
Abstract
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disorder with a broad spectrum of clinical presentations and association with multiple immunological abnormalities. Recent research of the Janus kinase (JAK)—signal transducer and activator of transcription (STAT) signaling pathway—revealed aberrant STAT signaling in inflammatory conditions and autoimmune diseases including SLE. STAT proteins are major components in interferon (IFN)-dependent gene expression and are responsible for signal transduction of over 50 cytokines, hormones, and growth factors regulating key cellular processes such as survival, proliferation, and differentiation. This review summarizes the present evidence from experimental animal models and patients with SLE for the involvement of STAT pathways in the pathogenesis of SLE underlining the role of different members of the STAT family. Genome-wide association studies provided evidence that variations in STAT4 gene are linked to the development of SLE in humans. First integration with genome-wide epigenomics data suggests that control of CD4+ T cell differentiation in which STATs play a major role may be an important component of the genetic contribution to disease susceptibility. Increased transcript and total protein STAT1 levels were described both in SLE T and B cells suggestive of the priming mechanisms that augment STAT1 signaling responses to IFN. STAT3 has a crucial role in Th17 differentiation, T follicular helper, and B cells, and STAT3 inhibition could represent a possible future therapeutic target in SLE. STAT5B appears to act as a critical modulator of human Treg development and function. While the imbalance between phosphorylated STAT5 and STAT3 in human SLE T cells was implicated in dysregulated IL-10 expression, Treg-specific deletion of STAT3 in mouse model even enhanced Th17-mediated inflammation. Finally, we present also a comprehensive analysis of studies investigating STAT signaling responses in conventional and regulatory subsets of SLE T and B cells and possible implications of STAT inhibition for clinical therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Most common diseases, including systemic lupus erythematosus (SLE), result from environmental factors interacting with genetic susceptibility variants. Genome-wide association studies (GWAS) have identified numerous loci that affect the susceptibility to common diseases and confirmed a genetic contribution to many autoimmune rheumatic diseases [1, 2]. GWAS have also provided insights into disease pathology and in certain diseases led to the identification of previously unrecognized pathogenic pathways [2]. Difficulty in assigning causality to a specific single nucleotide polymorphism (SNP) and identifying its effect on expression or function of a specific gene has limited their translational potential. GWAS have cataloged many SNPs associated with SLE, the significance of which has also proved elusive. Most genetic variations associated with SLE involve major histocompatibility complex antigens, type I interferon (IFN), and lymphocyte-signaling pathways [3, 4]. A common feature of GWAS is that the vast majority of signals (~90 %) lie outside traditional protein-coding gene sequences [5]. This suggested a role for regulatory variation as the 90 % of the human genome once dismissed as “junk” DNA was shown to have an important role in gene regulation. Recent studies found that most of the disease-associated variants occur in functional bits of DNA, known as enhancers, regulatory sequences that can bind with transcription factors to enhance the expression of remotely located genes [6–8]. Studies have further revealed that SNPs associated with SLE and other autoimmune diseases are enriched in B and T helper cell enhancer clusters or superenhancers (SEs) [9, 10]. Importantly, SEs were shown to be highly enriched for signal transducer and activator of transcription (STAT) family proteins, consistent with the findings that STAT proteins are essential for the establishment of the lineage-specific enhancer landscapes of differentiating cells [11, 12].
After outlining the principles of Janus kinase/STAT (JAK/STAT) signaling and the role of STATs in cell differentiation, this review summarizes the present evidence for the involvement of STAT pathways in the pathogenesis of SLE underlining the role of different members of the STAT family. We present also a comprehensive analysis of studies investigating STAT signaling responses in SLE CD4+ T and B cell subsets and possible implications of STAT inhibition for clinical therapy.
JAK-STAT Signaling Pathway
Research of the JAK/STAT signaling pathway revealed the complex mechanisms which allow cells to sense an array of the various extracellular signals and rapidly respond to them by controlling a target gene expression [13]. Successful signal transduction mediates cellular proliferation, differentiation, activation, maturation, cell migration, and survival of virtually all cell types and is therefore an important factor in human health and disease [14]. Aberrant signaling was linked to the immune disorders and neoplasias as well as to the inflammatory conditions and autoimmune diseases such as SLE [15–17].
STAT proteins were first identified in 1992 as DNA-binding proteins, major components in IFN-dependent gene expression [18]. Human STAT family currently consists of seven members (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, STAT6). Downstream of different transmembrane receptors, they aid signal transduction of over 50 cytokines, hormones, and growth factors [19, 20]. STATs are fairly large (750–850 amino-acid-long) proteins, consisting of different conserved functional domains, which together contribute to their distinct dual functional properties as transducers of signal and transcriptional factors. Two of those structures are not found in other families of transcriptional factors: an SRC homology 2 (SH2) domain and a highly conserved C-terminal tyrosine (Fig. 1), located around residue 700 in a transactivation domain (TAD), phosphorylation of which (P-Tyr) is crucial for STAT-mediated signaling in stimulated cells [19, 21, 22]. Upstream of the SH2 domain, in the center of the molecule, lays a DNA-binding domain (DBD), with β-sheets, folded similarly to other transcriptional factors, such as a nuclear factor kB (NF-kB) or p53. In an activated STAT dimer, DBD forms, together with the SH2 domain, a clamp-like structure, which encases the DNA and allows for some direct contact and binding to the DNA sequence [14, 16]. However, the DBD STATs can bind to two various DNA motifs: an IFN-γ-activated sequence (GAS, consensus motif: TTCN3GAA), used by STAT1, STAT3, STAT4, STAT5A, and STAT5B, or IFN-stimulated response elements (ISRE, consensus motif: AGTTTNCNTTTCC), used by STAT1 and STAT2. Exception is STAT6, which preferentially binds to a slightly different DNA motif: TTCN4GAA [16, 23–25]. In many genes, there are two STAT-binding sites, separated by approximately 20 base pairs, which facilitate formation of STAT tetramers (dimer-dimer pairs). They enhance a STAT-DNA interaction and are needed for a maximal transcriptional activity on some promoters [14, 26]. At the amino terminal of the protein lies a coiled-coil domain, which seems to participate in export of STATs from the nucleus [14].

Scheme of signal transducers and activators of transcription (STAT) domain structure. a An inactive STAT monomer with C-terminal tyrosine (Y) phosphorylation site. b Phosphorylation of a crucial tyrosine (Y) residue in the transactivation domain, which initiates STAT activation and dimerization. Two monomers connect through a reciprocal phosopho-Tyr-SH2 domain interaction. The transactivation domain of a certain STAT proteins contain a serine (S) residue, phosphorylation of which is thought to enhance transcriptional activity when phosphorylated
The canonical JAK/STAT pathway (Fig. 2) starts with a sequence of three consecutive tyrosine phosphorylations. Transmembrane ligand-activated cytokine receptors without kinase activity with noncovalently bound JAKs dimerize in response to binding of an extracellular ligand. This puts inactive JAKs in spatial proximity and enables their transphosphorylation and activation by distancing their inhibitory pseudokinase domains from kinase domains [16]. Next is a JAK-mediated phosphorylation of receptor tail, which then serves as docking site for two latent STAT monomers through their SH2 domain. Lastly, JAKs phosphorylate conserved C-terminal tyrosine residues of STAT proteins, which allow for their dimerization through reciprocal SH2 domains and translocation to the nucleus through a nuclear pore complex with the help of importins. In the nucleus, STATs dislodge importins and directly interact with a big and small grove of the DNA of response elements in the promoter of target genes which influences gene transcription [14, 20, 22, 25, 27]. Upon stimulation, STAT1, STAT3, STAT4, STAT5A, and STAT5B all form homodimers, but from the first studies of IFN-mediated signaling and identification of STAT1/STAT2 dimers (ISGF3 complex with IRF-9), it is known that heterodimers also exist [13, 16, 18, 24].

Janus kinase/signal transducers and activator of transcription (JAK/STAT) pathway. Binding of extracellular cytokine to the receptor initiates a signaling cascade. Receptor dimerization leads to cross-phosphorylation of JAKs on their tyrosine residues in transactivation domains. Activated JAKs phosphorylate receptor tails which enables docking of STATs through their SCR-homology domains (SH2) to the receptor. Phosphorylated STATs dimerize and move into the nucleus to activate transcription of cytokine-responsive genes. Negative regulators of STAT protein suppressors of cytokine signaling (SOCS) block STAT activation in the cytoplasm by binding to phosphorylated sites of receptor and/or JAK protein, thus turning off the initial signal at the source in cytoplasm. Protein tyrosine phosphatases (PTP) inhibit activation by direct dephosphorylation of activated proteins in cytoplasm and nucleus. Protein inhibitors of activated STAT (PIAS) associate with activated STAT dimers and prevent them from binding to DNA
Whole signal transduction is in normal cells completed when STATs become dephosphorylated by tyrosine phosphatases and are exported from the nucleus back to the cytoplasm. This process is facilitated by interactions between the coiled-coil and the DBD of STATs and exportins in less than an hour after the first stimulus, freeing them for next round of signaling [27]. There are four different types of JAKs (JAK1, JAK2, JAK3, TYK2) in human, but it seems that they do not have specificity for an exact STAT as various receptors can activate the same STAT protein even though they activate different JAKs. Primary specificity for the most STAT activation seems to be determined by distinct interactions between receptors and STATs, even though most receptors engage more than one STAT family member to a various degree. Signal transduction is also lineage-dependent, meaning that the same stimulus, which activates the same STATs, can modulate transcription of several genes in different cell types [16, 19].
Even though the canonical JAK/STAT signaling pathway is the most studied, at least STAT1, STAT3, and STAT5 can also be activated in a JAK-independent manner through other cytoplasmic tyrosine or serine kinases and through receptors with an intrinsic tyrosine kinase activity [14, 16, 19]. Several posttranslational modifications, other than tyrosine phosphorylation, can alter STAT function. One of the better known modifications is serine phosphorylation, which occurs at a second important phosphorylation site within the C-terminal of the molecule. With the serine phosphorylation, STATs participate in many other important pathways, such as ERK pathway, JNK pathway, p38MAPK pathway, mTOR pathway, protein kinase C delta pathway, and protein kinase R pathway [21, 28]. Other possible modifications of STATs include acetylation, methylation, and sumoylation. Acetylation and methylation can both promote and inhibit signal transduction, dependent on amino acid residue on which they occur: arginine methylation and lysine acetylation promote activation, and the role of lysine methylation is still ambiguous, whereas sumoylation is regarded as an exclusive negative regulator [16, 29]. Since 1997, it has been known that even unphosphorylated STATs can influence nuclear gene expression, probably through interactions with other transcriptional factors, such as NF-kB [14, 27, 30, 31]. Unphosphorylated STATs are also found to form distinct antiparallel dimers, located in the cytoplasm, which are thermodynamically as stable as phosphorylated STAT dimers but cannot translocate to the nucleus and could therefore be another form of latent STATs, waiting for activation [30, 32, 33].
Precise regulation of STAT activity, especially their inhibition and inactivation, ensuring that their activation is only transient, is critical for their biological function. Negative regulators, which inhibit the JAK/STAT pathway, can be divided into three major classes. Suppressor of cytokine signaling (SOCS) proteins block further receptor activation by binding to the phosphorylated receptor sites and/or JAK catalytic sites, thus turning off the initial signal at the source in cytoplasm [16, 25]. Phosphorylated STATs in the nucleus have relatively short half-life (≈15 min), which can be attributed to protein tyrosine phosphatases (PTPs) which regulate STATs by direct dephosphorylation in both cytoplasm and nucleus [17]. They can also bind to phosphorylated receptors, JAKs, or other upstream kinases and facilitate their dephosphorylation [22]. The third group inhibits transcriptional activity of the STATs separately from altering the phosphorylation status. Protein inhibitors of activated STAT (PIAS) associate with activated STAT dimers by their Zn-binding ring-finger domain in the center of the molecule, preventing them from binding to the DNA [34]. There is an emerging knowledge of other molecules like chaperons and noncoding RNAs (microRNA, long noncoding RNA) which seem to be important effectors of the JAK/STAT pathway [35–37]. Furthermore, it appears that STATs are both regulated by and act as epigenetic regulators, able to create an open chromatin at target loci, recruit acetyltransferases or methyltransferases, and drive widespread epigenetic changes [16].
The importance of STAT proteins and their role in organisms were first revealed by gene targeting in mice [13, 14, 18]. STAT1 and STAT2 knockout mice showed impaired responses to IFNs, providing better understanding of the role that IFNs play in innate as well as acquired immunity. Complete knockout of STAT3 mice resulted in embryonic lethality, illustrating the important role of STAT3 in the development of various lineages and tissues as well as regulation of cell proliferation, which has been later confirmed by tissue-specific STAT3 knockouts. STAT4 and STAT6 knockout mice exhibited impaired Th1/Th2 polarization owing to loss of IL-12 and IL-4 responsiveness, respectively. Knockout of STAT5A impaired prolactin-dependent mammary gland development and knockout of STAT5B failed to respond to growth hormone, therefore mice exhibited reduced growth [14, 38]. Even though knowledge, gained by mouse models, cannot be directly translated to humans, those experiments clearly showed the critical role of STATs in regulating intricate mechanisms of control over target gene expression by extracellular factors.
Dysfunction of STAT proteins has since been implicated in numerous human cancers and immune-related disorders [16, 22, 39, 40]. Moreover, results of studies of STAT expression and phosphorylation in T cells from healthy individuals revealed specific age-related changes. Significantly lower expression of total STAT1 protein in CD4+ T cells was found in adolescents than in children, which was associated with the increase in activated Tregs [41]. In another study, naïve CD4+ T cells from both older and young adults upregulated total STAT1 protein levels after T cell receptor (TCR) stimulation. However, TCR-activated naïve CD4+ T cells from older individuals displayed significantly lower STAT1 and STAT5 phosphorylation in response to IFN-α stimulation, while protein tyrosine kinase SHP-1, another STAT inhibitor, was not adequately excluded from the type I IFN receptor (IFNAR) signaling complex [42].
STATs in Control of Cell Differentiation
Cytokines, which are produced upon encounter of diverse microbial pathogens, instruct, together with TCR-induced signals, the generation of diverse types of effector CD4+ T cells. The balance with production of various types of regulatory T (Treg) cells is crucial for prevention of autoimmune pathology [43]. Cytokines activate members of the STAT family, which in turn control T helper (Th) cell differentiation [44–46] (Fig. 3). When IL-12 binds its receptor, STAT4 is activated. Phosphorylated STAT4 dimer translocates to the nucleus, where it binds to the DNA-binding sites in the regulatory elements of target genes and activates expression of the Th1 signature cytokine IFN-γ [44–47]. This in turn has an autocrine effect through IFN-γ-induced STAT1 phosphorylation, nuclear translocation, and transcription of Th1-specific genes, including T-box-containing protein, T-bet (encoded by Tbx21) [44–46, 48, 49]. STAT1 and STAT4, which also negatively regulates the genes favoring Th2 cell differentiation, are regarded as lineage-specific pioneer factors, as they initiate lineage specification in Th1 cells [44, 46]. Meanwhile, activation of STAT6 by IL-4 culminates in expression of the Th2 lineage-specific transcription factor GATA-binding protein-3 (GATA-3) [44–46, 50, 51]. STAT3 is activated by IL-6 leading to Th17 differentiation [52]. Binding site motifs for these STATs are present in the regulatory regions of the genes encoding the lineage-specific transcription factors: T-bet for Th1, GATA-3 for Th2, and retinoic acid-related orphan nuclear hormone receptor RORγτ for Th17 [44–46, 53]. In addition to IL-17-producing Th17 cells, relatively new subtypes of effector CD4+ T cells are IL-9-producing Th9 cells and IL-22-producing Th22 cells [54, 55]. Th9 cell differentiation in naïve CD4+ T cells is initiated by transforming growth factor (TGF)-β, which stimulates the expression of PU.1 and IL-4, which activates STAT6 and IRF4 expression [43, 46]. The molecular mechanisms and transcriptional factors involved in Th22 cell differentiation are poorly characterized. Ligand-activated transcription factor aryl hydrocarbon receptor (AhR) is considered to be a key factor in differentiation of Th22 cells, which is induced by IL6 and tumor necrosis factor (TNF) [43, 46].

A simplified scheme of CD4+ T cell differentiation, showing STAT and lineage-specific transcription factors, cytokines required for differentiation, and the signature cytokine for each subset. Naïve CD4+ T cells exit the thymus and, depending on the nature of antigen stimulation signal, differentiate into specialized T helper (Th) subsets under the influence of cytokines. Signaling downstream of cytokine receptors leads to activation of STAT transcription factors which prepare the chromatin landscape for the expression of lineage-specific transcription factors. In addition, naturally occurring Treg exits the thymus as a distinct lineage. Each subset is characterized by the production of key signature cytokine. Considerable plasticity was shown between these subsets, as illustrated by the dashed arrows. TF transcription factor; STAT signal transducer and activator of transcription; iTreg induced regulatory T cell; nTreg naturally occurring regulatory T cell; Thf T follicular helper cell
IL6 and IL21 induce differentiation of follicular T helper (Tfh) cells, which express C-X-C type chemokine receptor, CXCR5, and lineage-specific transcription factor BCL6 [56]. STAT3 depletion significantly reduced the CXCR5+ Tfh cells both in human and mouse [46, 57].
STAT5 activates transcription of FoxP3, which is expressed in natural Treg (nTreg) cells derived from thymus and adaptive or induced Treg (iTreg) cells [58, 59]. While iTreg can be induced from CD4+ T cells upon treatment with TGF- β and IL-2 is important in this process, STAT5 influences also the Treg survival by regulating expression of the IL-2 receptor α-chain (CD25) and the anti-apoptotic gene Bcl-2 [58–61].
Although Th cell lineages have distinct phenotypes, there is considerable heterogeneity and plasticity between subsets [45]. For example, Th17 cells can produce the Th1 cytokine IFN-γ, while Treg may acquire a phenotype, which is proinflammatory rather than suppressive [62–64]. This instability in Treg contributes to, rather than constrains, inflammation and has been implicated in the pathogenesis of autoimmunity [63, 64]. Cell identity is dependent on the existence of enhancer elements, and clusters of enhancers termed SEs are highly enriched at Th lineage-defining genes [65, 66]. A major breakthrough for enhancer biology was the discovery, facilitated by next-generation sequencing technology, that the presence of unique combinations of histone modifications-epigenetic changes maps the enhancer elements of each cell type [66, 67]. SEs are enriched for lineage specifying transcription factors and the transcriptional co-activators, including the histone acetyl transferase p300 [66]. While in Th1 and Th2 cells the majority of differentially active enhancers were STAT4- or STAT6-dependent, in Th17 cells STAT3 had a major role in p300 recruitment [12, 68]. In addition, the reconstitution of STAT4- and STAT6-deficient cells with linage-specifying T-bet and GATA3 failed to recover the active enhancer landscapes. This argues for a primary role of STATs as environmental sensors in dictating global enhancer landscape and Th cell identity [12].
Several disorders of primary immunodeficiency have been linked to known mutations of STATs [16, 40]. Although they have revealed the unique functions of STAT genes and related signaling pathways in T cells [40, 69], contributions of individual STATs to the development and effector function of primary human B cells remain largely unknown. It has been appreciated for many years that cytokines can activate specific JAK/STAT signaling pathways in B cells [70, 71]. Recently, it was shown that B cell-intrinsic STAT3 is necessary and sufficient, whereas STAT1 is dispensable, for their establishment of long-lived, protective, antigen-specific antibody responses [72]. Furthermore, the threshold of STAT3 activation required for differentiation into antibody-secreting plasma cells was found to be lower in memory compared with naïve B cells [73].
STAT1
STAT1 is a transcription factor predominately involved in the signal transduction by either type I, type II, or type III IFNs [16, 24, 40, 74]. While IFN-γ predominately signals through creating a STAT1/STAT1 homodimer, other types of IFNs activate a complex of transcription factors composed of STAT1, STAT2, and IRF9 [16, 24]. In addition to regulating Th1 cell-specific cytokine production, STAT1 signaling functions partly by controlling the proliferation and apoptosis of immune cells [74–76]. Patients with loss-of-function STAT1 mutations are prone to mycobacterial and viral infections. In contrast, patients with gain-of-function (GOF) STAT1 mutations, which lead to STAT1 hyperactivation and defective nuclear dephosphorylation, are prone to fungal infections [40, 74]. Such genetic lesion underlying chronic mucocutaneous candidiasis is associated with exaggerated IFN-γ signaling, which inhibits IL-17 transcription, resulting in susceptibility to fungal infections [77, 78]. These patients with increased IFN-γ signaling are also at risk for autoimmune disease [40, 79]. STAT1 GOF mutations have been reported in association with immune dysregulation, polyendocrinopathy, and enteropathy, X-linked (IPEX)-like syndrome with normal Treg frequency and function [79].
In MRL/lpr murine model of SLE, high endogenous levels of both total STAT1 and phosphorylated STAT1 proteins were demonstrated in lymphocytes [80, 81]. With the use of phospho-specific flow cytometry, a low increase in basal STAT1 phosphorylation in MRL/lpr murine model over the disease course was found which was larger in B cells than in T cells [82].
In the pristane-induced mouse model of SLE, characterized by IFNAR-dependent autoantibodies and glomerulonephritis [83], STAT1 and IRF9 were required for the production of IgG autoantibodies. In addition, upregulation of Toll-like receptor (TLR)7 by IFN-α was greatly reduced in STAT1–/– B cells, which were impaired in activation through both TLR7 and TLR9 [84].
A recent study in MRL/lpr model found that STAT1–/–, but not IRF9–/– or IFNAR2–/– mice, developed interstitial nephritis characterized by infiltration with RORγT+ lymphocytes. While fewer IFN-γ-producing CD4+ T cells were observed in spleen and lymph nodes obtained from STAT1–/– mice, IFNAR2–/– and IRF9–/– mice had significantly larger populations of IFN-γ-producing CD4+ T cells both in spleens and lymph nodes. In spite of evident interstitial renal disease and abnormal renal function, STAT1–/– mice had decreased proteinuria, glomerulonephritis, and autoantibody production. Shunting of STAT phosphorylation from STAT1 to STAT3 was also demonstrated [85].
Role of STAT1 was evaluated also in several clinical studies in patients with SLE. In a cohort study of 101 patients with SLE, it was observed that patients with anemia displayed significantly elevated transcript levels of STAT1 in peripheral blood mononuclear cells (PBMC) compared with nonanemic patients, which was not related to the disease activity or presence of lupus nephritis [86]. In a recent study, STAT1 messenger RNA (mRNA) levels in PBMC appeared as an expression enhancer of CCL2 and CXCL10 chemokines as indicated by the significantly stronger correlation of CCL2 and CXCL10 with IFN signature score in a subgroup of patients with SLE characterized by high STAT1 expression [87].
Over-expression of STAT1 mRNA was found also in T cells from SLE patients, and the increased transcript levels of STAT1 were significantly associated with the presence of lupus nephritis [88]. Increased levels of total STAT1 protein were also shown in PBMC from SLE patients, which correlated significantly with disease activity and with the expression of IFN-inducible genes [89]. The expression of total STAT1 protein was significantly increased also in B cells from SLE patients compared with healthy controls [90]. In contrast, no basal STAT1 activation was detected by phospho-specific flow cytometry in T and B cells from SLE patients [91].
Overall, significantly increased transcript and total protein STAT1 levels were described both in SLE T and B cells, in mice models, and clinical studies in patients with SLE, which is suggestive of the priming mechanisms that augment STAT1 signaling responses to IFNs [92]. As over-expression of a group of interferon-stimulated genes (ISGs) by PBMC from SLE patients, termed the IFN signature, is seen in SLE patients, STAT1 has been implicated in SLE pathogenesis by its role in the IFN signaling [93, 94]. Results from pristane-treated mice, which also exhibit a strong IFN signature [95], suggest that type I IFN-dependent STAT1 signaling is upstream of TLR signaling in the activation of autoreactive B cells. However, in MRL/lpr mouse model, crosstalk between type I and type II IFNs was found, as in the absence of type I IFN signaling proteins, a compensatory increase in IFN-γ signaling was observed, which may be mediated by STAT1 shunting. In addition, interactions between STAT1 and STAT3 and their influence on Th17 cells appeared to be crucial in determining the manifestations of lupus-like disease in MRL/lpr model. Association of STAT1 expression only with some disease manifestations was described in SLE patients. Of note, in previous studies, fluctuations in the IFN signature were shown not to mirror rapid clinical changes in SLE patients [96, 97]. In a chronic autoimmune disease, such as SLE, it is likely that type I and II IFN-induced STAT1 signaling changes over time and, depending on its differential effects on subsets of B and T cells, might even predominantly activate or suppress immune responses during relapses and remissions. The absence of basal STAT1 phosphorylation described in SLE patients suggests that STAT1 may not be constitutively activated in their blood T and B cells in general.
STAT2
STAT2 is predominantly involved in the signal transduction of type I IFNs (i.e., IFN-α and IFN-β). As these IFNs are mainly thought to engage in the response to viral infections, STAT2 signaling is important for the induction of antiviral effects [40, 74]. In addition to defective antiviral responses, STAT2-null mice and STAT2-null cell lines show also blunted apoptotic effects of IFN-α and IFN-β [40, 98].
It was shown that both IFN-α and IFN-γ induce the formation of the ISGF3 complex and bind to ISRE and GAS, respectively [99]. However, existence of STAT1-independent type IFN signaling pathway, where STAT2/IRF9 can potentially substitute for a role of ISGF3, was also shown [100]. Deficiency of STAT2 in humans profoundly affects type I IFN signaling but not type II IFN-γ signaling, explaining the associated clinical phenotype of susceptibility to viral infection described only in one family with STAT2 deficiency [40, 101].
Constitutive over-expression of STAT2 gene among other members of the IFN signaling pathway (STAT1 and IRF7) was described as part of the IFN signature in B6.NZMSle1/Sle2/Sle3 lupus-prone mice model compared with nonautoimmune mice [102]. In vitro, myeloid dendritic cells from lupus-prone mice constitutively over-expressed IFN-responsive genes such as IFN-β, Oas-3, Mx-1, ISG-15, and CXCL10. In vivo, myeloid dendritic cells and to a lesser extent T and B cells from young prediseased lupus-prone mice also expressed the IFN signature. Of note, the in vivo IFN signature was similar from young, preautoimmune mice and from mice with high titers of autoantibodies, suggesting that the IFN signature in myeloid dendritic cells preceded disease onset [102].
In a human study, PBMC from SLE patients but not healthy controls displayed constitutive phosphorylation of JAK1 and STAT2, even in the absence of disease activity [103].
Therefore, in addition to constitutive over-expression of STAT2 gene as part of IFN signature in mouse model, constitutive activation of STAT2 was found in patients with SLE. However, in contrast to STAT1, the role of STAT2 in SLE has been poorly investigated. Due to different involvement of STAT1 and STAT2 in the signal transduction of type I and type II IFNs, studies of both STATs can provide complete answers to the important question of relative involvement of different types of IFNs in SLE pathogenesis.
Although treatment of MRL/lpr mice with a monoclonal anti-IFNAR antibody ameliorates disease at early time points, at later stages of disease, type I IFN signaling-independent pathways overcome the therapeutic effects [104]. As mentioned above, increased IFN-γ production was demonstrated in the secondary lymphoid organs of IFNAR2–/– mice, which still have prominent autoantibody production and have marginally increased kidney pathology, suggesting that crosstalk between type I and type II IFNs is important in MRL/lpr mice [85].
In a recent human study, autoantibody positivity was shown to coincide or follow type II IFN dysregulation and elevated IFN-α activity occurred shortly before SLE classification [105].
STAT3
Although STAT3 mediates signaling through at least six classes of receptors and has crucial functions in various tissues, T cell-specific deletion of STAT3 mainly affects the expression of IL-17 and IL-21 and consequently results in decreased severity of several autoimmune disease models [67, 71, 106–109]. Th17 cells are the main producers of cytokines from IL-17 family, which mediate the recruitment of neutrophils, enhance B cell functions, and induce release of other proinflammatory cytokines, which altogether promote tissue damage in various autoimmune diseases, including SLE [43]. In addition to the crucial role of STAT3 in Th17 differentiation, a role for STAT3 in regulating T cell proliferation and survival was shown in STAT3–/– T cells, which have delayed proliferation and poor clonal expansion, particularly in the setting of inflammation [67, 107].
As discussed above, STAT3 is important also in the differentiation of Tfh, which provides help to B cells within the germinal center [46, 56, 57]. Elevated expression and/or production of IL-21 and circulating Tfh-like cells were shown in an extensive array of human autoimmune diseases, including SLE [110, 111].
In addition, STAT3 has critical functions in B cells because of its role in signaling by IL-6, IL-10, and IL-21, which was shown to stimulate antibody production through a STAT3-dependent pathway [40, 72].
Global gene expression analysis identified aberrant activation of STAT3 signaling in B cells among the molecular pathways that were dysregulated by the Sle1ab genomic interval on murine chromosome 1, which mediates the loss of immune tolerance to chromatin resulting in antinuclear antibody (ANA) production in the lupus-prone NZM2410 mouse [112]. In addition, in vitro treatment of splenic mononuclear cells isolated from ANA-positive Sle1ab mice with anti-IL-6 antibodies or JAK2 inhibitor AG490 suppressed ANA production in short-term culture, indicating that the IL-6 signaling pathway was essential to the production of autoantibodies [112].
While gene expression microarray data of bone marrow mononuclear cells from SLE patients showed increased expression of STAT3 gene, splenic B cells from 5-month-old NZB/NZW F1 lupus mice also showed activation of STAT3 [113].
On the other hand, no detectable basal activation of STAT3 over the disease course was found using phospho-specific flow cytometry in T cells or B cells in MRL/lpr murine model [82]. Basal activation-phosphorylation of STAT3 was however shown in T cells from SLE patients in a recent study by using phospho-specific flow cytometry [91].
In another study, T cells from patients with SLE were shown to display increased levels of total and phosphorylated STAT3 [114]. Consistent with a role of STAT3 signaling downstream of chemokine receptors, silencing of STAT3 expression using a small interfering RNA approach resulted in decreased chemokine-provoked SLE T cell migration [114].
STAT3 inhibition in lupus-prone MRL/lpr mice using the small-molecule inhibitor Stattic delayed the onset of autoantibody production and kidney disease. Inhibitor treatment reduced lymphadenopathy and the numbers of total T cells and Tfh, but not Th17 cells. Stattic-treated T cells exhibited decreased proliferation and a decrease in ability to migrate to chemokine CXCL12 in vitro [115].
The aforementioned studies demonstrated that inhibition of STAT3 signaling not only suppressed ANA production in vitro but also delayed autoantibody production in vivo and the onset of kidney damage in mouse models of SLE. Although the majority of effects seemed to be intrinsic to the T cells, future experiments using conditional knockout models could address the impact that this treatment may have on B cells. While interactions between STAT1 and STAT3 found in STAT1–/– MRL/lpr mouse were shown to influence number of Th17 cells, STAT3 inhibition had an impact on the number of Tfh cells, which may serve to amplify the effect of treatment by dampening the help provided by T cells to autoreactive B cells. In contrast to the absence of basal STAT1 activation, basal phosphorylation of STAT3 was shown in T cells from SLE patients and silencing of STAT3 expression decreased their chemokine-provoked migration. Therefore, although fewer studies have documented SLE-associated differences in STAT3 than STAT1 signaling, their results suggested that STAT3 inhibition could represent a therapeutic target in SLE.
STAT4
The postulated involvement of STAT4 in autoimmune diseases is predominantly linked to the essential role of STAT4 as a crucial mediator of IL-12 function that regulates the differentiation of Th1 cells and their inflammatory responses [43, 76]. The signature of IFN-γ, their main cytokine with potent proinflammatory impact, has long been correlated with having detrimental effects in several autoimmune diseases including SLE [116–118]. It is still contradictory whether Th1 cells only play a pathogenic role in autoimmune diseases or they also contribute to protective and anti-inflammatory immune responses. For example, in experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS), animals lacking IFN-γ developed more severe disease compared with IFN-γ-sufficient controls [43, 119, 120]. In contrast, mice deficient in the STAT4 gene were protected from developing EAE [121].
However, transgenic STAT4 knockout mice which showed relatively reduced levels of IFN-γ had more severe nephritis and worse mortality in the lupus-like New Zealand mixed 2328 and 2410 murine models in the absence of high levels of IgG anti-DNA [122, 123]. On the other hand, treatment with a STAT4-specific antisense oligonucleotide temporarily ameliorated disease in MRL/lpr mice with advanced nephritis [124].
Since Remmers et al. reported that variant allele of STAT4 was associated with increased risk of developing SLE and RA, the association between STAT4 SNP variants and SLE has been replicated in a number of GWAS in European and Asian populations [125–129]. Associations of different STAT4 risk alleles with severe renal insufficiency in lupus nephritis as well as with ishemic cerebrovascular events and anti-phospholipid antibodies were reported [130, 131]. As the genetic associations with STAT4 involve SNP variants located within the gene’s third intron, it was suggested that variants located in the large STAT4 intron could influence the gene’s transcription rate by disrupting a transcription factor binding site or a binding site for modified histone proteins [125, 132]. It was likely that there were more disease-associated SNPs in the variant haplotype, potentially even SNPs outside the third intron of STAT4, which could have functional consequences. Multiple haplotypic associations across STAT4 with SLE were identified in different racial groups [133]. Lamana et al. reported correlated association between minor allele of rs7574865 and higher STAT4 mRNA and protein expression [134].
The part of the immune system in which the dysfunction actually occurs, however, was not readily apparent. T cells and particularly CD4+ T cells seemed to be the most likely culprits by the role of STAT4 in promoting these cells to differentiate into inflammatory subsets. However, STAT4 can be activated by type I IFNs and variant STAT4 in other cell subsets could also contribute to disease risk [135]. The SNP rs7574865 in the third intron was indeed associated with increased sensitivity to IFN-α signaling, as evident from its simultaneous association with both lower serum IFN-α activity and greater IFN-α-induced gene expression in PBMCs [136]. The same risk variant was also associated with a particular SLE phenotype that has younger age of onset (<30 years) and is characterized by a higher frequency of nephritis and anti-double-stranded DNA autoantibodies [133].
As discussed above, an important aspect of the pathogenesis of SLE is the IFN signature, and polymorphisms in TYK2, whose gene product is activated by type I IFNs, IL-12, and other cytokines, have also been reported to be associated with SLE [137, 138].
In addition, in multiple studies, a variant allele of IRF5, whose gene product is involved in induction of IFN-α, has been found to be associated with increased risk of developing SLE [137–140].
Although one study demonstrated that the STAT4 SNP, rs7582694, which correlates with the production of anti-double-stranded DNA antibodies, has a multiplicative risk effect of 1.82 with two independent risk alleles of IRF5, another study using 30 tagged SNPs revealed no significant interaction effects between SNPs in STAT4 and IRF5 [139, 140].
Interestingly, data from the genetic and epigenetic fine mapping of causal autoimmune disease variants show for candidate causal SLE STAT4 SNPs top enhancer in stimulated CD4+ T cells and for candidate causal IRF5 SNPs top enhancer in CD14+ monocytes [6]. On the other hand, data from Hnisz et al. show IRF5 on the list of genes associated with SLE SNP-containing superenhancers in B cells [9].
Although contrasting roles of STAT4 in lupus-like autoimmunity were shown in different mouse models, large-scale GWAS studies have provided evidence that variations in STAT4 gene are linked to the development of SLE in humans. While STAT4 knockout mice, which showed relatively reduced levels of IFN-γ had even more severe nephritis in one mouse model, inhibition of STAT4 in another model at least temporarily ameliorated disease. Recent mapping of candidate causal STAT4 SNPs to enhancers of stimulated CD4+ T cells suggests that future studies of STAT4 signaling should focus on this subset.
STAT5
STAT5 has two isoforms, STAT5A and STAT5B, which are encoded by distinct genes and have 96 % similarity in sequence and overlapping functions in diverse tissues [40, 67, 141]. Combined deficiency of both genes in mice profoundly affects all hematopoietic cells, and the few peripheral T cells that develop in mice deficient in both STAT5A and STAT5B show defects in IL-2 receptor-α expression and have an activated phenotype, leading to the development of autoimmunity [40, 67, 142, 143].
STAT5A sufficiency cannot adequately compensate for STAT5B defects, and human STAT5B deficiency, a rare autosomal recessive primary immunodeficiency, is characterized also by autoimmunity associated with reduction of Treg [40, 144]. Treg subset of CD4+ T cells was shown to suppress effector T cell function and attenuate immune responses also against self-antigens [145]. Whether there is differential regulation between STAT5A and STAT5B of human iTreg in the periphery versus nTreg development in the thymus has not been determined yet. However, differentially regulated expression of FoxP3 and IL-2R was found in STAT5B knockdown, but not STAT5A knockdown T cells [146]. In homozygous STAT5B-deficient patients, reduced FoxP3 expression was associated also with impaired regulatory function of STAT5B-null Treg cells [146]. In addition, in homozygous STAT5B-deficient patients, FoxP3+ cells predominately displayed CD45RO+ memory phenotype with expression of proliferation marker Ki-67 [146].
In the absence of exogenous stimulation, no detectable activation of STAT5 over the disease course was found in CD4+ T cells or B cells in MRL/lpr murine model [82].
In contrast, in B and T cells from SLE patients, phosphorylated STAT5 levels were increased above healthy controls and patients with rheumatoid arthritis [91]. In addition, based on basal phosphorylation of STAT5 and STAT3, SLE samples clustered into two main groups, which were associated with the SLE Disease Activity Index 2000 [91].
In both studies utilizing phospho-specific flow cytometry, monoclonal antibody recognizing the phosphorylated tyrosine (Y694) of STAT5A was used.
As recent studies have shown that STAT5B, not STAT5A, is a critical modulator of human Treg development and function, it seems prudent to further explore the role of STAT5B signaling also in the future studies in patients with SLE.
It was shown that STAT3 and STAT5 bind to multiple common sites across the locus encoding IL-17. In addition, STAT3-driven IL-17 transcription was blocked by STAT5 in T cells [147]. As increased phosphorylation of both STAT3 and STAT5 was described in T cells from SLE patients, further studies are needed to determine whether they engage in this type of head-to-head competition also in SLE T cells.
Of note, in a recent study, authors dissected molecular events contributing to increased IL-10 expression in SLE T cells and found enhanced recruitment of STAT3 to the IL10 promoter and the intronic enhancer element (I-SRE), which mediated the recruitment of histone acetyltransferase p300 to those regions, instructing epigenetic remodeling [148]. In SLE T cells, chromatin remodeling was enhanced as a result of increased STAT3 phosphorylation, resulting in a replacement of STAT5 at the I-SRE, suggesting that targeting the imbalance between phosphorylated STAT3 and STAT5 in SLE T cells could correct dysregulated cytokine expression [148].
STAT6
Due to the actions of STAT6 and its downstream targets in pathological Th2 cell responses such as asthma and allergy, both mouse and human systems have been studied in terms of STAT6 functions [67, 149–151]. The importance of STAT6 for Th2 cell differentiation has been well established in STAT6–/– mice, where the expression of Th2 cytokines including IL-4, IL-5, and IL-13 was diminished [152–154]. In human cells, STAT6 mediates the expression of more than 80 % of IL-4-regulated genes, a higher proportion than was reported in previous studies using mouse cells [151]. Based on the ability of Th2 cells to suppress cell-mediated or Th1 models of disease, Th2 cells were initially described as anti-inflammatory, but a number of studies established their role in the pathogenesis of antibody-mediated autoimmune diseases [43]. Their cytokines were implicated also in the immunopathology of SLE [155–158]. Additionally, interplay of STAT6 with PU.1 and IRF4 is critical in the generation of the recently described Th9 subset of CD4+ T cells, which was also shown to be involved in pathology of SLE [43, 54, 159, 160].
When phospho-specific flow cytometry was used in MRL/lpr murine model, a low but detectable increase in basal STAT6 phosphorylation was reported over the disease course that was larger in B cells than in T cells [82].
In Lyn–/– mice which develop lupus-like autoimmune disease and have coexistent intrinsic alergic traits, STAT6 deficiency ameliorated atopy and spontaneous peritoneal eosinophilia; however, autoimmune disease was markedly exacerbated [161].
In contrast, in the lupus-like New Zealand mixed 2328 and 2410 murine models, STAT6 deficiency ameliorates kidney disease, particularly glomerulosclerosis, despite the presence of high levels of IgG anti-double-stranded DNA (dsDNA) antibodies [122, 123].
It was suggested that IL-4 might directly promote extracellular matrix deposition in the glomeruli, as also the blockade of IL-4 by antibody treatment ameliorated glomerulosclerosis and delayed or even prevented the development of end-stage renal disease, despite the presence of high levels of IgG anti-dsDNA antibodies [122].
As for STAT4, contrasting roles in SLE like autoimmunity in different mice models were shown also for STAT6. Increased levels of basal STAT6 phosphorylation in B and T cells from MRL/lpr mouse suggest constitutive activation of STAT6 signaling. Unfortunately, STAT6 signaling was not yet studied in B and T cell subsets from SLE patients, although one study reported association between STAT6 gene polymorphisms and SLE in Chinese patients [162].
SLE B and CD4+ T Cell Subset Specific Differences in STAT Signaling Responses
In addition to basal STAT activation, recent studies analyzed also STAT signaling responses to stimulation with cytokines in various subsets of immune cells. The group from Stanford which developed the method of phospho-specific flow cytometry [163] used it also to analyze cytokine-induced STAT phosphorylation during the progression of SLE [82]. STAT signaling responses to stimulation with 10 cytokines were measured in five immune cell types from the MRL/lpr mice. Among many SLE-associated STAT signaling changes, they reported increased CD4+ T cell STAT3 responses to IL-10 and STAT5 to IL-15 stimulation. In addition to decreased STAT1 response to IL-6, SLE CD4+ T cells displayed decreased STAT6 response to IL-4, which remained comparatively strong in B cells. Decreased STAT1 responses to IFN-α, IFN-γ, and IL-21 were found in both CD4+ T cells and B cells, which displayed also decreased STAT3 response to IL-10. Along with decrease in STAT1 responses to cytokines with age of MRL/lpr mice, they also found increased intracellular expression of SOCS-1 protein, suggesting that IFN signaling during the progression of lupus-like disease in mice induces negative feedback regulation of STAT1. Later study utilizing phospho-specific cytometry also found significantly decreased STAT1, STAT3, and STAT5 responses to IFN-α in T cells and B cells from SLE patients, compared with healthy controls [91]. However, longitudinal study would be required to understand if the IFN-driven feedback regulation of STATs, described in mouse model, has a role in transiting between flare and remission in patients with SLE.
In addition to decreased IFN-α-induced responses, STAT3 response to IL6 was also significantly decreased in T cells from SLE patients [91].
In another study, significantly decreased STAT3 responses to IL-21 and significantly increased expression of SOCS1 were found in CD4+ T cells from SLE patients compared with healthy controls [164]. In addition, STAT3 responses to IL-21 were also decreased in B cells from SLE patients. However, percentage of memory CD4+CD45RO+ T cells producing IL-21 was significantly increased in SLE patients. Downregulation of IL-21 signaling, mediating key pathological signals both in T and B cells, was interpreted as an attenuation of previously increased levels of STAT3 phosphorylation due to increased and tonic IL-21 stimulation [164]. Therefore, by using phospho-specific flow cytometry, specific differences in STAT3 signaling were found in the CD4+ subset of T cells from SLE patients, which may be important also in the autocrine loop for IL-21 production via STAT3 phosphorylation [165, 166]. STAT3 is involved in differentiation of Tfh cells, defined by their IL-21 production, which was shown to be increased in CD4+ T cells from SLE patients in previous studies [56, 57, 167]. Together with Tfh responses, B cell-intrinsic STAT3 signaling, which was shown to be critical for their differentiation into antibody-producing plasma cells, may be important in the context of autoimmunity [72, 73, 168–170].
Complex roles of STAT3 were shown also in Treg cells [67, 171, 172]. IL-6 inhibits FoxP3 expression, an effect that depends on STAT3 [171]. However, upon Treg-specific ablation of STAT3, which is critical for Th17 differentiation, the ability of Treg cells to constrain a pathogenic Th17 cell response was selectively impaired, whereas suppression of Th1 or Th2 cell responses remained intact [172].
Function of the newly defined STAT3-dependent Th17-specific regulatory T cells (Treg17) was studied also in the context of SLE with Treg-specific deletion of STAT3 in mice in which SLE was induced by intraperitoneal injection of pristane [173]. Lack of Treg17 cells in these mice caused selectively enhanced peritoneal Th17 inflammation and also resulted in aggravated pulmonary vasculitis with increased percentages of Th17 cells and significantly higher mortality. Overshooting Th17 responses in the absence of Treg17 cells after pristane injection were also associated with the aggravation of lupus nephritis [173]. Results of both studies underline the importance of studying intrinsic activation of STAT3 in Treg as it endows these cells with the ability to specifically suppress Th17 cell responses.
Significant differences in numerous TCR-induced as well as STAT signaling responses were described recently between Treg and conventional FoxP3-CD4+ T cells in mouse [174]. In addition, T cell signaling network analysis revealed distinct differences in TCR-induced signaling between resting-naïve and activated effector Treg subsets [175, 176] from healthy donors [177].
Although T cell signaling abnormalities in SLE, particularly downstream of TCR, have been thoroughly studied [178], relatively few studies have documented SLE-associated differences in T cell STAT signaling. In addition, analyzes performed at the level of total T cells may fail to capture SLE-associated alterations specific to a given T cell subset as shown by aforementioned studies utilizing phospho-specific flow cytometry.
Recent studies reported significantly increased percentage of FoxP3+Helios+ Treg cells in SLE patients compared with healthy and disease controls, which was positively correlated with disease activity [179, 180].
Basal STAT5 signaling was investigated in CD4+ T cell subsets from SLE patients, and increased basal STAT5A phosphorylation levels were found in conventional FoxP3- CD4+ T cells as well as in Foxp3+Helios+ Treg, which predominately displayed memory phenotype with increased Ki-67 expression. It was therefore suggested that such Treg might be expanded in active SLE through γ-chain signaling cytokines [180].
Differential STAT5 responsiveness to IL-7 and IL-2 common-γ chain family cytokines was shown by Tregs and FoxP3-conventional CD4+ T cells from healthy donors [181].
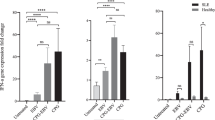
IL-2-induced signaling was shown to be crucial also for Treg function [61, 182]. Increased frequency of FoxP3+ Treg lacking expression of the IL-2 receptor α-chain (CD25) was found in blood samples from patients with SLE [183–185]. To quantify the impact of CD25 expression on nuclear translocation of phosphorylated STAT5 (pSTAT5) in FoxP3+ Tregs from SLE patients, we stimulated whole blood samples with recombinant human IL-2. Imaging flow cytometry was used to compare pSTAT5 nuclear translocation between CD25+ and CD25− subset of SLE FoxP3+ Tregs after IL-2 stimulation (Fig. 4). Despite lower levels of STAT5 phosphorylation in CD25− subset, pSTAT5 was predominately localized to nucleus after IL-2 stimulation also in this subset of SLE FoxP3+ cells.

Nuclear localization of phosphorylated STAT5 (pSTAT5) in CD25+ and CD25− subset of FOXP3+CD4+ T cells from SLE patient after IL-2 stimulation. EDTA-anticoagulated whole blood sample from patient with SLE was stimulated with 100 ng/mL recombinant human IL-2 (PeproTech, Rocky Hill, NJ, USA) for 15 min at 37 °C. BD Phosflow technology was used according to the manufacturer’s instructions (BD Biosciences, San Jose, CA, USA) to stain cells with anti-CD4, anti-CD25, anti-FOXP3, antibody recognizing specific phosphorylated STAT5A tyrosine-pSTAT5 (Y694), and 20 ng/mL 7-AAD (All BD Biosciences). Image files of cells were collected for each sample using the ImageStream X imaging flow cytometer (Amnis, Seattle, WA, USA) and analyzed using IDEAS software (Amnis) for colocalization of pSTAT5 within the 7-AAD-stained nucleus. Representative histograms of 7-AAD/pSTAT5 similarity scores, correlating 7-AAD nuclear stain with the pSTAT5 signal in CD25+FOXP3+ (blue) and CD25-FOXP3+ (yellow) subset of CD4+ T cells gated on FOXP3 versus CD25 dot plot shown below. The higher the similarity score, the more nuclear localization is visualized in the example images of cells shown right: pSTAT5 protein (green) is localized to the nucleus stained with 7-AAD (red) in both subsets of FOXP3+ (blue) cells: CD25+ (yellow) and CD25− shown below
Recently, also human CD19+CD24hiCD38hi B cells were shown to possess regulatory capacity, as they suppressed Th1 and Th17 cell differentiation and converted conventional CD4+CD25− T cells into Treg cells via the release of IL-10 [186, 187]. Regulatory B (Breg) cells from SLE patients were shown to lack the suppressive capacity of their healthy counterparts [187]. Phospho-specific flow cytometry was used to analyze STAT3 signaling response to CD40 ligation in the limited numbers of CD24hiCD38hi B cells obtainable within peripheral blood samples. In vitro stimulation of PBMC from healthy donors with agonistic CD40 monoclonal antibodies led to significantly higher increase in STAT3 phosphorylation in CD24hiCD38hi B cells compared with other B cell subsets defined by CD24 and CD38 expression [187]. No significant increases in the phosphorylation of STAT3 in any B cell subset from SLE patients were detected. Furthermore, authors observed consistently lower basal STAT3 phosphorylation in CD24hiCD38hi B cells from SLE patients compared with healthy controls. Therefore, in contrast to results of the aforementioned study, which analyzed total B cells from SLE patients and found increased basal STAT3 phosphorylation, the subset of SLE B cells with regulatory potential displayed even lower basal STAT3 activation compared with healthy controls [91, 187]. In addition, by using phospho-specific flow cytometry, authors clearly demonstrated that STAT3 is phosphorylated in CD24hiCD38hi B cells from healthy donors in response to CD40 ligation. Results of their study also suggested that STAT3 signaling response is impaired in CD24hiCD38hi B cells from SLE patients, which also produced less IL-10 [187].
Signals controlling the generation of Breg cells were not defined until recently. Menoni et al. described a regulatory feedback mechanism in which plasmacytoid dendritic cells (pDCs) drove the differentiation of CD19+CD24hiCD38hi B cells into IL-10-producing CD24+CD38hi Breg cells via the release of IFN-α and CD40 engagement, and Breg cells conversely restrained IFN-α production by pDCs via IL-10 release [90].
pDCs from SLE patients promoted plasmablast differentiation but failed to induce Breg cells. In addition, control of pDC IFN-α production by CD24+CD38hi B cells was impaired and frequency of single IL-10-producing B cells was reduced in SLE patients. SLE CD24+CD38hi B cells, but not other B cell subsets, displayed altered responses to IFN-α stimulation. STAT3, but not STAT1 phosphorylation after IFN-α stimulation, was significantly decreased in SLE CD24+CD38hi B cells compared with healthy CD24+CD38hi B cells [90]. Both studies show that STAT signaling analyses performed at the level of total B cells would fail to capture SLE-associated alterations specific to the Breg cell subset.
Defining differences in STAT signaling between conventional and regulatory subsets of SLE B and T cells can help to unravel how present immunosuppressive medication interacts with intracellular signaling networks and will be important in predicting the specificity of potential STAT signaling inhibitors in the future.
Conclusion
The complexities of JAK-STAT signaling as well as CD4+ T and B cell differentiation are still emerging. However, first integration of genome-wide epigenomics data with data from GWASs on SLE suggests that control of CD4+ T cell cytokine identity in which STATs play a major role may be an important component of the genetic contribution to SLE disease susceptibility. While interactions between STAT1 and STAT3 found in mouse models were shown to influence number of Th17 cells, STAT3 inhibition had an impact on the number of Tfh. Unlike JAKs, whose kinase domains are a pharmacological target of JAKinibs, STATs do not have catalytic activity. Inhibition of STATs, tested in mouse models of SLE, was achieved by interfering with STAT4 binding to DNA and by using the small molecule, which prevents binding tyrosine-phosphorylated peptide motifs to STAT3 SH2 domains. While treatment with a STAT4-specific antisense oligonucleotide given to MRL/lpr mice with advanced nephritis only temporarily ameliorated disease, inhibition of STAT3 only delayed lupus nephritis in MRL/lpr mice. This raises the possibility of either incomplete STAT inhibition or activation of other inflammatory pathways in the absence of STAT4- and STAT3-mediated activation. Indeed, STAT4 knockout mice with decreased Th1 response showed even more severe nephritis and Treg-specific deletion of STAT3 resulted in enhanced Th17 inflammation and aggravation of lupus nephritis. Therefore, consequence of differential JAK/STAT signaling in different cell subsets makes it difficult to draw straightforward conclusions from applying inhibitors of single molecules. In addition, the translation of findings from murine models into clarification of the human situation is also difficult as most human data concern STAT4 genetic associations and STAT1 expression studies. As shown by results of recent studies, characterization of STAT signaling responses in relevant T and B cell subsets is likely to reveal new aspects of the regulation of immune response and immunopathology of SLE.
References
Altshuler D, Daly MJ, Lander ES (2008) Genetic mapping in human disease. Science 322:881–888
Parkes M, Cortes A, van Heel DA, Brown MA (2013) Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet 14:661–673
Ghodke-Puranik Y, Niewold TB (2015) Immunogenetics of systemic lupus erythematosus: a comprehensive review. J Autoimmun 64:125–136
Rullo OJ, Tsao BP (2013) Recent insights into the genetic basis of systemic lupus erythematosus. Ann Rheum Dis 72(Suppl 2):ii56–ii61
Tak YG, Farnham PJ (2015) Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin 8:57
Farh KK, Marson A, Zhu J et al (2015) Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518:337–343
Maurano MT, Humbert R, Rynes E et al (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337:1190–1195
Miguel-Escalada I, Pasquali L, Ferrer J (2015) Transcriptional enhancers: functional insights and role in human disease. Curr Opin Genet Dev 33:71–76
Hnisz D, Abraham BJ, Lee TI et al (2013) Super-enhancers in the control of cell identity and disease. Cell 155:934–947
Parker SC, Stitzel ML, Taylor DL et al (2013) Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A 110:17921–17926
Vahedi G, Kanno Y, Furumoto Y et al (2015) Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 520:558–562
Vahedi G, Takahashi H, Nakayamada S et al (2012) STATs shape the active enhancer landscape of T cell populations. Cell 151:981–993
Stark GR, Darnell JE Jr (2012) The JAK-STAT pathway at twenty. Immunity 36:503–514
Levy DE, Darnell JE Jr (2002) Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3:651–662
Li HS, Watowich SS (2014) Innate immune regulation by STAT-mediated transcriptional mechanisms. Immunol Rev 261:84–101
Villarino AV, Kanno Y, Ferdinand JR, O'Shea JJ (2015) Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol 194:21–27
Bohmer FD, Friedrich K (2014) Protein tyrosine phosphatases as wardens of STAT signaling. Jakstat 3, e28087
Darnell JE Jr, Kerr IM, Stark GR (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421
Darnell JE Jr (1997) STATs and gene regulation. Science 277:1630–1635
Liongue C, Ward AC (2013) Evolution of the JAK-STAT pathway. Jakstat 2, e22756
Decker T, Kovarik P (2000) Serine phosphorylation of STATs. Oncogene 19:2628–2637
Dorritie KA, Redner RL, Johnson DE (2014) STAT transcription factors in normal and cancer stem cells. Adv Biol Regul 56:30–44
Ehret GB, Reichenbach P, Schindler U et al (2001) DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J Biol Chem 276:6675–6688
Au-Yeung N, Mandhana R, Horvath CM (2013) Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. Jakstat 2, e23931
Abroun S, Saki N, Ahmadvand M, Asghari F, Salari F, Rahim F (2015) STATs: an old story, yet mesmerizing. Cell J 17:395–411
Vinkemeier U, Cohen SL, Moarefi I, Chait BT, Kuriyan J, Darnell JE Jr (1996) DNA binding of in vitro activated Stat1 alpha, Stat1 beta and truncated Stat1: interaction between NH2-terminal domains stabilizes binding of two dimers to tandem DNA sites. EMBO J 15:5616–5626
Reich NC (2013) STATs get their move on. Jakstat 2, e27080
Saleiro D, Platanias LC (2015) Intersection of mTOR and STAT signaling in immunity. Trends Immunol 36:21–29
Woo CH, Shishido T, McClain C et al (2008) Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res 102:538–545
Yang J, Stark GR (2008) Roles of unphosphorylated STATs in signaling. Cell Res 18:443–451
O'Shea JJ, Notarangelo LD, Johnston JA, Candotti F (1997) Advances in the understanding of cytokine signal transduction: the role of Jaks and STATs in immunoregulation and the pathogenesis of immunodeficiency. J Clin Immunol 17:431–447
Zhong M, Henriksen MA, Takeuchi K et al (2005) Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc Natl Acad Sci U S A 102:3966–3971
Wenta N, Strauss H, Meyer S, Vinkemeier U (2008) Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc Natl Acad Sci U S A 105:9238–9243
Porritt RA, Hertzog PJ (2015) Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol 36:150–160
Lui PY, Jin DY, Stevenson NJ (2015) MicroRNA: master controllers of intracellular signaling pathways. Cell Mol Life Sci 72:3531–3542
Witte S, Muljo SA (2014) Integrating non-coding RNAs in JAK-STAT regulatory networks. Jakstat 3, e28055
Bocchini CE, Kasembeli MM, Roh SH, Tweardy DJ (2014) Contribution of chaperones to STAT pathway signaling. Jakstat 3, e970459
Schindler CW (2002) Series introduction. JAK-STAT signaling in human disease. J Clin Invest 109:1133–1137
Ward AC, Touw I, Yoshimura A (2000) The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood 95:19–29
O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A (2015) The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med 66:311–328
Holcar M, Goropevsek A, Ihan A, Avcin T (2015) Age-related differences in percentages of regulatory and effector T lymphocytes and their subsets in healthy individuals and characteristic STAT1/STAT5 signalling response in helper T lymphocytes. J Immunol Res 2015:352934
Li G, Ju J, Weyand CM, Goronzy JJ (2015) Age-associated failure to adjust type I IFN receptor signaling thresholds after T cell activation. J Immunol 195:865–874
Raphael I, Nalawade S, Eagar TN, Forsthuber TG (2015) T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 74:5–17
Zhu J, Paul WE (2010) Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev 238:247–262
Bonelli M, Shih HY, Hirahara K et al (2014) Helper T cell plasticity: impact of extrinsic and intrinsic signals on transcriptomes and epigenomes. Curr Top Microbiol Immunol 381:279–326
Tripathi SK, Lahesmaa R (2014) Transcriptional and epigenetic regulation of T-helper lineage specification. Immunol Rev 261:62–83
Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM (1993) Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science 260:547–549
Girdlestone J, Wing M (1996) Autocrine activation by interferon-gamma of STAT factors following T cell activation. Eur J Immunol 26:704–709
Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH (2000) A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100:655–669
Sokol CL, Barton GM, Farr AG, Medzhitov R (2008) A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol 9:310–318
Zheng W, Flavell RA (1997) The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89:587–596
Park H, Li Z, Yang XO et al (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6:1133–1141
Ivanov II, McKenzie BS, Zhou L et al (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133
Veldhoen M, Uyttenhove C, van Snick J et al (2008) Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol 9:1341–1346
Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H (2009) Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol 10:864–871
Ma CS, Deenick EK (2014) Human T follicular helper (Tfh) cells and disease. Immunol Cell Biol 92:64–71
Ma CS, Avery DT, Chan A et al (2012) Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood 119:3997–4008
Toda A, Piccirillo CA (2006) Development and function of naturally occurring CD4+CD25+ regulatory T cells. J Leukoc Biol 80:458–470
Chen W, Jin W, Hardegen N et al (2003) Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 198:1875–1886
Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA (2007) IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol 178:2018–2027
Mahmud SA, Manlove LS, Farrar MA (2013) Interleukin-2 and STAT5 in regulatory T cell development and function. Jakstat 2, e23154
Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E (2013) Cutting edge: the pathogenicity of IFN-gamma-producing Th17 cells is independent of T-bet. J Immunol 190:4478–4482
Lochner M, Peduto L, Cherrier M et al (2008) In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J Exp Med 205:1381–1393
Zhou X, Bailey-Bucktrout SL, Jeker LT et al (2009) Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol 10:1000–1007
Nguyen ML, Jones SA, Prier JE, Russ BE (2015) Transcriptional enhancers in the regulation of T cell differentiation. Front Immunol 6:462
Witte S, O'Shea JJ, Vahedi G (2015) Super-enhancers: asset management in immune cell genomes. Trends Immunol 36:519–526
O'Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y (2011) Genomic views of STAT function in CD4+ T helper cell differentiation. Nat Rev Immunol 11:239–250
Ciofani M, Madar A, Galan C et al (2012) A validated regulatory network for Th17 cell specification. Cell 151:289–303
O'Shea JJ, Murray PJ (2008) Cytokine signaling modules in inflammatory responses. Immunity 28:477–487
Leonard WJ (2001) Cytokines and immunodeficiency diseases. Nat Rev Immunol 1:200–208
Murray PJ (2007) The JAK-STAT signaling pathway: input and output integration. J Immunol 178:2623–2629
Avery DT, Deenick EK, Ma CS et al (2010) B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med 207:155–171
Deenick EK, Avery DT, Chan A et al (2013) Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J Exp Med 210:2739–2753
Miklossy G, Hilliard TS, Turkson J (2013) Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov 12:611–629
Adamkova L, Souckova K, Kovarik J (2007) Transcription protein STAT1: biology and relation to cancer. Folia Biol (Praha) 53:1–6
Szabo SJ, Sullivan BM, Peng SL, Glimcher LH (2003) Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol 21:713–758
Liu L, Okada S, Kong XF et al (2011) Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 208:1635–1648
van de Veerdonk FL, Plantinga TS, Hoischen A et al (2011) STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 365:54–61
Uzel G, Sampaio EP, Lawrence MG et al (2013) Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol 131:1611–1623
Hadj-Slimane R, Chelbi-Alix MK, Tovey MG, Bobe P (2004) An essential role for IFN-alpha in the overexpression of Fas ligand on MRL/lpr lymphocytes and on their spontaneous Fas-mediated cytotoxic potential. J Interferon Cytokine Res 24:717–728
Dong J, Wang QX, Zhou CY, Ma XF, Zhang YC (2007) Activation of the STAT1 signalling pathway in lupus nephritis in MRL/lpr mice. Lupus 16:101–109
Hale MB, Krutzik PO, Samra SS, Crane JM, Nolan GP (2009) Stage dependent aberrant regulation of cytokine-STAT signaling in murine systemic lupus erythematosus. PLoS One 4, e6756
Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L (2009) Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol 30:455–464
Thibault DL, Chu AD, Graham KL et al (2008) IRF9 and STAT1 are required for IgG autoantibody production and B cell expression of TLR7 in mice. J Clin Invest 118:1417–1426
Yiu G, Rasmussen TK, Ajami B et al (2015) Development of Th17-associated interstitial kidney inflammation in lupus-prone mice lacking the gene encoding STAT-1. Arthritis Rheumatol
Dominguez-Gutierrez PR, Ceribelli A, Satoh M, Sobel ES, Reeves WH, Chan EK (2014) Positive correlation of STAT1 and miR-146a with anemia in patients with systemic lupus erythematosus. J Clin Immunol 34:171–180
Dominguez-Gutierrez PR, Ceribelli A, Satoh M, Sobel ES, Reeves WH, Chan EK (2014) Elevated signal transducers and activators of transcription 1 correlates with increased C-C motif chemokine ligand 2 and C-X-C motif chemokine 10 levels in peripheral blood of patients with systemic lupus erythematosus. Arthritis Res Ther 16:R20
Lu MC, Lai NS, Chen HC et al (2013) Decreased microRNA(miR)-145 and increased miR-224 expression in T cells from patients with systemic lupus erythematosus involved in lupus immunopathogenesis. Clin Exp Immunol 171:91–99
Karonitsch T, Feierl E, Steiner CW et al (2009) Activation of the interferon-gamma signaling pathway in systemic lupus erythematosus peripheral blood mononuclear cells. Arthritis Rheum 60:1463–1471
Menon M, Blair PA, Isenberg DA, Mauri C (2016) A regulatory feedback between plasmacytoid dendritic cells and regulatory B cells is aberrant in systemic lupus erythematosus. Immunity 44:683–697
Huang X, Guo Y, Bao C, Shen N (2011) Multidimensional single cell based STAT phosphorylation profiling identifies a novel biosignature for evaluation of systemic lupus erythematosus activity. PLoS One 6, e21671
Ivashkiv LB, Donlin LT (2014) Regulation of type I interferon responses. Nat Rev Immunol 14:36–49
Baechler EC, Batliwalla FM, Karypis G et al (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100:2610–2615
Liang Y, Xu WD, Yang XK et al (2014) Association of signaling transducers and activators of transcription 1 and systemic lupus erythematosus. Autoimmunity 47:141–145
Zhuang H, Szeto C, Han S, Yang L, Reeves WH (2015) Animal models of interferon signature positive lupus. Front Immunol 6:291
Landolt-Marticorena C, Bonventi G, Lubovich A et al (2009) Lack of association between the interferon-alpha signature and longitudinal changes in disease activity in systemic lupus erythematosus. Ann Rheum Dis 68:1440–1446
Petri M, Singh S, Tesfasyone H et al (2009) Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus 18:980–989
Gamero AM, Young MR, Mentor-Marcel R et al (2010) STAT2 contributes to promotion of colorectal and skin carcinogenesis. Cancer Prev Res (Phila) 3:495–504
Matsumoto M, Tanaka N, Harada H et al (1999) Activation of the transcription factor ISGF3 by interferon-gamma. Biol Chem 380:699–703
Abdul-Sater AA, Majoros A, Plumlee CR et al (2015) Different STAT transcription complexes drive early and delayed responses to type I IFNs. J Immunol 195:210–216
Hambleton S, Goodbourn S, Young DF et al (2013) STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci U S A 110:3053–3058
Sriram U, Varghese L, Bennett HL et al (2012) Myeloid dendritic cells from B6.NZM Sle1/Sle2/Sle3 lupus-prone mice express an IFN signature that precedes disease onset. J Immunol 189:80–91
Ramirez-Velez G, Medina F, Ramirez-Montano L et al (2012) Constitutive phosphorylation of interferon receptor A-associated signaling proteins in systemic lupus erythematosus. PLoS One 7, e41414
Baccala R, Gonzalez-Quintial R, Schreiber RD, Lawson BR, Kono DH, Theofilopoulos AN (2012) Anti-IFN-α/β receptor antibody treatment ameliorates disease in lupus-predisposed mice. J Immunol 189:5976–5984
Munroe ME, Lu R, Zhao YD et al (2016) Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis. doi:10.1136/annrheumdis-2015-208140
Yang XO, Panopoulos AD, Nurieva R et al (2007) STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 282:9358–9363
Durant L, Watford WT, Ramos HL et al (2010) Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 32:605–615
Harris TJ, Grosso JF, Yen HR et al (2007) Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol 179:4313–4317
Liu X, Lee YS, Yu CR, Egwuagu CE (2008) Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J Immunol 180:6070–6076
Zhu Y, Zou L, Liu YC (2016) T follicular helper cells, T follicular regulatory cells and autoimmunity. Int Immunol 28:173–179
Moens L, Tangye SG (2014) Cytokine-Mediated regulation of plasma cell generation: IL-21 takes center stage. Front Immunol 5:65
Liu K, Liang C, Liang Z, Tus K, Wakeland EK (2005) Sle1ab mediates the aberrant activation of STAT3 and Ras-ERK signaling pathways in B lymphocytes. J Immunol 174:1630–1637
Nakou M, Bertsias G, Stagakis I et al (2010) Gene network analysis of bone marrow mononuclear cells reveals activation of multiple kinase pathways in human systemic lupus erythematosus. PLoS One 5, e13351
Harada T, Kyttaris V, Li Y, Juang YT, Wang Y, Tsokos GC (2007) Increased expression of STAT3 in SLE T cells contributes to enhanced chemokine-mediated cell migration. Autoimmunity 40:1–8
Edwards LJ, Mizui M, Kyttaris V (2015) Signal transducer and activator of transcription (STAT) 3 inhibition delays the onset of lupus nephritis in MRL/lpr mice. Clin Immunol 158:221–230
Balomenos D, Rumold R, Theofilopoulos AN (1998) Interferon-gamma is required for lupus-like disease and lymphoaccumulation in MRL-lpr mice. J Clin Invest 101:364–371
Schwarting A, Wada T, Kinoshita K, Tesch G, Kelley VR (1998) IFN-gamma receptor signaling is essential for the initiation, acceleration, and destruction of autoimmune kidney disease in MRL-Fas(lpr) mice. J Immunol 161:494–503
Haas C, Ryffel B, Le Hir M (1998) IFN-gamma receptor deletion prevents autoantibody production and glomerulonephritis in lupus-prone (NZB x NZW)F1 mice. J Immunol 160:3713–3718
Ferber IA, Brocke S, Taylor-Edwards C et al (1996) Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol 156:5–7
Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA (1996) IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol 157:3223–3227
Chitnis T, Najafian N, Benou C et al (2001) Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest 108:739–747
Singh RR, Saxena V, Zang S et al (2003) Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J Immunol 170:4818–4825
Jacob CO, Zang S, Li L et al (2003) Pivotal role of Stat4 and Stat6 in the pathogenesis of the lupus-like disease in the New Zealand mixed 2328 mice. J Immunol 171:1564–1571
Menke J, Bork T, Kutska B et al (2011) Targeting transcription factor Stat4 uncovers a role for interleukin-18 in the pathogenesis of severe lupus nephritis in mice. Kidney Int 79:452–463
Remmers EF, Plenge RM, Lee AT et al (2007) STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 357:977–986
Han JW, Zheng HF, Cui Y et al (2009) Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 41:1234–1237
Graham RR, Cotsapas C, Davies L et al (2008) Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet 40:1059–1061
Harley JB, Alarcon-Riquelme ME, Criswell LA et al (2008) Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 40:204–210
Gateva V, Sandling JK, Hom G et al (2009) A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 41:1228–1233
Bolin K, Sandling JK, Zickert A et al (2013) Association of STAT4 polymorphism with severe renal insufficiency in lupus nephritis. PLoS One 8, e84450
Svenungsson E, Gustafsson J, Leonard D et al (2010) A STAT4 risk allele is associated with ischaemic cerebrovascular events and anti-phospholipid antibodies in systemic lupus erythematosus. Ann Rheum Dis 69:834–840
Korman BD, Kastner DL, Gregersen PK, Remmers EF (2008) STAT4: genetics, mechanisms, and implications for autoimmunity. Curr Allergy Asthma Rep 8:398–403
Namjou B, Sestak AL, Armstrong DL et al (2009) High-density genotyping of STAT4 reveals multiple haplotypic associations with systemic lupus erythematosus in different racial groups. Arthritis Rheum 60:1085–1095
Lamana A, Lopez-Santalla M, Castillo-Gonzalez R et al (2015) The minor allele of rs7574865 in the STAT4 gene is associated with increased mRNA and protein expression. PLoS One 10, e0142683
Cho SS, Bacon CM, Sudarshan C et al (1996) Activation of STAT4 by IL-12 and IFN-alpha: evidence for the involvement of ligand-induced tyrosine and serine phosphorylation. J Immunol 157:4781–4789
Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, Niewold TB (2009) Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-alpha in lupus patients in vivo. J Immunol 182:34–38
Hellquist A, Jarvinen TM, Koskenmies S et al (2009) Evidence for genetic association and interaction between the TYK2 and IRF5 genes in systemic lupus erythematosus. J Rheumatol 36:1631–1638
Connolly JJ, Hakonarson H (2012) Role of cytokines in systemic lupus erythematosus: recent progress from GWAS and sequencing. J Biomed Biotechnol 2012:798924
Sigurdsson S, Nordmark G, Garnier S et al (2008) A risk haplotype of STAT4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Hum Mol Genet 17:2868–2876
Abelson AK, Delgado-Vega AM, Kozyrev SV et al (2009) STAT4 associates with systemic lupus erythematosus through two independent effects that correlate with gene expression and act additively with IRF5 to increase risk. Ann Rheum Dis 68:1746–1753
Lin JX, Leonard WJ (2000) The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 19:2566–2576
Akira S (1999) Functional roles of STAT family proteins: lessons from knockout mice. Stem Cells 17:138–146
Antov A, Yang L, Vig M, Baltimore D, Van Parijs L (2003) Essential role for STAT5 signaling in CD25+CD4+ regulatory T cell homeostasis and the maintenance of self-tolerance. J Immunol 171:3435–3441
Cohen AC, Nadeau KC, Tu W et al (2006) Cutting edge: decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol 177:2770–2774
Piccirillo CA, d'Hennezel E, Sgouroudis E, Yurchenko E (2008) CD4+Foxp3+ regulatory T cells in the control of autoimmunity: in vivo veritas. Curr Opin Immunol 20:655–662
Jenks JA, Seki S, Kanai T et al (2013) Differentiating the roles of STAT5B and STAT5A in human CD4+ T cells. Clin Immunol 148:227–236
Yang XP, Ghoreschi K, Steward-Tharp SM et al (2011) Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol 12:247–254
Hedrich CM, Rauen T, Apostolidis SA et al (2014) Stat3 promotes IL-10 expression in lupus T cells through trans-activation and chromatin remodeling. Proc Natl Acad Sci U S A 111:13457–13462
Chen Z, Lund R, Aittokallio T, Kosonen M, Nevalainen O, Lahesmaa R (2003) Identification of novel IL-4/Stat6-regulated genes in T lymphocytes. J Immunol 171:3627–3635
Tuomela S, Rautajoki KJ, Moulder R, Nyman TA, Lahesmaa R (2009) Identification of novel Stat6 regulated proteins in IL-4-treated mouse lymphocytes. Proteomics 9:1087–1098
Elo LL, Jarvenpaa H, Tuomela S et al (2010) Genome-wide profiling of interleukin-4 and STAT6 transcription factor regulation of human Th2 cell programming. Immunity 32:852–862
Kaplan MH, Schindler U, Smiley ST, Grusby MJ (1996) Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 4:313–319
Shimoda K, van Deursen J, Sangster MY et al (1996) Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature 380:630–633
Takeda K, Tanaka T, Shi W et al (1996) Essential role of Stat6 in IL-4 signalling. Nature 380:627–630
Dean GS, Tyrrell-Price J, Crawley E, Isenberg DA (2000) Cytokines and systemic lupus erythematosus. Ann Rheum Dis 59:243–251
Theofilopoulos AN, Koundouris S, Kono DH, Lawson BR (2001) The role of IFN-gamma in systemic lupus erythematosus: a challenge to the Th1/Th2 paradigm in autoimmunity. Arthritis Res 3:136–141
Luzina IG, Knitzer RH, Atamas SP et al (1999) Vasculitis in the Palmerston North mouse model of lupus: phenotype and cytokine production profile of infiltrating cells. Arthritis Rheum 42:561–568
Nakajima A, Hirose S, Yagita H, Okumura K (1997) Roles of IL-4 and IL-12 in the development of lupus in NZB/W F1 mice. J Immunol 158:1466–1472
Leng RX, Pan HF, Ye DQ, Xu Y (2012) Potential roles of IL-9 in the pathogenesis of systemic lupus erythematosus. Am J Clin Exp Immunol 1:28–32
Ouyang H, Shi Y, Liu Z et al (2013) Increased interleukin 9 and CD4+IL-9+ T cells in patients with systemic lupus erythematosus. Mol Med Rep 7:1031–1037
Lau M, Tsantikos E, Maxwell MJ, Tarlinton DM, Anderson GP, Hibbs ML (2012) Loss of STAT6 promotes autoimmune disease and atopy on a susceptible genetic background. J Autoimmun 39:388–397
Yu HH, Liu PH, Lin YC et al (2010) Interleukin 4 and STAT6 gene polymorphisms are associated with systemic lupus erythematosus in Chinese patients. Lupus 19:1219–1228
Krutzik PO, Irish JM, Nolan GP, Perez OD (2004) Analysis of protein phosphorylation and cellular signaling events by flow cytometry: techniques and clinical applications. Clin Immunol 110:206–221
Rasmussen TK, Andersen T, Bak RO et al (2015) Overexpression of microRNA-155 increases IL-21 mediated STAT3 signaling and IL-21 production in systemic lupus erythematosus. Arthritis Res Ther 17:154
Caprioli F, Sarra M, Caruso R et al (2008) Autocrine regulation of IL-21 production in human T lymphocytes. J Immunol 180:1800–1807
Nurieva R, Yang XO, Martinez G et al (2007) Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 448:480–483
Dolff S, Abdulahad WH, Westra J et al (2011) Increase in IL-21 producing T-cells in patients with systemic lupus erythematosus. Arthritis Res Ther 13:157
Pratama A, Vinuesa CG (2014) Control of TFH cell numbers: why and how? Immunol Cell Biol 92:40–48
Vinuesa CG, Linterman MA, Goodnow CC, Randall KL (2010) T cells and follicular dendritic cells in germinal center B-cell formation and selection. Immunol Rev 237:72–89
Ettinger R, Kuchen S, Lipsky PE (2008) The role of IL-21 in regulating B-cell function in health and disease. Immunol Rev 223:60–86
Bettelli E, Carrier Y, Gao W et al (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441:235–238
Chaudhry A, Rudra D, Treuting P et al (2009) CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326:986–991
Kluger MA, Melderis S, Nosko A et al (2015) Treg17 cells are programmed by Stat3 to suppress Th17 responses in systemic lupus. Kidney Int 89:158–166
Yan D, Farache J, Mathis D, Benoist C (2015) Imbalanced signal transduction in regulatory T cells expressing the transcription factor FoxP3. Proc Natl Acad Sci U S A 112:14942–14947
Miyara M, Yoshioka Y, Kitoh A et al (2009) Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 30:899–911
Sakaguchi S, Miyara M, Costantino CM, Hafler DA (2010) FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10:490–500
Kalland ME, Oberprieler NG, Vang T, Tasken K, Torgersen KM (2011) T cell-signaling network analysis reveals distinct differences between CD28 and CD2 costimulation responses in various subsets and in the MAPK pathway between resting and activated regulatory T cells. J Immunol 187:5233–5245
Moulton VR, Tsokos GC (2015) T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Invest 125:2220–2227
Golding A, Hasni S, Illei G, Shevach EM (2013) The percentage of FoxP3+Helios+ Treg cells correlates positively with disease activity in systemic lupus erythematosus. Arthritis Rheum 65:2898–2906
Alexander T, Sattler A, Templin L et al (2013) Foxp3+ Helios+ regulatory T cells are expanded in active systemic lupus erythematosus. Ann Rheum Dis 72:1549–1558
Dupont G, Demaret J, Venet F et al (2014) Comparative dose-responses of recombinant human IL-2 and IL-7 on STAT5 phosphorylation in CD4+FOXP3- cells versus regulatory T cells: a whole blood perspective. Cytokine 69:146–149
Barron L, Dooms H, Hoyer KK et al (2010) Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J Immunol 185:6426–6430
Horwitz DA (2010) Identity of mysterious CD4+CD25-Foxp3+ cells in SLE. Arthritis Res Ther 12:101
Zhang B, Zhang X, Tang FL, Zhu LP, Liu Y, Lipsky PE (2008) Clinical significance of increased CD4+CD25-Foxp3+ T cells in patients with new-onset systemic lupus erythematosus. Ann Rheum Dis 67:1037–1040
Bonelli M, Goschl L, Bluml S et al (2014) CD4(+)CD25(-)Foxp3(+) T cells: a marker for lupus nephritis? Arthritis Res Ther 16:104
Flores-Borja F, Bosma A, Ng D et al (2013) CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med 5:173ra123
Blair PA, Norena LY, Flores-Borja F et al (2010) CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity 32:129–140
Acknowledgments
This work was partially supported by the University Medical Center Ljubljana Research Grant 20140208.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Goropevšek, A., Holcar, M. & Avčin, T. The Role of STAT Signaling Pathways in the Pathogenesis of Systemic Lupus Erythematosus. Clinic Rev Allerg Immunol 52, 164–181 (2017). https://doi.org/10.1007/s12016-016-8550-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-016-8550-y