
Abstract
Contemporary genetic research has provided evidences that angioedema represents a diverse family of disorders related to kinin metabolism, with a much greater genetic complexity than was initially considered. Convincing data have also recently been published indicating that the clinical heterogeneity of hereditary angioedema due to C1 inhibitor deficiency (classified as C1-INH-HAE) could be attributed at least in part, either to the type of SERPING1 mutations or to mutations in genes encoding for enzymes involved in the metabolism and function of bradykinin. Alterations detected in at least one more gene (F12) are nowadays considered responsible for 25 % of cases of hereditary angioedema with normal C1-INH (type III hereditary angioedema (HAE), nlC1-INH-HAE). Interesting data derived from genetic approaches of non-hereditary angioedemas indicate that other immune pathways might be implicated in the pathogenesis of HAE. More than 125 years after the recognition of the hereditary nature of HAE by Osler, the heterogeneity of clinical expressions, the genetics of this disorder, and the genotype-phenotype relationships, still presents a challenge that will be discussed in this review. Large scale, in-depth genetic studies are expected not only to answer these emerging questions but also to further elucidate many of the unmet aspects of angioedema pathogenesis. Uncovering genetic biomarkers affecting the severity of the disease and/or the effectiveness of the various treatment modalities might lead to the prevention of attacks and the optimization of C1-INH-HAE management that is expected to provide a valuable benefit to the sufferers of angioedema.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rapid changes in the treatment of hereditary angioedema (HAE) have accelerated during the past decade, owing to the research which led to a better understanding of the pathogenesis and the genetics of the disease. Under the light of these advances, HAE is nowadays considered a heterogeneous disorder with a complex pathophysiology. Two decades after the initial recognition that mutations of the SERPING1 gene encoding for C1 inhibitor (C1-INH) are responsible for HAE due to C1-INH deficiency (C1-INH-HAE) [1, 2], a new clinically indistinguishable type of HAE was described associated with mutations in the F12 gene encoding for coagulation factor XII (FXII; Hageman factor (FXII-HAE)) [3]. Despite the fact that SERPING1 was defined the primary gene encoding to C1-INH production, in a small proportion of patients with C1-INH-HAE, no SERPING1 alterations can be detected even after meticulous analysis of the coding region [4–6]. Moreover, the vast majority of patients with a clinical history suggestive of HAE but with normal C1-INH function have no mutation in the genes associated with HAE (SERPING1, F12; unknown HAE, U-HAE) [7, 8]. Interestingly, accumulating evidence indicates that defects in genes encoding proteins involved in kinin generation or its catabolism, other than SERPING1 and F12, may be involved in the pathogenesis of HAE and may modify its clinical expression [9, 10]. Thus, nearly a century after the recognition of the hereditary nature of HAE, the genetics of this disorder presents with a challenging complexity that will be thoroughly discussed in this review.
C1-INH-HAE Genetics
The Allelic Heterogeneity of C1-INH-HAE
SERPING1 gene (serpin peptidase inhibitor, clade G (C1 inhibitor), member 1) (OMIM no. 606860; GenBank NM_000062.2) extends over a 17,159-bp genomic region located on chromosome 11q12-q13.1 (Fig. 1). The gene comprises eight exons and seven introns, presents an unusual promoter with no TATA sequence but a TdT-like initiator and a polypurine-polypyrimidine tract, and gives rise to a messenger RNA (mRNA) product of 1827 bp excluding the poly(A) tail [11]. By sequence analogies, the encoded protein, C1-INH, was identified as a member of the protein superfamily of serine protease inhibitors (Serpin). Its reactive site, identified as P1 site by analogy with α1-antitrypsin, is located in exon 8 at Arg444. C1-INH is highly glycosylated (26–33 % of the total mass). The glycosylated sites are located within a stretch of 100 amino acids at the N-terminal end of the molecule and appear to be irrelevant to the protein function, although removal of the sugars increases its catabolic rate [12]. The N-terminal end represents the non serpin-like domain that is highly divergent among different species suggesting that they accept extensive variations both in primary sequence and in glycosylation pattern (Fig. 2) [14–16]. Over 450 different SERPING1 mutations related with C1-INH-HAE have been detected up to now, scattered over all exons and exon/intron boundaries of the gene with higher concentration in exon 8, 5, and 6 and low concentration in the aminoterminal end. Most of them are listed in the Human Gene Mutation Database (Online Mendelian Inheritance in Man—OMIM ID 106100) [17] and a database specific to this disease (HAEdb, hae.enzim.hu) [18]. Recently retrieved data from HAEdb show that the majority of C1-INH-HAE-related SERPING1 mutations are missense mutations (34 %) followed by frameshift alterations and small indels (31 %), large gene rearrangements (17 %), splice-site defects (10 %), nonsense mutations (7 %), and regulatory mutations (1 %). Most of these mutations including nonsense mutations and indels lead to premature truncation of protein synthesis causing a translational frame shift. Usually, mRNAs with premature stop codons are eliminated by nonsense-mediated mRNA decay [19]. If truncated proteins are translated at all, they are non-functional and/or are rapidly degraded. Mutations resulting in the production of truncated or misfolded proteins that are not secreted efficiently occur throughout the gene and are responsible for the common type 1 C1-INH-HAE. This C1-INH-HAE type accounts for 80–85 % of all cases and is defined by reduced plasma C1-INH concentrations leading to low C1-INH function.

The location of the SERPING1 gene on chromosome 11 and its structure. Black lines connecting boxes represent introns, white boxes represent untranslated regions (UTRs), and arrows denote the Alu repeats

The five crucial regions for the proper function of C1-INH are presented on the crystal structure of the C-terminal serpin domain (residues 113–478) of recombinant non-glycosylated C1-INH (PDB 2OAY) [13] generated using PyMOL Molecular Graphics System; in yellow, a small portion of the N-terminal domain (residues 97–112). The blue color marks the reactive-center loop (RCL), inside of which the residue Arg444 is shown in red. The P15-P9 portion of the RCL is called the hinge region that is essential for the RCL mobility. At the top of the A β-sheet is the breach region and near its center is the shutter domain. The gate region is composed of s3C and s4C strands. The breach, shutter, and gate regions drive the initial insertion and passing of RCL in the β-sheet A of the serpin. Missense mutations affecting these regions of the protein are most probably pathogenic
Single amino acid substitutions, located mainly at exon 8 that decode the reactive center as well as two critical hinge regions upstream (proximal hinge) and downstream (distal hinge) the reactive site of C1-INH, lead to the less common but clinically indistinguishable type 2 C1-INH-HAE (15–20 % of cases). The majority of these mutations represent mutations located in residue Arg444, according to traditional amino acid numbering [20], which corresponds to residue Arg466 according to HUGO recommendations (where the methionine at position 1 has the number 1, while in traditional system the number −22) [21]. Only one mutation, a deletion of Lys251/Lys273 that creates a potential new glycosilation site, causing type 2 C1-INH-HAE has been detected far from the reactive-center region [22]. All these mutations lead to the production of a dysfunctional C1-INH protein that results in low C1-INH function despite normal levels of antigenic C1-INH [23].
C1-INH-HAE is transmitted as an autosomal dominant trait [1, 2]. Very rare cases of homozygous C1-INH-deficient patients have been described, mostly in consanguineous parents [24–26], while two cases of gonadal mosaicism have also been reported [27, 28]. However, on the contrary to what is expected in a dominantly inherited disease, heterozygous patients present with C1-INH plasma levels ranging from 5 to 30 % of the normal level [29]. The mechanisms that prevent the remaining wild-type allele from generating C1-INH plasma levels around 50 % are yet to be elucidated. This discrepancy has been attributed to an increased catabolic rate [30] and/or a decreased rate of C1-INH synthesis, with concomitant downregulation of the normal allele at least in some mutations [31–33]. At last, it is worthy to be mentioned that SERPING1 exhibits also a unique kind of phenotypic heterogeneity. Beyond mutations associated with C1-INH-HAE, this gene harbors a number of single nucleotide polymorphisms (SNPs), which have been associated with other diseases. One of the few common missense variants in the coding region, the polymorphism rs4926 [c.1438G>A, p.Val480Met], is associated with nasal carriage of Staphylococcus aureus [34], while six common intronic SNPs (especially the rs2511989) are strongly associated with age-related macular degeneration (AMD) in Caucasians [35]. Among them, the rs2511988 [c.1030-20A>G], located into intron 6, upstream of the 3′ splice site of exon 7, has been proposed to be more prone to functional alterations [36]. However, such an association was not confirmed in Chinese [37] and Japanese populations [38]. Similarly, a SERPING1 mutation (replacement of Ala443 with Val) affecting complement regulation but preserving kallikrein inhibitory activity has been described in one family. None of the members of this family had angioedema but were presented with systemic lupus erythematosus (SLE) possibly due to acquired deficiency in C2 and C4 [39].
The Mutagenic Lability of SERPING1
The pronounced allelic heterogeneity as well as the fact that approximately 20–25 % of all unrelated C1-INH-HAE cases have no apparent family history of C1-INH-HAE (i.e. sporadic cases) and present de novo SERPING1 mutations with the same mutational spectrum as the familial cases [40], make the SERPING1 gene a prime example of mutagenic lability. Investigation of SERPING1 mutations has revealed several key features about the DNA itself, as well as protein structure-function relationships that may explain this phenomenon [41, 42]. SERPING1’s proximity to the centromere is expected to be the main reason for its high mutation rate, since centromeric regions are considered the most rapidly evolving compartments in the eukaryotic genome. Another feature of the SERPING1 gene to which its high mutation spectrum has been attributed, is the high incidence of DNA repetitive elements. The 7 introns of the gene contain 17 Alu repeat sequences [43] which represent “hotspots” for non-homologous recombination events that may cause partial deletions or duplications of the gene [44] (Fig. 1). Large mutations related to clusters of Alu repeats in various orientations, particularly in introns 4 and 6, represent a major source of genetic instability of the SERPING1 gene. An additional structural feature of the SERPING1 gene potentially responsible for the higher than average mutation rates is the high frequency of CpG sites. Like Alu elements, CpG sites also represent mutational hotspots as they are prone to spontaneous deamination. The CpG dinucleotide in the first two positions of codon 444/466 encoding the central arginine (CGC) of the reactive center of C1-INH seems to be a frequent target for recurrent amino acid substitutions [45]. Moreover, the region encoding the reactive center, like in some other Serpins, contains both primary and secondary structure DNA polymerase-pause sites, which enhance its rates of mutation and evolution.
Interestingly, some of SERPING1 alterations represent extremely rare mutagenesis events. Such alterations are the dinucleotide mutation g.8502_03TC>AA resulting in the missense mutation V233E [46], as well as the recently described substitution of two consecutive nucleotides TC to AA resulting in a termination codon (F225X) [47]. Dinucleotide substitution is an extremely rare mutagenesis mechanism in eukaryotes usually resulting in a missense mutation. Considering other genes and diseases, causative dinucleotide substitutions have been described in less than 1 % of Factor IX gene associated with hemophilia B, in Keratin 10 gene causing annular epidermolytic ichthyosis, and in the RET proto-oncogene resulting in the aggressive type 2B of multiple endocrine neoplasia syndrome (MEN-2B) [47].
The Pathogenicity of Single Amino Acid Substitutions
How mutations causing single amino acid substitutions almost invariably result in a loss of C1-INH inhibitory activity remains as yet an intriguing enigma. Evidently, misfolding of serpins resulting in serpin polymerization that causes retention or impaired secretion of the mutated protein is a common molecular event highly relevant to pathologic processes of serpinopathies [48]. Actually, there are reports of C1-INH oligomerization which leads to lose of its inhibitory activity, like in most serpins with proximal-hinge mutations. As such, point mutations have been described affecting the C-terminal region relative to the reactive center causing partial C1-INH secretion due to intracellular multimerization [49]. Similarly, polymerization has been observed when the C1-INH molecule carries a mutation in the reactive-center proximal hinge [50]. However, contrary to other serpinopathies, these mutants are not converted to a substrate but exist in both monomeric and multimeric forms. Moreover, no conformation-dependent consequences of an intracellular accumulation of C1-INH polymers indicating misfolded-protein stress have been related so far to C1-INH mutations, other than its deficiency in the plasma.
More recently, Madsen et al. [51] demonstrated the presence of polymerized C1-INH in the plasma of C1-INH-HAE patients carrying certain SERPING1 mutations. The pathogenetic role of these polymers remains as yet unclear. However, given the inability of polymers to control target proteases and the potent activation of the kallikrein kinin system induced by misfolded proteins [52], it is probable that they can potentiate the formation of bradykinin. Unequivocally, further research is required towards a better understanding of the ability of C1-INH mutants to form and accumulate intracellularly polymers that are secreted thereafter into the blood, or to assemble polymers in the blood stream, in order to reveal the molecular mechanisms of the extended phenotypic variability of C1-INH-HAE [53].
Another interesting point in regard with the pathogenicity of single amino acid substitutions is related to the differential control exerted by certain C1-INH mutants (R378C and I271T) on the protease C1s as opposed to kallikrein [25, 54]. It is worth to be noticed here that commonly used methods using C1s as target are unable to effectively evaluate C1-INH function in such cases, where the use of a newly introduced method based on a contact phase protease target is preferable [54].
Notwithstanding the above explanations, questions are still emerging in regard to the causal nature of all the reported SERPING1 missense mutations. C1-INH structure-function correlates do not always explain satisfactorily the linkage between specific mutations and the disease phenotypes. Unfortunately, appropriate functional or segregation studies cannot be carried out due to the large number of private mutations and the unavailability of relatives. Regarding functional studies, this complexity is further accentuated by findings suggesting that some mutations may have different consequences in different cell types [55].
The Molecular Analysis of SERPING1 Gene
Currently, the molecular analysis of SERPING1 initiates with the prioritized amplification of all exons and the exon/intron boundaries by PCR and the detection of mutations by direct sequencing. If no mutation were found by sequencing, or in case a detected missense mutation would be proven benign for protein function by bioinformatic tools, further analysis for the identification of large gene rearrangements is performed [4]. For this purpose, two different techniques are widely used, namely the long-range PCR and the multiplex ligation-dependent probe amplification (MLPA). In long-range PCR, segments of the SERPING1 gene encompassing at least 7–15 kb are amplified using common oligonucleotide primers, specific Taq polymerase mixes, and long-time PCR conditions [4, 5, 56]. MLPA is a high-throughput, sensitive technique for detecting copy number variations in genomic sequences [57] usually performed by a commercially available kit (SALSA MLPA probemix P243-A2 SERPING1 kit, MRC-Holland, Amsterdam, The Netherlands). Targeted analysis by simple and widely accessible methods, like polymerase chain reaction restriction fragment length polymorphism (PCR-RFLP), is not applicable in SERPING1 molecular studies; such molecular approaches are useful in the molecular studies of families with known SERPING1 mutations by modifying the DNA sequence in order to be recognized by restriction enzymes.
The cumbersome and time consuming molecular analysis of SERPING1 gene prevents sometimes the detection of the causal genetic defect due to an incomplete study of the gene. Assuming that a mutation detected after sequencing the initially examined exon(s) of the gene is the causal one, further analysis is usually not taking place. This practice, however, hampers the identification of mutations that possibly coexist in positions next to the initially analyzed or large gene rearrangements. In order to clarify such a case, we had recently submitted to bioinformatics analysis all missense mutations deposited in HAEdb and we found that at least one of them was benign, thus making its causative effect disputable. The reanalysis of SERPING1 in this case uncovered an additional genetic damage that was deleterious for the protein function (nonsense mutation), obviously representing the causative alteration [4]. This case also indicates that multiple single amino acid substitutions might coexist in the same patient with as yet unknown consequences to both C1-INH production and function and possibly to the disease phenotype.
Besides the above, serious questions regarding the adequacy of conventional molecular analysis in uncovering the causal genetic alterations arise from the fact that in about 5 % of C1-INH-HAE cases no mutation could be detected in the coding region of SERPING1 [4–6]. This finding indicates that, in some patients, a causative defect modifying C1-INH expression may be located in an intronic or an untranslated region of the gene. Actually, such functional intronic alterations have been recently reported [55, 58, 59]. In any case, the comprehensive analysis of SERPING1 must be affirmed before attributing the disorder to a possibly deleterious effect of a detected missense mutation and before reporting the result of a genetic analysis as negative.
Bearing in mind the above arguments and shortcomings, it remains elusive whether the presence of all SERPING1 alterations considered so far responsible for HAE, are isolated or are they expressed in tandem with either functional alterations on genes involved in the function or degradation of bradykinin, or whether they display SERPING1 polymorphisms and mutations with as yet unknown functionality.
Genotype-Phenotype Correlations in C1-INH-HAE
The clinical expression of C1-INH-HAE is characterized by a great variability. Features of the disease, like the age of disease onset, frequency and triggers of attacks, severity and localization of edema, prodromal signs and symptoms, and the need for long-term treatment, vary broadly even among members of the same family sharing the same mutation [23, 60, 61]. Some patients suffer frequent, life-threatening attacks, while others experience a mild disease course, whereas as much as 14 % of carriers of SERPING1 mutation may remain symptom free throughout their entire lives [56, 62]. Evidently, about 5 % of the adult C1-INH-deficient patients are identified only after their child has been diagnosed, and despite carrying and transmitting the mutation to their children, may remain asymptomatic. Therefore, the factors influencing this heterogeneous clinical expression represent one of the oldest and unsolved mysteries of the disease that needs to be solved since the optimal therapy is still based on individualization of patients’ history [63]. Following the initial detection of SERPING1 mutations and their consequences on the complex structure of C1-INH, it was assumed that certain genetic alterations might result in specific disease phenotypes. The very first evidence for such a non-linear genotype-phenotype correlation was derived from studies indicating that the presence of one mutated allele affects mRNA transcription and protein secretion, leads to downregulation of the normal allele and, finally, to C1-INH plasma levels ranging from 10 to 30 % of normal [32, 33, 64]. In fact, C1-INH levels are not correlated to the clinical presentation of the disease irrespective of the type of SERPING1 mutation. Almost all heterozygotes present with plasma C1-INH concentration lower than the expected 50 %. In isolated reports of homozygous C1-INH-HAE patients, suffering from a severe clinical disease course, their heterozygous family members were symptom free [24, 25, 65], which offers further evidence to support a possible relationship between certain mutation types and disease phenotype. Over the recent decades, the increasing number of mutations associated with a low prevalence of the disease did not allow the confirmation of a strong correlation between certain SERPING1 mutations and the clinical phenotype of C1-INH-HAE. Thus, it was generally accepted that there is little correlation between the type of SERPING1 mutations and disease phenotype [53, 66]. Subsequent studies attempted to correlate the type of SERPING1 mutations with the severity of the disease but reached to conflicting results [9, 67, 68]. Interestingly, however, in the largest study ever in this field, Speletas et al. [4] examined 265 C1-INH-HAE patients from 4 European countries (including the study cohort of one of previous studies [9]) and showed that patients carrying SERPING1 missense mutations have a significantly lower probability of manifesting C1-INH-HAE attacks before age 10 than those with all other SERPING1 defects (i.e., regulatory, nonsense, splice defects, frameshift, large indels). Bearing in mind that early onset of C1-INH-HAE symptoms is predictive of higher severity of the disease course [69, 70], carriage of missense mutations may predict milder HAE-C1-INH clinical course. In accordance to these results, a similar genotype-phenotype correlation has been very recently reported in a small cohort of Serbian patients [71].
The intra-familial heterogeneity of C1-INH-HAE prompted further studies aiming to investigate the effect of common SERPING1 polymorphisms and/or of alterations in genes encoding other proteins involved in the pathogenesis of the disease. So far no correlation was found between the severity of C1-INH-HAE and the p.V480M polymorphism (c.1438G>A, rs4926) in the SERPING1 gene [72, 73], while conflicting results were obtained regarding the association with the C allele of another common SERPING1 polymorphism (c.-21T>C, rs28362944) [74, 75]. However, Duponchel et al. [55] showed that transfections with the minigene construct carrying the C variant at position c.-21 consistently yielded a weak product lacking exon 2 in the transfected cell lines, suggesting that this allele might contribute to the lower expression of the normal protein and, hence, to the occurrence of more severe disease. Similarly, older studies attempting to correlate polymorphisms in other proteins with C1-INH-HAE severity had failed to provide definitive results. The interesting finding of Blasko et al. [76] that the biannual attack rate is lower in patients carrying three or four copies of C4B gene encoding the B isotype of the C4 protein, has not been confirmed as yet. An common functional polymorphism of the BDKR2 gene encoding B2 bradykinin receptor, which is located in the 5′UTR of the gene and characterized by the absence of 9-base pair repeat sequencing (B2R-9 allele, rs71103505), has been associated with increased C1-INH-HAE severity only in one study where it was found to be consistently present in the most symptomatic cases (21 patients) [77]. Finally, no correlation was found between the severity of C1-INH-HAE and the functional polymorphisms in the genes of bradykinins’ B1 receptor (BDKR1), in the ACE gene or in the gene of mannose-binding lectin (MBL2) [78, 79].
Recently however, Bors et al. [9] reported a strong correlation between the severity of the disease and the F12-46C/T polymorphism (c.-4C/T, rs1801020). Thereafter, Speletas et al. [10] confirmed this result analyzing 258 C1-INH-HAE patients from 113 unrelated European families (including the study cohort of Bors et al. [9]). In this study, the carriage of F12-46C/T polymorphism was found strongly associated with a 7-year delay in disease onset, and significantly but negatively associated with the need for long-term treatment, both independently of the type of SERPING1 mutations. The F12-46C/T polymorphism is located in the promoter region of the F12 gene, four nucleotides before the initiation codon (ATG, Methionine), creating a new initiation codon (ATG) for transcription of the mRNA and a frameshift that produces a truncated protein. The T allele destroys the Kozak’s consensus sequence (GCCAGCCATGG) for translation initiation signaling and prevents proper recognition of the translation initiation site [80, 81]. Thus, the above finding could be attributed to the effect that the polymorphism has on the synthesis of FXII and, secondarily, on the production of bradykinin downstream the contact system. Another interesting feature of C1-INH-HAE is that the proportion of the various types of disease-related gene alterations was recently found to be different between different geographical regions [4]. Missense mutations and small indels represent 30 and 28 %, respectively, of all SERPING1 alterations observed in Hungarian patients, while the corresponding percentages among Romanian patients are 50 and 14 %. Taken together with the fact that the disease may follow a variable course in the different life periods of the same patient, this finding indicate a possible implication of environmental factors either in the mutagenesis or in the epigenetic regulation of the SERPING1 gene.
Genetics of FXII-HAE
nlC1-INH-HAE (type III HAE) is a new entity described in 2000 simultaneously by Bork et al. [82] and Binkley and Davis [83]. This rare kind of inherited (and familial) angioedema is exhibited by similar clinical manifestations to C1-INH-HAE, but with otherwise normal plasma levels of functional C1-INH, and without causal mutations at the SERPING1 locus [82]. In some of the affected females, clinical angioedema attacks can be attributed to exposure to estrogens (i.e., contraceptives, hormone replacement therapy). A subset of about 300 reported cases of (≈25 % of all estrogen-dependent HAEs) have been attributed to alterations in the F12 gene (OMIM no. 610619) [84]. The F12 gene has been mapped to chromosome 5 (5q33-qter) and comprised 13 introns and 14 exons covering 12 kb [85]. Several loss-of-function mutations in the F12 gene associated with FXII deficiency (Hageman’s disease) have been described [86]. Beyond these, four F12 alterations have been identified so far which, according to their co-segregation patterns, were assumed to be causal for HAE with normal C1-INH function. Firstly, two distinct missense mutations located on the exon 9 have been described, resulting in threonine-to-lysine (Thr328Lys) and threonine-to-arginine (Thr328Arg) substitutions. Moreover, a large deletion of 72 bp (c.971_1018+24del72) located at the exon 9/intron 9 border was identified in two unrelated families of Turkish origin that is involved in the proline-rich region of the FXII in which the above two substitutions are located [87, 88]. More recently, a duplication of 18 bp (c.892_909dup) causing the repeated presence of six amino acids (p.298–303) in the same region of factor XII was also described in a Hungarian family [89]. All the above four mutations are located in the proline-rich linker peptide between the Kringle and trypsin-like serine protease (Tryp-SPc) domains of the FXII protein.
The Thr328Lys substitution has been identified in clinically affected and symptom-free individuals from numerous families [3, 90], whereas the Thr328Arg replacement has been found only in two German families [3]. After the discovery of these mutations, it was suggested that they confer a putative gain of FXII function leading to upregulation of contact system activation and therefore may account for increased bradykinin formation. Actually, patients with FXII-HAE who are carriers of the Thr328Lys mutation present with an increased amidolytic FXII activity, but with normal plasma levels of the protein [91]. On the contrary, however, Bork et al. [92] have shown that there is no difference between FXII-HAE patients with the Thr328Lys mutation and their healthy probands neither in FXII surface activation by silicon dioxide nor in kallikrein-like activity with and without activation by dextran sulfate, indicating that the Thr328Lys mutation does not cause a gain of function of FXII. Thus, the role of the F12 mutations in the generation of FXII-HAE and the underlying pathophysiology of this HAE type remains still poorly understood. Notwithstanding, the response of some patient’s attacks to bradykinin receptor antagonist (icatibant) [93] supports the assumption that contact pathway dysregulation is involved, with bradykinin being the primary mediator of this type of angioedema. Interestingly, after examining a large cohort of women with anti-histamine-resistant angioedema, negative family history and normal C1-INH activity, Bork et al. [94] have also found that 3.4 % of them carried the Thr328Lys mutation in a heterozygous form, indicating that FXII-HAE can exist even in unaffected family members. More recently, Gelincik et al. [95] reported two more cases of idiopathic non-histaminergic angioedema (InH-AAE) carrying F12 mutations (c1681-1G/A—intron 13 and c1027G/C—exon 10). Although the above data may indicate a putative a gain-of-function mutations in patients with FXII-HAE, both of these mutations have been previously identified in association with FXII deficiency. Moreover, it was confirmed that one of the detected alterations was a de novo mutation. It remains to be elucidated whether these alterations are actually causal and are not coincident with some other protein damage.
The Special Case of ACEi-AAE
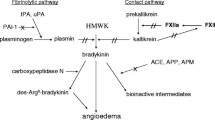
The pathophysiology of angiotensin-converting enzyme inhibitor-associated angioedema (ACEi-AAE) is not fully known, but several lines of evidence indicate that a blockade of the bradykinin and substance P degradation by ACE (kininase II), and perhaps impaired metabolism of bradykinin and des-Arg(9)-bradykinin by aminopeptidase P (APP) might be the primary mechanism of this disorder [96]. As both the frequency and the severity of ACEi-AAE appear to be rising over the past several decades, efforts have been made towards uncovering genetic markers that could predict the risk of ACE inhibitor-exposed individuals. Through a pedigree analysis and a case–control study, Duan et al. [97] found that the genetic SNP variant encoding for APP, C-2399A (rs3788853) for XPNPEP2, is indeed associated with a reduction in APP activity and a higher incidence of ACEi-AAE. Subsequently, Woodard-Grice et al. [98] pointed out that this SNP has been associated with APP activity and with the emergence of ACEi-induced AAE in men, but not in women. More recently, Cilia La Corte et al. [99] identified, including the above, 3 SNPs (c.-2399C-A, c.-1612G-T, c.-393G-A) that are significantly associated with plasma APP activity, the ATG haplotype of which was associated with decreased plasma APP activity and contributes to the development of ACEi-AAE. Moreover, the above mentioned rs71103505 polymorphism along with another SNP located in 5′UTR of BDKRB2 gene, namely the rs1799722 (g.96671139C>T, c.-191C>T, also known as C-58T) have been also associated with ACEi-AAE [100], in accordance with the previous observation of Lung et al. [77] regarding the correlation of rs71103505 polymorphism with the severity of C1-INH-HAE. The −9/+9 polymorphism (rs71103505) has been associated with bradykinin-mediated vasodilation, as well as bradykinin-mediated tissue plasminogen activator (t-PA) release in men, during ACE inhibition [101]. More recently, Pare et al. [102] by both a replicated genome-wide association study (GWAS) and a candidate gene approach including the largest reported sample of ACEi-AAE and ACE inhibitor-exposed controls, was unable to confirm the above correlation. On the contrary, they detected an association between increased risk of ACEi-AAE and a polymorphism in intron 1 of the gene encoding for neprilysin (MME, rs989692). Neprilysin (neutral endopeptidase, EC 3.4.24.11 (NEP)) is a membrane-bound metallopeptidase that colocalizes with ACE and metabolizes a number of vasodilator and vasoconstrictor peptides, including bradykinin [103]. Most interesting, however, were the findings of this study implicating inflammatory pathways in the pathogenesis ACEi-AAE. Consistent associations were detected between ACEi-AAE and polymorphisms in two other genes involved in immune regulation, i.e. the rs500766 polymorphism in the PRKCQ gene encoding for protein kinase C θ (PKCθ) and the rs2724635 polymorphism in the ETS variant gene 6 (ETV6), also known as TEL (or translocation ETS leukemia). PKCθ is involved in the activation of T lymphocytes [104]. ETV6 is a transcriptional repressor that is disrupted by translocation in 26–47 % of cases of childhood pre-B-cell acute lymphocytic leukemias, but also in some cases of T cell leukemias [105, 106]. ETV6 also regulates interleukin 18 (IL-18), IL-10, and IL-4 cytokines [106, 107] involved in the clonal expansion of Th1 (IL-18 and IL-10) and Th2 (IL-4) subsets of CD4+ helper T cells [108].
A Link Between HAE and Autoimmune Diseases
The possible involvement of immune regulation in the pathogenesis ACEi-AAE, as indicated by the above study, could imply that immune-mediated mechanisms may be implicated in HAE. Despite the fact that the link between autoimmune manifestations and HAE remains debated [109], Triggianese et al. [110] recently reported that the prevalence of autoimmune diseases among their 143 HAE patients as high as 4.2 %. Moreover, they claim that HAE patients express enhanced production of autoantibodies due most probably to an increased activation of B cells, found to be associated with a high expression of TLR-9. In favor of the involvement of immunoregulatory mechanisms in HAE, is another piece of evidence indicating that HAE patients have higher levels of circulating IL-17, and growth factors FGFb and GM-CSF compared with healthy individuals, which becomes further elevated during clinical attacks [111]. Currently, no further genetic data are available to demonstrate a possible linkage between autoimmunity and HAE.
The Role of Kinin Catabolic Enzymes Deficiencies
Carboxypeptidase N (CPN) is an important enzyme that regulates biologically active peptides including complement anaphylatoxins and kinins by removing carboxy-terminal arginine or lysine [112]. In 2004, a unique form of HAE has been described associated with CPN deficiency [113–115]. The proband of the affected family displayed an 11-year history of angioedema attacks occurring about once weekly and lasting 24 h. Plasma CPN activity that was as low as 20 % of normal, remained unchanged during attacks, concurrently with increase in C3a and histamine levels. Several family members were clinically affected, expressing a combination of angioedema or chronic urticaria, as well as hay fever and asthma, and had slightly depressed serum CPN, suggesting an autosomal recessive inheritance. More than two decades after the publication of this case, Cao and Hegele [116] sequenced the CPN1 gene which encodes the catalytic subunit of CPN, in the archival genomic DNA of the proband and identified three mutations. The first was a frameshift alteration in exon 1 due to a single G insertion at nucleotide 385 (385fsInsG) that should result in a truncated protein with little to no active site. This mutation was not present in the 128 normal Caucasians screened, and therefore was assumed to be extremely rare. The second was a missense mutation in exon 3 that predicted substitution of aspartic acid for the wild-type glycine at amino acid 178 (Gly178Asp) which is a highly conserved amino acid in different species and many human carboxypeptidases. This mutation had an allelic frequency of 0.0078 among the 128 normal Caucasian subjects, suggesting that it was also rare. This aspartic acid 178 mutation may produce a protein product with reduced activity, thereby explaining why this individual had 21 % CPN activity compared with 50 % that would be expected in a single allelic exon 1 frameshift mutation. The third polymorphism, in intron 1, did not affect the coding sequence, suggesting that it did not contribute to the CPN deficient phenotype. Interestingly, the CPN1 harbors five nonsynonymous mutations and several genomic alterations (especially in 3′ UTR-region), associations and functional consequences of which are still unknown. More recently, Willemse et al. [117] while exploring the role of CPN in the development of ACEi-AAE, identified a patient with a complete carboxypeptidase N deficiency (3 % of the normal serum CPN activity). The patient suffered from a moderate facial angioedema attacks, 2 years after initiating an ACEi, and her attacks ceased subsequent to discontinuation of the drug and H1-antihistaminics. In a series of 162 patients with suspected bradykinin-mediated angioedema, Dessart et al. [118] detected 21 % with non-iatrogenic defective kininase activity and 30 % with idiopathic increased kinin formation. Isolated or combined deficiencies in three major enzymes involved in kinin degradation (kininases), namely CPN, ACE, and aminopeptidase P (APP) were found. Seven out of their 20 C1-INH-HAE patients, had additionally a significant deficiency in one or several kinin catabolic enzymes (three of APP, one of CPN, and three of ACE). Two patients with an ACE (kininase II) deficiency were related, while one asymptomatic individual with both deficiency of C1-INH and APP was detected. A kinin catabolism deficiency was detected in five out of the six patients with ACEi-AAE, normal C1-INH and no F12 gene mutation (two of ACE deficiency and three of APP associated to the SNP c.-2399C>A of the XPNPEP2 gene). Moreover, 34 patients with normal C1-INH had a kinin catabolism deficiency, with enzymatic activities below the low reference value. Of these, 10 displayed CPN, 15 APP, and 12 ACE deficiencies associated with the SNP c.-2399C>A of the XPNPEP2 gene.
Future Implications
The emerging picture for 2016 is that HAEs represent a family of diverse disorders of the contact system-kinin (mainly bradykinin) metabolism, with a much greater genetic complexity than the discovery of SERPING1 gene could offer. Recent achievements provide convincing evidences that many of the obscure issues, like the genetic damage(s) underlying U-HAE and normal C1-INH-HAE, and the detection of genetic markers of disease severity, are close to be elucidated. In order to achieve this goal, large-scale studies and the use of contemporary genetic analysis approaches, like genome-wide association studies or next-generation sequencing, are required. However, neither a detailed genetic study nor any other isolated approaches can effectively address the complexity characterizing the clinical expressions of HAE. Classifying and diagnosing patients at specific clinical risks can be reached only by using advanced tools, which will combine clinical, genetic, transcriptomic, and biochemical approaches. Precision medicine is a much promising tool towards the achievement of this goal [119]. Once a satisfactory level of predictability was reached, it will allow to introduce appropriate modifications in the current prognostic and therapeutic approach. This will certainly upgrade the prevention and timely management of the disease, reduce life-threatening conditions, and improve patients’ quality of life and minimize of the burden of the disease. Moreover, the performance of robust clinical trials for drugs under investigation, which at the moment presents great difficulties due to the peculiar nature of the disease, will be facilitated. Beyond these, the accumulated evidence indicates that, in the near future, genetics will also facilitate understanding many of the unresolved aspects of HAEs pathogenesis. In depth genetic studies will uncover disease associations with mutations in genes encoding proteins other than those apparently involved in bradykinin metabolism and function. Possible associations of this kind might give answers in unexplained issues of the disease or might suggest the implication of additional pathways in its pathogenesis. For example, more than 30 years ago it was observed that HAE is provoked by olfactory stimuli [120], while recent evidence indicates that HAE patients present an impaired sense of smell [121]. The physical closeness of SERPING1 gene span to a large olfactory receptor cluster on chromosome 11q13 [122] cannot exclude a high recombination frequency and the possibility of this olfactory region to be in linkage disequilibrium with SERPING1, with its mutations exerting functional roles on SERPING1-linked olfactory receptor cluster and vice versa [109]. Similarly, a genome-wide screen might localize high-affinity estrogen response elements (EREs) in genes encoding for proteins involved in HAE pathogenesis and shed light to the gender hormone-mediated effect that remains one of the most complex features of HAE. EREs are specific DNA sequences mediating the stimulation of target genes expression in response to estrogens. Over 70.000 EREs have been identified in human genome [123]. The F12 promoter contains at position −44/−31 a palindrome similar, but not identical, to an ERE together with four hemisite EREs between positions −1314 and −608 [124]. The presence of these elements underlies the mechanism by which estrogens enhance FXII concentrations in plasma, and possibly is associated with estrogen-mediated features of not only the FXII-HAE but, to a different degree, of all HAE types. Unknown, however, remains the role of EREs localized within a range of −10 to +5 kb from mRNA 5′-ends in SERPING1 gene as well as in other HAE-related genes, like C4B and MBL2 [125].
Diagnostic Issues
Genetic testing is not included in the first-line diagnostic approach recommended for patients with angioedema, since the vast majority of angioedema cases can be diagnosed on the basis of family history, clinical picture, and complement tests (antigenic and functional levels of C1-INH, C1q, and C4) [126]. However, these may not always indicate the appropriate form of angioedema, while genetic testing can be proven a valuable tool for improving diagnostic accuracy and, finally, the adequacy of patient care [127]. In general, genetic testing might be useful in case of high clinical suspicion of HAE where the complement tests are inconclusive. Such typical examples are doubtful cases necessitating differentiation between the hereditary and the acquired forms of angioedema. More interesting is the confirmation of diagnosis upon the first presentation of the disease in children with a positive family history of C1-INH-HAE. It has been shown that patients with early onset of the disease have more severe disease course than those with later onset [69, 70]. Moreover, since gastrointestinal symptoms are a common presenting symptom in children with C1-INH-HAE, there is a greater need for proper differential diagnosis [128]. Notwithstanding that the clinical appearance of the disease is very unusual before the second year of age, the antigenic and functional C1-INH levels are not reliable indicators of C1-INH-HAE at this age, as their reference ranges are much lower compared with the adult reference ranges [129, 130]. Therefore, the confirmation of C1-INH-HAE diagnosis before the second year of age, might be complemented by genetic analysis. Moreover, bearing in mind that missense SERPING1 mutations [4] and the carriage of the F12-46C/T functional polymorphism [10] are significantly associated with the age at C1-INH-HAE onset, genetic analysis of the offspring of an affected parent might contribute to better genetic counseling. Detection of F12 mutations is the only test to date to confirm the diagnosis of FXII-HAE. Identification of pre-symptomatic individuals in pedigrees with an established diagnosis of FXII-HAE should be a priority in order to avoid exogenous triggers, such as estrogens (e.g., oral contraceptives in young women) and avoid fatal attacks. Pregestational genetic testing for specific mutations by chorionic villous sampling or amniocentesis might be helpful in prenatal diagnosis of C1-INH-HAE or FXII-HAE. In established pregnancies, it could be considered only in cases of an affected parent carrying a known genetic defect in SERPING1 or F12 gene but has ethical implications. Since not all genetic defects of the SERPING1 gene detected in HAE patients are pathogenic, prenatal diagnosis must be considered only when the parent’s mutation is undoubtedly disease-causing [4]. Given that SERPING1 or F12 defects may result in a nonfatal and manageable disease in the offspring, it should be considerd that modern advances in therapy have significantly improved the quality of life of the patients. Prenatal diagnosis should be decided by the parents only after an appropriate genetic counseling and the considerations of benefits versus risks. Taking into account that no SERPING1 mutations were detected in 8–10 % of cases [4] and that in up to 75 % of estrogen-associated HAE no F12 mutations are found, preimplantation genetic diagnosis might be an option in families with C1-INH-HAE or FXII-HAE since it allows the selection of healthy embryos [131].
References
Cicardi M, Igarashi T, Kim MS, Frangi D, Agostoni A, Davis AE 3rd (1987) Restriction fragment length polymorphism of the C1 inhibitor gene in hereditary angioneurotic edema. J Clin Invest 80:1640–1643
Stoppa-Lyonnet D, Tosi M, Laurent J, Sobel A, Lagrue G, Meo T (1987) Altered C1 inhibitor genes in type I hereditary angioedema. N Engl J Med 317:1–6
Dewald G, Bork K (2006) Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun 343:1286–1289
Speletas M, Szilagyi A, Psarros F, Moldovan D, Magerl M, Kompoti M et al (2015) Hereditary angioedema: molecular and clinical differences among European populations. J Allergy Clin Immunol 135:570–573
Pappalardo E, Caccia S, Suffritti C, Tordai A, Zingale LC, Cicardi M (2008) Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: functional and structural correlates. Mol Immunol 45:3536–3544
Csuka D, Szilágyi Á, Farkas H (2015) Hereditary angioedema due to C1-inhibitor deficiency—from a genetic point of view. Hered Genet 4:e112
Zuraw BL, Bork K, Binkley KE, Banerji A, Christiansen SC, Castaldo A et al (2012) Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc 33(Suppl 1):145–156
Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, Caballero T, HAWK Under the Patronage of EAACI et al (2014) Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy 69:602–616
Bors A, Csuka D, Varga L, Farkas H, Tordai A, Füst G, Szilagyi A (2013) Less severe clinical manifestations in patients with hereditary angioedema with missense C1INH gene mutations. J Allergy Clin Immunol 131:1708–1711
Speletas M, Szilagyi A, Csuka D, Koutsostathis N, Psarros F, Moldovan D et al (2015) F12-46C/T polymorphism as modifier of the clinical phenotype of hereditary angioedema. Allergy 70:1661–1664
Davis AE 3rd, Whitehead AS, Harrison RA, Dauphinais A, Bruns GA, Cicardi M, Rosen FS (1986) Human inhibitor of the first component of complement, C1: characterization of cDNA clones and localization of the gene to chromosome 11. Proc Natl Acad Sci U S A 83:3161–3165
Bock SC, Skriver K, Nielsen E, Thogersen HC, Wiman B, Donaldson VH, Eddy RL, Marrinan J, Radziejewska E, Huber R et al (1986) Human C1 inhibitor: primary structure, cDNA cloning, and chromosomal localization. Biochemistry 25:4292–4301
Beinrohr L, Harmat V, Dobó J, Lörincz Z, Gál P, Závodszky P (2007) C1 inhibitor serpin domain structure reveals the likely mechanism of heparin potentiation and conformational disease. J Biol Chem 282:21100–21109
Russell JA, Whaley K, Heaphy S (1997) The sequence of a cDNA encoding functional murine C1-inhibitor protein. Biochim Biophys Acta 1352:156–160
Lener M, Vinci G, Duponchel C, Meo T, Tosi M (1998) Molecular cloning, gene structure and expression profile of mouse C1 inhibitor. Eur J Biochem 254:117–122
Carugati A, Pappalardo E, Zingale LC, Cicardi M (2001) C1-inhibitor deficiency and angioedema. Mol Immunol 38:161–173
Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN (2014) The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 133:1–9
Kalmár L, Hegedüs T, Farkas H, Nagy M, Tordai A (2005) HAEdb: a novel interactive, locus-specific mutation database for the C1 inhibitor gene. Hum Mutat 25:1–5
Amrani N, Sachs MS, Jacobsen A (2006) Early nonsense: mRNA decay solves a translational problem. Nat Rev Mol Cell Biol 7:415–425
Carter PE, Duponchel C, Tosi M, Fothergill JE (1991) Complete nucleotide sequence for the C1 inhibitor with an unusually high density of Alu elements. Eur J Biochem 197:301–308
Antonarakis SE, The Nomenclature Working Group (1998) Recommendations for a nomenclature system for human gene mutations. Hum Mutat 11:1–3
Parad RB, Kramer J, Strunk RC, Rosen FS, Davis AE (1990) Dysfunctional C1 inhibitor Ta: deletion of Lys-251 results in acquisition of an N-glycosylation site. Proc Natl Acad Sci U S A 87:6786–6790
Longhurst H, Cicardi M (2012) Hereditary angio-oedema. Lancet 379:474–481
Blanch A, Roche O, Urrutia I, Gamboa P, Fontán G, López-Trascasa M (2006) First case of homozygous C1 inhibitor deficiency. J Allergy Clin Immunol 118:1330–1335
Lopez-Lera A, Favier B, de la Cruz RM, Garrido S, Drouet C, López-Trascasa M (2010) A new case of homozygous C1-inhibitor deficiency suggests a role for Arg378 in the control of kinin pathway activation. J Allergy Clin Immunol 126:1307–1310
Bafunno V, Divella C, Sessa F, Tiscia GL, Castellano G, Gesualdo L, Margaglione M, Montinaro V (2013) De novo homozygous mutation of the C1 inhibitor gene in a patient with hereditary angioedema. J Allergy Clin Immunol 132:748–750
Yu TC, Shyur SD, Huang LH, Wen DC, Li JS (2007) Paternal mosaicism and hereditary angioedema in a Taiwanese family. Ann Allergy Asthma Immunol 99:375–379
Guarino S, Perricone C, Guarino MD, Giardina E, Gambardella S, Rosaria D’Apice M, Bulli C, Perricone R, Novelli G (2006) Gonadal mosaicism in hereditary angioedema. Clin Genet 70:83–85
Cicardi M, Igarashi T, Rosen FS, Davis AE 3rd (1987) Molecular basis for the deficiency of complement 1 inhibitor in type I hereditary angioneurotic edema. J Clin Invest 79:698–702
Quastel M, Harrison R, Cicardi M, Alper CA, Rosen FS (1983) Behavior in vivo of normal and dysfunctional C1 inhibitor in normal subjects and patients with hereditary angioneurotic edema. J Clin Invest 71:1041–1046
Kramer J, Rosen FS, Colten HR, Rajczy K, Strunk RC (1993) Transinhibition of C1 inhibitor synthesis in type I hereditary angioneurotic edema. J Clin Invest 91:1258–1262
Ernst SC, Circolo A, Davis AE 3rd, Gheesling-Mullis K, Fliesler M, Strunk RC (1996) Impaired production of both normal and mutant C1 inhibitor proteins in type I hereditary angioedema with a duplication in exon 8. J Immunol 157:405–410
Pappalardo E, Zingale LC, Cicardi M (2004) C1 inhibitor gene expression in patients with hereditary angioedema: quantitative evaluation by means of real-time RT-PCR. J Allergy Clin Immunol 114:638–644
Emonts M, de Jongh CE, Houwing-Duistermaat JJ, van Leeuwen WB, de Groot R, Verbrugh HA, Hermans PW, van Belkum A (2007) Association between nasal carriage of Staphylococcus aureus and the human complement cascade activator serine protease C1 inhibitor (C1INH) valine vs. methionine polymorphism at amino acid position 480. FEMS Immunol Med Microbiol 50:330–332
Ennis S, Jomary C, Mullins R, Cree A, Chen X, Macleod A, Jones S, Collins A, Stone E, Lotery A (2008) Association between the SERPING1 gene and age-related macular degeneration: a two-stage case–control study. Lancet 372:1828–1834
Kralovicova J, Vorechovsky I (2009) SERPING1 rs2511988 and age-related macular degeneration. Lancet 373:461–462
Lu F, Zhao P, Fan Y, Tang S, Hu J, Liu X et al (2010) An association study of SERPING1 gene and age-related macular degeneration in a Han Chinese population. Mol Vis 16:1–6
Nakata I, Yamashiro K, Yamada R, Gotoh N, Nakanishi H, Hayashi H et al (2011) Association between the SERPING1 gene and age-related macular degeneration and polypoidal choroidal vasculopathy in Japanese. PLoS One 6:e19108
Zahedi R, Wisnieski J, Davies AE 3rd (1997) Role of the P2 residue of complement 1 inhibitor (Ala443) in determination of target protease specificity: inhibition of complement and contact system proteases. J Immunol 159:983–988
Pappalardo E, Cicardi M, Duponchel C, Carugati A, Choquet S, Agostoni A, Tosi M (2000) Frequent de novo mutations and exon deletions in the C1 inhibitor gene of patients with angioedema. J Allergy Clin Immunol 106:1147–1154
Pappalardo E, Zingale LC, Terlizzi A, Zanichelli A, Folcioni A, Cicardi M (2002) Mechanisms of C1-inhibitor deficiency. Immunobiology 205:542–551
Cicardi M, Zingale L, Zanichelli A, Pappalardo E, Cicardi B (2005) C1 inhibitor: molecular and clinical aspects. Springer Semin Immunopathol 27:286–298
Tosi M, Duponchel C, Bourgarel P, Colomb M, Meo T (1986) Molecular cloning of human C1 inhibitor: sequence homologies with α1-antitrypsin and other members of the serpins superfamily. Gene 42:265–272
Stoppa-Lyonnet D, Carter PE, Meo T, Tosi M (1990) Clusters of intragenic Alu repeats predispose the human C1 inhibitor locus to deleterious rearrangements. Proc Natl Acad Sci U S A 87:1551–1555
Skriver K, Radziejewska E, Silbermann JA, Donaldson VH, Bock SC (1989) CpG mutations in the reactive site of human C1 inhibitor. J Biol Chem 264:3066–3071
Roche O, Blanch A, Duponchel C, Fontan G, Tosi M, Lopez-Trascasa M (2005) Hereditary angioedema: the mutation spectrum of SERPING1/C1NH in a large Spanish cohort. Hum Mutat 26:135–44
Speletas M, Boukas K, Papadopoulou-Alataki E, Tsitsami E, Germenis AE (2009) Hereditary angioedema in Greek families caused by novel and recurrent mutations. Hum Immunol 70:925–929
Davies MJ, Lomas DA (2008) The molecular aetiology of the serpinopathies. Int J Biochem Cell Biol 40:1273–1286
Eldering E, Verpy E, Roem D, Meo T, Tosi M (1995) COOH-terminal substitutions in the serpin C1 inhibitor that cause loop overinsertion and subsequent multimerization. J Biol Chem 270:2579–2587
Aulak KS, Eldering E, Hack CE, Lubbers YP, Harrison RA, Mast A, Cicardi M, Davis AE 3rd (1993) A hinge region mutation in C1-inhibitor (Ala436→Thr) results in nonsubstrate-like behavior and in polymerization of the molecule. J Biol Chem 268:18088–18094
Madsen DE, Hansen S, Gram J, Bygum A, Drouet C, Sidelmann JJ (2014) Presence of C1-inhibitor polymers in a subset of patients suffering from hereditary angioedema. PLoS One 9:e112051
Maas C, Govers-Riemslag JW, Bouma B, Schiks B, Hazenberg BP, Lokhorst HM et al (2008) Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest 118:3208–3218
Cugno M, Zanichelli A, Foieni F, Caccia S, Cicardi M (2009) C1-inhibitor deficiency and angioedema: molecular mechanisms and clinical progress. Trends Mol Med 15:69–78
Ghannam A, Sellier P, Defendi F, Favier B, Charignon D, López-Lera A, López-Trascasa M, Ponard D, Drouet C (2015) C1 inhibitor function using contact-phase proteases as target: evaluation of an innovative assay. Allergy 70:1103–1111
Duponchel C, Djenouhat K, Frémeaux-Bacchi V, Monnier N, Drouet C, Tosi M (2006) Functional analysis of splicing mutations and of an exon 2 polymorphic variant of SERPING1/C1NH. Hum Mutat 27:295–296
Roche O, Blanch A, Caballero T, Sastre N, Callejo D, López-Trascasa M (2005) Hereditary angioedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain. Ann Allergy Asthma Immunol 94:498–503
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G (2002) Relative quantifications of 40 nucleic acid sequences by multiplex ligation dependent probe amplification. Nucleic Acids Res 30:e57
Kang HR, Yim EY, Oh SY, Chang YS, Kim YK, Cho SH, Min KU, Kim YY (2006) Normal C1 inhibitor mRNA expression level in type I hereditary angioedema patients: newly found C1 inhibitor gene mutations. Allergy 61:260–264
Colobran R, Pujol-Borrell R, Hernández-González M, Guilarte M (2014) A novel splice site mutation in the SERPING1 gene leads to haploinsufficiency by complete degradation of the mutant allele mRNA in a case of familial hereditary angioedema. J Clin Immunol 34:521–523
Bork K, Davis-Lorton M (2013) Overview of hereditary angioedema caused by C1-inhibitor deficiency: assessment and clinical management. Eur Ann Allergy Clin Immunol 45:7–16
Zuraw BL (2008) Hereditary angioedema. N Engl J Med 359:1027–1036
Davis AE 3rd (2008) Hereditary angioedema: a current state-of-the-art review, III: mechanisms of hereditary angioedema. Ann Allergy Asthma Immunol 100:S7–S12
Cardarelli W (2013) Managed care implications of hereditary angioedema. Am J Manag Care 19(7 Suppl):s119–24
Kramer J, Rosen FS, Colten HR, Kramer J, Rosen FS, Colten HR (1993) Transinhibition of C1 inhibitor synthesis in type I hereditary angioneurotic edema. J Clin Invest 91:1258–1262
Verpy E, Biasotto M, Brai M, Misiano G, Meo T, Tosi M (1996) Exhaustive mutation scanning by fluorescence-assisted mismatch analysis discloses new genotype-phenotype correlations in angiodema. Am J Hum Genet 59:308–319
Agostoni A, Aygören-Pürsün E, Binkley KE, Blanch A, Bork K, Bouillet L et al (2004) Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol 114(3 Suppl):S51–S131
Xu YY, Zhi YX, Yin J, Wang LL, Wen LP, Gu JQ, Guan K, Craig T, Zhang HY (2012) Mutational spectrum and geno-phenotype correlation in Chinese families with hereditary angioedema. Allergy 67:1430–1436
Bafunno V, Bova M, Loffredo S, Divella C, Petraroli A, Marone G, Montinaro V, Margaglione M, Triggiani M (2014) Mutational spectrum of the C1 inhibitor gene in a cohort of Italian patients with hereditary angioedema: description of nine novel mutations. Ann Hum Genet 78:73–82
Farkas H (2010) Pediatric hereditary angioedema due to C1-inhibitor deficiency. Allergy, Asthma Clin Immunol 6:18
Bork K, Meng G, Staubach P, Hardt J (2006) Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 119:267–274
Andrejević S, Korošec P, Šilar M, Košnik M, Mijanović R, Bonači-Nikolić B, Rijavec M (2015) Hereditary angioedema due to C1 inhibitor deficiency 1 in Serbia: two novel mutations and evidence of genotype-phenotype correlation. PLoS One 10(11): e0142174
Kalmár L, Bors A, Farkas H, Vas S, Fandl B, Varga L, Füst G, Tordai A (2003) Mutation screening of the C1 inhibitor gene among Hungarian patients with hereditary angioedema. Hum Mutat 22:498
Martinho A, Mendes J, Simões O, Nunes R, Gomes J, Dias Castro E et al (2013) Mutations analysis of C1 inhibitor coding sequence gene among Portuguese patients with hereditary angioedema. Mol Immunol 53:431–434
Cumming SA, Halsall DJ, Ewan PW, Lomas DA (2003) The effect of sequence variations within the coding region of the C1 inhibitor gene on disease expression and protein function in families with hereditary angiooedema. J Med Genet 40:e114
Bygum A, Fagerberg CR, Ponard D, Monnier N, Lunardi J, Drouet C (2011) Mutational spectrum and phenotypes in Danish families with hereditary angioedema because of C1 inhibitor deficiency. Allergy 66:76–84
Blaskó B, Széplaki G, Varga L, Ronai Z, Prohászka Z, Sasvari-Szekely M, Visy B, Farkas H, Füst G (2007) Relationship between copy number of genes (C4A, C4B) encoding the fourth component of complement and the clinical course of hereditary angioedema (HAE). Mol Immunol 44:2667–2674
Lung CC, Chan EK, Zuraw BL (1997) Analysis of an exon 1 polymorphism of the B2 bradykinin receptor gene and its transcript in normal subjects and patients with C1 inhibitor deficiency. J Allergy Clin Immunol 99:134–146
Freiberger T, Vyskocilová M, Kolárová L, Kuklínek P, Krystůfková O, Lahodná M, Hanzlíková J, Litzman J (2002) Exon 1 polymorphism of the B2BKR gene does not influence the clinical status of patients with hereditary angioedema. Hum Immunol 63:492–494
Freiberger T, Grombiříková H, Ravčuková B, Jarkovský J, Kuklínek P, Kryštůfková O, Hanzlíková J et al (2011) No evidence for linkage between the hereditary angiooedema clinical phenotype and the BDKR1, BDKR2, ACE or MBL2 gene. Scand J Immunol 74:100–106
Kaplan AP, Joseph K (2014) Pathogenic mechanisms of bradykinin mediated diseases: dysregulation of an innate inflammatory pathway. Adv Immunol 121:41–89
Kanaji T, Okamura T, Osaki K, Kuroiwa M, Shimoda K, Hamasaki N, Niho Y (1998) A common genetic polymorphism (46 C to T substitution) in the 5′-untranslated region of the coagulation factor XII gene is associated with low translation efficiency and decrease in plasma factor XII level. Blood 91:2010–2014
Bork K, Barnstedt S, Koch P, Traupe H (2000) Hereditary angioedema with normal C1-inhibitor activity in women. Lancet 356:213–217
Binkley K, Davis AE III (2000) Clinical, biochemical, and genetic characterization of a novel estrogen-dependent inherited form of angioedema. J Allergy Clin Immunol 106:546–550
Riedl MA (2013) Hereditary angioedema with normal C1-INH (HAE type III). J Allergy Clin Immunol Pract 1:427–432
Royle NJ, Nigli M, Cool D, MacGillivray RT, Hamerton JL (1988) Structural gene encoding human factor XII is located at 5q33-qter. Somat Cell Mol Genet 14:217–221
Schloesser M, Zeerleder S, Lutze G, Halbmayer WM, Hofferbert S, Hinney B et al (1997) Mutations in the human factor XII gene. Blood 90:3967–3977
Bork K, Wulff K, Meinke P, Wagner N, Hardt J, Witzke G (2011) A novel mutation in the coagulation factor 12 gene in subjects with hereditary angioedema and normal C1-inhibitor. Clin Immunol 141:31–35
Bork K, Wulff K, Hardt J, Witzke G, Lohse P (2014) Characterization of a partial exon 9/intron 9 deletion in the coagulation factor XII gene (F12) detected in two Turkish families with hereditary angioedema and normal C1 inhibitor. Haemophilia 20:e372–e375
Kiss N, Barabas E, Varnai K, Halász A, Varga LÁ, Prohászka Z, Farkas H, Szilágyi Á (2013) Novel duplication in the F12 gene in a patient with recurrent angioedema. Clin Immunol 149:142–145
Moreno AS, Valle SO, Levy S, França AT, Serpa FS, Arcuri HA, Palma MS et al (2015) Coagulation factor XII gene mutation in Brazilian families with hereditary angioedema with normal C1 inhibitor. Int Arch Allergy Immunol 166:114–120
Cichon S, Martin L, Hennies HC, Müller F, Van Driessche K, Karpushova A, Stevens W et al (2006) Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am J Hum Genet 79:1098–1104
Bork K, Kleist R, Hardt J, Witzke G (2009) Kallikrein-kinin system and fibrinolysis in hereditary angioedema due to factor XII gene mutation Thr309Lys. Blood Coagul Fibrinolysis 20:325–332
Bouillet L, Boccon-Gibod I, Ponard D, Drouet C, Cesbron JY, Dumestre-Perard C, Monnier N, Lunardi J, Massot C, Gompel A (2009) Bradykinin receptor 2 antagonist (icatibant) for hereditary angioedema type III attacks. Ann Allergy Asthma Immunol 103:448
Bork K, Wulff K, Witzke G, Stanger C, Lohse P, Hardt J (2013) Antihistamine-resistant angioedema in women with negative family history: estrogens and F12 gene mutations. Am J Med 126:1142.e9–1142.e14
Gelincik A, Demir S, Olgaç M, Karaman V, Toksoy G, Çolakoğlu B, Büyüköztürk S, Uyguner ZO (2015) Idiopathic angioedema with F12 mutation: is it a new entity? Ann Allergy Asthma Immunol 114:154–156
Hoover T, Lippmann M, Grouzmann E, Marceau F, Herscu P (2010) Angiotensin converting enzyme inhibitor induced angio-oedema: a review of the pathophysiology and risk factors. Clin Exp Allergy 40:50–61
Duan QL, Nikpoor B, Dube MP, Molinaro G, Meijer IA, Dion P, Rochefort D et al (2005) A variant in XPNPEP2 is associated with angioedema induced by angiotensin I-converting enzyme inhibitors. Am J Hum Genet 77:617–26
Woodard-Grice AV, Lucisano AC, Byrd JB, Stone ER, Simmons WH, Brown NJ (2010) Sex-dependent and race-dependent association of XPNPEP2 C-2399A polymorphism with angiotensin-converting enzyme inhibitor-associated angioedema. Pharmacogenet Genomics 20:532–536
Cilia La Corte AL, Carter AM, Rice GI, Duan QL, Rouleau GA, Adam A, Grant PJ, Hooper NM (2011) A functional XPNPEP2 promoter haplotype leads to reduced plasma aminopeptidase P and increased risk of ACE inhibitor-induced angioedema. Hum Mutat 32:1326–13231
Moholisa RR, Rayner BR, Patricia Owen E, Schwager SL, Stark JS, Badri M, Cupido CL, Sturrock ED (2013) Association of B2 receptor polymorphisms and ACE activity with ACE inhibitor-induced angioedema in black and mixed-race South Africans. J Clin Hypertens 15:413–419
Van Guilder GP, Pretorius M, Luther JM, Byrd JB, Hill K, Gainer JV, Brown NJ (2008) Bradykinin type 2 receptor BE1 genotype influences bradykinin-dependent vasodilation during angiotensin-converting enzyme inhibition. Hypertension 51:454–459
Pare G, Kubo M, Byrd JB, McCarty CA, Woodard-Grice A, Teo KK, Anand SS et al (2013) Genetic variants associated with angiotensin-converting enzyme inhibitor-associated angioedema. Pharmacogenet Genomics 23:470–478
Roques BP, Noble F, Dauge V, Fournie-Zaluski MC, Beaumont A (1993) Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev 45:87–146
Salek-Ardakani S, So T, Halteman BS, Altman A, Croft M (2004) Differential regulation of Th2 and Th1 lung inflammatory responses by protein kinase C theta. J Immunol 173:6440–6447
Bohlander SK (2005) ETV6: a versatile player in leukemogenesis. Semin Cancer Biol 15:162–174
Boily G, Larose J, Langlois S, Sinnett D (2007) Identification of transcripts modulated by ETV6 expression. Br J Haematol 136:48–62
Sakurai T, Yamada T, Kihara-Negishi F, Teramoto S, Sato Y, Izawa T, Oikawa T (2003) Effects of overexpression of the Ets family transcription factor TEL on cell growth and differentiation of K562 cells. Int J Oncol 22:1327–1333
Bocsi J, Richter M, Hambsch J, Barten MJ, Dahnert I, Schneider P, Tárnok A (2006) Transient Th1/Th2 disbalance indicates postoperative effusions and edema after cardiopulmonary bypass in children. Cytometry A 69:165–168
Triggianese P, Chimenti MS, Toubi E, Ballanti E, Guarino MD, Perricone C, Perricone R (2015) The autoimmune side of hereditary angioedema: insights on the pathogenesis. Autoimmun Rev 14:665–669
Triggianese P, Guarino MD, Ballanti E, Chimenti MS, Perricone R (2014) Hereditary angioedema and autoimmunity. Isr Med Assoc J 16:622–624
Salemi M, Mandalà V, Muggeo V, Misiano G, Milano S, Colonna-Romano G, Arcoleo F, Cillari E (2015) Growth factors and IL-17 in hereditary angioedema. Clin Exp Med
Matthews KW, Mueller-Ortiz SL, Wetsel RA (2004) Carboxypeptidase N: a pleiotropic regulator of inflammation. Mol Immunol 40:785–793
Mathews KP, Pan PM, Gardner NJ, Hugli TE (1980) Familial carboxypeptidase N deficiency. Ann Intern Med 93:443–445
Mathews KP, Curd JG, Hugli TE (1986) Decreased synthesis of serum carboxypeptidase N (SCPN) in familial SCPN deficiency. J Clin Immunol 6:87–91
Mathews KP (1986) Deficiencies in regulator proteins 4. Anaphylatoxin inactivator. Prog Allergy 39:344–351
Cao H, Hegele RA (2003) DNA polymorphism and mutations in CPN1, including the genomic basis of carboxypeptidase N deficiency. J Hum Genet 48:20–22
Willemse JL, Chen D, Hendriks DF (2008) Major carboxypeptidase N deficiency. Clin Chim Acta 389:181–182
Dessart P, Defendi F, Humeau H, Nicolie B, Sarre ME, Charignon D, Ponard D, Cichon S, Drouet C, Martin L (2015) Distinct conditions support a novel classification for bradykinin-mediated angio-oedema. Dermatology 230:324–331
Reardon S (2015) Precision-medicine plan raises hopes. Nature 517:540
Tannenbaum H (1982) Angioedema provoked by olfactory stimuli. Can Med Assoc J 127:735–736
Perricone C, Agmon-Levin N, Shoenfeld N, de Carolis C, Guarino MD et al (2011) Evidence of impaired sense of smell in hereditary angioedema. Allergy 66:149–154
Buettner JA, Glusman G, Ben-Arie N, Ramos P, Lancet D, Evans GA (1998) Organization and evolution of olfactory receptor genes on human chromosome 11. Genomics 53:56–68
O’Lone R, Frith MC, Karlsson EK, Hansen U (2004) Genomic targets of nuclear estrogen receptors. Mol Endocrinol 18:1859–1875
Farsetti A, Misiti S, Citarella F, Farsetti A, Pontecorvi A, Fantoni A (1995) Molecular basis of estrogen regulation of Hageman factor XII gene expression. Endocrinology 136:5076–5083
Bourdeau V, Deschênes J, Metivier R, Nagai Y, Nguyen D, Hudson T, White J, Gannon F, Mader S (2004) Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol Endocrinol 18:1411–1427
Craig TJ, Bernstein JA, Farkas H, Bouillet L, Boccon-Gibod I (2014) Diagnosis and treatment of bradykinin-mediated angioedema: outcomes from an angioedema expert consensus meeting. Int Arch Allergy Immunol 165:119–127
Weiler CR, van Dellen RG (2006) Genetic test indications and interpretations in patients with hereditary angioedema. Mayo Clin Proc 81:958–972
Caballero T (2013) Angio-oedema due to hereditary C1 inhibitor deficiency in children. Allergol Immunopathol (Madr) 41:45–53
Nielsen EW, Johansen HT, Holt J, Mollnes TE (1994) C1 inhibitor and diagnosis of hereditary angioedema in newborns. Pediatr Res 35:184–187
Roach B, Kim Y, Jerome E, Michael AF (1981) Influence of age and sex on serum complement components in children. Am J Dis Child 135:918–920
Bautista-Llácer R, Alberola TM, Vendrell X, Fernández E, Pérez-Alonso M (2010) Case report: first successful application of preimplantation genetic diagnosis for hereditary angiooedema. Reprod Biomed Online 21:658–662
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Germenis, A.E., Speletas, M. Genetics of Hereditary Angioedema Revisited. Clinic Rev Allerg Immunol 51, 170–182 (2016). https://doi.org/10.1007/s12016-016-8543-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-016-8543-x