KL (2017) The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol 13(5):320. https://doi.org/10.1038/nrrheum.2017.51
Eming R, Rech J, Barth S, Kalden JR, Schuler G, Harrer T, Hertl M (2006) Prolonged clinical remission of patients with severe pemphigus upon rapid removal of desmoglein-reactive autoantibodies by immunoadsorption. Dermatology 212(2):177–187. https://doi.org/10.1159/000090659
Langenhan J, Dworschak J, Saschenbrecker S, Komorowski L, Schlumberger W, Stocker W, Westermann J, Recke A, Zillikens D, Schmidt E, Probst C (2014) Specific immunoadsorption of pathogenic autoantibodies in pemphigus requires the entire ectodomains of desmogleins. Exp Dermatol 23(4):253–259. https://doi.org/10.1111/exd.12355
Schmidt E, Spindler V, Eming R, Amagai M, Antonicelli F, Baines JF, Belheouane M, Bernard P, Borradori L, Caproni M, Di Zenzo G, Grando S, Harman K, Jonkman MF, Koga H, Ludwig RJ, Kowalczyk AP, Muller EJ, Nishie W, Pas H, Payne AS, Sadik CD, Seppanen A, Setterfield J, Shimizu H, Sinha AA, Sprecher E, Sticherling M, Ujiie H, Zillikens D, Hertl M, Waschke J (2017) Meeting report of the pathogenesis of pemphigus and pemphigoid meeting in Munich, September 2016. J Invest Dermatol 137(6):1199–1203. https://doi.org/10.1016/j.jid.2017.01.028
Galichet A, Borradori L, Muller EJ (2014) A new light on an old disease: adhesion signaling in pemphigus vulgaris. J Invest Dermatol 134(1):8–10. https://doi.org/10.1038/jid.2013.439
Walter E, Vielmuth F, Rotkopf L, Sardy M, Horvath ON, Goebeler M, Schmidt E, Eming R, Hertl M, Spindler V, Waschke J (2017) Different signaling patterns contribute to loss of keratinocyte cohesion dependent on autoantibody profile in pemphigus. Sci Rep 7(1):3579. https://doi.org/10.1038/s41598-017-03697-7
Egu DT, Walter E, Spindler V, Waschke J (2017) Inhibition of p38MAPK signaling prevents epidermal blistering and alterations of desmosome structure induced by pemphigus autoantibodies in human epidermis. Br J Dermatol. https://doi.org/10.1111/bjd.15721
Sweeney SE (2009) The as-yet unfulfilled promise of p38 MAPK inhibitors. Nat Rev Rheumatol 5(9):475–477. https://doi.org/10.1038/nrrheum.2009.171
Schultz HY, Diaz LA, Sirois DA, Werth VP, Grando SA (2011) Generating consensus research goals and treatment strategies for pemphigus and pemphigoid: the 2010 JC Bystryn pemphigus and pemphigoid meeting. J Invest Dermatol 131(7):1395–1399. https://doi.org/10.1038/jid.2011.120
Pacheco-Tovar D, Lopez-Luna A, Herrera-Esparza R, Avalos-Diaz E (2011) The caspase pathway as a possible therapeutic target in experimental pemphigus. Autoimmune Dis 2011:563091. https://doi.org/10.4061/2011/563091
Li N, Zhao M, Wang J, Liu Z, Diaz LA (2009) Involvement of the apoptotic mechanism in pemphigus foliaceus autoimmune injury of the skin. J Immunol 182(1):711–717
Schmidt-Hieber M, Dabrowski R, Aicher B, Lohneis P, Busse A, Tietze-Buerger C, Reufi B, Thiel E, Blau IW (2012) In vitro effects of perifosine, bortezomib and lenalidomide against hematopoietic progenitor cells from healthy donors. Investig New Drugs 30(4):1396–1403. https://doi.org/10.1007/s10637-011-9705-6
van der Helm LH, Bosman MC, Schuringa JJ, Vellenga E (2015) Effective targeting of primitive AML CD34+ cells by the second-generation proteasome inhibitor carfilzomib. Br J Haematol 171(4):652–655. https://doi.org/10.1111/bjh.13418
Kaur K, Kalra S, Kaushal S (2014) Systematic review of tofacitinib: a new drug for the management of rheumatoid arthritis. Clin Ther 36(7):1074–1086. https://doi.org/10.1016/j.clinthera.2014.06.018
Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, de Jong A, Harel S, DeStefano GM, Rothman L, Singh P, Petukhova L, Mackay-Wiggan J, Christiano AM, Clynes R (2014) Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med 20(9):1043–1049. https://doi.org/10.1038/nm.3645
Merrill JT, Burgos-Vargas R, Westhovens R, Chalmers A, D'Cruz D, Wallace DJ, Bae SC, Sigal L, Becker JC, Kelly S, Raghupathi K, Li T, Peng Y, Kinaszczuk M, Nash P (2010) The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 62(10):3077–3087. https://doi.org/10.1002/art.27601
de Paoli FV, Nielsen BD, Rasmussen F, Deleuran B, Sondergaard K (2014) Abatacept induces clinical improvement in patients with severe systemic sclerosis. Scand J Rheumatol 43(4):342–345. https://doi.org/10.3109/03009742.2013.812238
Emery P, Burmester GR, Bykerk VP, Combe BG, Furst DE, Barre E, Karyekar CS, Wong DA, Huizinga TW (2015) Evaluating drug-free remission with abatacept in early rheumatoid arthritis: results from the phase 3b, multicentre, randomised, active-controlled AVERT study of 24 months, with a 12-month, double-blind treatment period. Ann Rheum Dis 74(1):19–26. https://doi.org/10.1136/annrheumdis-2014-206106
Mease PJ, Gottlieb AB, van der Heijde D, FitzGerald O, Johnsen A, Nys M, Banerjee S, Gladman DD (2017) Efficacy and safety of abatacept, a T-cell modulator, in a randomised, double-blind, placebo-controlled, phase III study in psoriatic arthritis. Ann Rheum Dis 76(9):1550–1558. https://doi.org/10.1136/annrheumdis-2016-210724
Narbutt J, Lukamowicz J, Bogaczewicz J, Sysa-Jedrzejowska A, Torzecka JD, Lesiak A (2008) Serum concentration of interleukin-6 is increased both in active and remission stages of pemphigus vulgaris. Mediat Inflamm 2008:875394. https://doi.org/10.1155/2008/875394
Elhai M, Meunier M, Matucci-Cerinic M, Maurer B, Riemekasten G, Leturcq T, Pellerito R, Von Muhlen CA, Vacca A, Airo P, Bartoli F, Fiori G, Bokarewa M, Riccieri V, Becker M, Avouac J, Muller-Ladner U, Distler O, Allanore Y, Eustar (2013) Outcomes of patients with systemic sclerosis-associated polyarthritis and myopathy treated with tocilizumab or abatacept: a EUSTAR observational study. Ann Rheum Dis 72 (7):1217–1220. doi:https://doi.org/10.1136/annrheumdis-2012-202657
Khanna D, Denton CP, Merkel PA, Krieg T, Le Brun FO, Marr A, Papadakis K, Pope J, Matucci-Cerinic M, Furst DE, Investigators D, Investigators D (2016) Effect of Macitentan on the development of new ischemic digital ulcers in patients with systemic sclerosis: DUAL-1 and DUAL-2 randomized clinical trials. JAMA 315(18):1975–1988. https://doi.org/10.1001/jama.2016.5258
Acknowledgements
We are grateful to Günther Henkel and Irina Nuhn for the clinical photographs. This review was supported by a grant of the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation-FOR 2497) to TS, RE and MH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
When necessary, informed consent has been collected from patients.
Rights and permissions
About this article

Cite this article
Pollmann, R., Schmidt, T., Eming, R. et al. Pemphigus: a Comprehensive Review on Pathogenesis, Clinical Presentation and Novel Therapeutic Approaches. Clinic Rev Allerg Immunol 54, 1–25 (2018). https://doi.org/10.1007/s12016-017-8662-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-017-8662-z