
Abstract
Innate immunity is a critical partner in the regulation of inflammation and some mutations in genes implied in innate immunity pathways can cause genetic disorders characterized by seemingly unprovoked self-limited inflammatory attacks. These rare conditions are collectively named “hereditary periodic fever syndromes” (HPFS), and protean pathogenetic mechanisms combined with several clinical phenotypes characterize at least four distinct conditions: (1) familial Mediterranean fever, which is the prototype and the most widely recognized among HPFS, inherited as an autosomal recessive disorder showing recurrent dysregulated inflammatory processes, caused by an abnormal interaction between cytoskeleton and inflammasome, a key-signaling platform that releases interleukin-1β (IL-1β); (2) the group of cryopyrin-associated periodic syndrome, which upsets directly the production of IL-1β, with a dominant pattern of inheritance; (3) tumor necrosis factor receptor-associated periodic syndrome, which is an autosomal dominant disorder subverting the functions and traffic of a cell membrane protein; and (4) mevalonate kinase deficiency, which is an autosomal recessive metabolic disorder halting the biosynthesis of cholesterol. MEFV, NLRP3, TNFRSF1A, and MVK are respectively the four causing genes of these conditions, all resulting in excessive IL-1β signaling, though the encoded proteins act at different levels in cytoskeletal filament organization, apoptosis, and activation of the IL-1β-structured inflammasome. The differential diagnosis of HPFS can be challenging, as there are no universally accepted diagnostic algorithms, and near half of patients may have a specific disease without any genetic pathogenetic variant identified. Herein, we outline the most relevant aspects of HPFS at the crossroads between clinical medicine and immunology and all the most recent advances in their treatment, as the increasing use of IL-1 antagonists has achieved unexpected clinical results in a large number of patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the appearance of the first eukaryotic cells, different host responses have evolved in order to secure cellular integrity, homeostasis, and survival against invading pathogens. The basic physiological reactions in virtually all animals are carried out by innate immunity cells: their crucial importance is highlighted by the fact that greater than 95 % of the animal kingdom does not have T or B cells and yet displays potent resistance to pathogens [1]. Even when T and B cells appeared in vertebrates, their adaptive responses remained dependant on macrophage-like cells, which enhanced their ability to recognize pathogens through different kinds of receptors, and finally act as specialized killers [2, 3]. The complex circuitry of immune cells, like macrophages, dendritic cells, natural killer cells, and neutrophils, can promote inflammation that is fundamental for protection against pathogens, but in parallel it has also been implicated in the pathogenesis of rare disorders characterized by seemingly unprovoked and self-limited bouts of inflammation, called hereditary periodic fever syndromes (HPFS): the regulatory mechanisms of innate immunity are subverted in this family of diseases, all characterized by the huge release of proinflammatory signaling proteins, such as interleukin-1β (IL-1β) [4]. The term “autoinflammatory” was coined to underscore the absence of pathogens, circulating autoantibodies, or self-reactive T cells in HPFS [5]. To date, nearly all mutations that have been linked to HPFS disrupt inflammatory signaling pathways within the innate immune system: this disruption generates a vast spectrum of systemic inflammatory signs affecting multiple organs, which derive by the activation of the inflammasome, a multimeric cytosolic protein complex regulating the proteolytic processing of proIL-1β into the biologically active IL-1β [6]. Figure 1 reveals that the inflammasome is vital in the innate immunity, showing its activation in macrophages and dendritic cells: the oversecretion of IL-1β by the inflammasome plays a role in all HPFS. IL-1β, secreted by stimulated monocytes, macrophages, and dendritic cells, and to a lesser degree by several other cell types, including neutrophils, keratinocytes, epithelial or endothelial cells, smooth muscle cells, and fibroblasts, is considered the eldest multi-functional proinflammatory cytokine with protean effects in nearly all body organs, either alone or in combination with other chemokines [7]. Since its cloning in the early 1980s [8], the discovery of IL-1β heterogeneous biological activities has significantly increased our understanding of the inner mechanism of many diseases, including HPFS. ProIL-1β is biologically inactive and must be converted to the 17-kDa IL-1β in order to function, under a specific inflammasome-mediated mechanism involving caspase 1 and a tight feedback-control operated by several naturally occurring inhibitors, such as IL-1 receptor antagonist (IL-1Ra) and other soluble receptors [9]. The most appropriate treatment for HPFS must be tailored to the single patient, based on the severity of clinical phenotypes, which can vary greatly, and during the last two decades several IL-1-targeting agents have been developed. Nevertheless, organ damage in terms of hearing loss, blindness, kidney failure, joint restriction, osteoporosis, infertility, growth failure, pubertal delay, cognitive impairment, or serosal scarring may occur if treatment is not provided in due time [10].

The inflammasome is a vital player in the innate immunity, and the activation of NLRP3-inflammasome in macrophages and dendritic cells has been extensively studied. The NLRP3 protein belongs to the family of nucleotide-binding and oligomerization domain-like receptors (NLRs) and is unique among the NLR family members, as it contains a C-terminal extension composed of two distinct domains: a FIIND domain and a caspase activation/recruitment domain (CARD). All the NLR family members have similar structures with a pyrin domain (PYD) and a CARD, combined with a C-terminal ligand binding leucine-rich repeat domain (LRR) and a central nucleotide-binding and oligomerization domain (NACHT). Caspase-1, formerly called interleukin-1β (IL-1β) converting enzyme, is synthesized as an inactive zymogen (pro-caspase-1) that undergoes autocatalytic processing after an appropriate trigger, such as different pathogen-associated molecular patterns (PAMPs), i.e., liposaccharides, peptidoglycans, bacterial nucleic acids, or damage-associated molecular pattern molecules (DAMPs), i.e., crystals of monosodium urate, mitochondrial DNA, S100 proteins, etc. In the absence of immune activators, an internal interaction occurs between the NACHT domain and LRRs, suppressing the interplay between NLRP3 and ASC (apoptosis-associated speck-like protein containing a C-terminal CARD), and preventing the inflammasome assembly. The second step of inflammasome activation is the oligomerization and subsequent assembly of NLRP3, its interaction with ASC and Cardinal protein (CARD inhibitor of NFκB-activating ligands), and recruitment/activation of pro-caspase-1 into this protein complex. The final result of NLRP3 activation is the secretion of mature and active IL-1β after direct cleavage operated by caspase-1 of proIL-1β
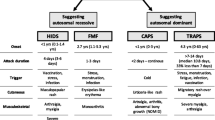
This review depicts four HPFS, in which a genetically mediated excessive production of IL-1β is variably linked to their pathogenesis, explaining why the use of IL-1 blockade is consistently associated with the resolution of their heterogeneous inflammatory manifestations. Table 1 provides a general overview of these disorders.
Familial Mediterranean Fever
Familial Mediterranean fever (FMF) is the most common among HPFS worldwide, an autosomal recessive inherited disease caused by loss-of-function mutations in the MEFV gene, which mostly affects Eastern Mediterranean populations, i.e. Turkish, Armenian, Arabian, and Sephardic Jewish people [11]. Pyrin is the protein of 781 amino acids encoded by the MEFV gene, expressed primarily in the myeloid cell lineage, which modulates cell susceptibility to apoptosis through the regulation of inflammation and specifically IL-1β production [12]. How the mutated variants of pyrin affect apoptotic processes and lead to an inappropriate or prolonged inflammatory response are still incompletely understood. More than 300 variants throughout the MEFV gene have been identified, with M694V, M680I, and E148Q as the most common [13]. The membranous synovial and serosal linings are primary targets of the inflammatory cascade in FMF and explain articular, thoracic, and abdominal pain displayed by the majority of patients: children and adults usually experience self-limited attacks characterized by 1-to-3-days’ duration episodes of fever with joint or serosal inflammation, which recur approximately every month. Severe abdominal and chest pain are the most typical symptoms occurring in more than 90 and 40 % of patients, respectively. Suggestive for FMF diagnosis is the presence of recurrent erysipelas-like rash on the skin of inferior limbs [14]. Patients with a classic phenotype who have also been genetically confirmed to have MEFV mutations are defined as phenotype I, and those who have no clinical signs but the required genotype are referred to as phenotype III patients, while phenotype II is when patients develop AA amyloidosis without any previous attacks typical of FMF [15]. The development of systemic amyloidosis, due to the deposition of a cleavage product, serum amyloid A (SAA), one of the acute reactants produced during disease flares, is the ominous long-term complication of FMF: the products of amyloidogenesis can be stored in different organs, mostly in kidney, and symptomatic amyloidosis clinically affecting one or more organs, confirmed by examination of tissue sections by Congo red dye, is still observed in a relevant subset of patients [16]. Diagnosis of FMF remains clinical and requires information about family history and response to colchicine, an alkaloid derived from several members of the lily plant family, including meadow saffron, since specific laboratory tests of confirmation are not available [17]. According to the so-called Tel Hashomer criteria, a definitive diagnosis of FMF requires the presence of two major criteria (recurrent febrile episodes accompanied by serositis, presence of AA amyloidosis, or favorable response to colchicine) or one major and two minor criteria (recurrent febrile episodes, erysipelas-like skin rash, and FMF in a first-degree relative); genetic diagnosis can be confirmed by the presence of two mutations in the MEFV gene, but also heterozygous carriers can show an incomplete and even typical FMF expression [18]. In fact, FMF has been traditionally considered an autosomal recessive disease, but many heterozygous patients were noted to display recurrent subclinical inflammatory processes, and some reports of families with seemingly dominant inheritance have been published, evoking the hypothesis that FMF is not fully recessive [19]. The detection of a single heterozygous mutation, in the presence of clear FMF symptoms, may be a sufficient prerequisite for a colchicine trial, as a full response to colchicine is a diagnostic requisite [20].
Tumor Necrosis Factor Receptor-Associated Periodic Syndrome
Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) was originally described in a family of Irish and Scottish pedigree in which longlasting fevers, skin rashes, and muscular and abdominal pain recurred: it was initially thought to be a tumor necrosis factor (TNF) receptor-mediated disease, resulting from the failure of the TNF receptor to insert itself into cell membranes [21]. The genetic basis of this autosomal dominant condition was discovered later and associated with the TNF receptor 1, encoded by the TNFRSF1A gene [22]. TRAPS is caused by TNFRSF1A missense mutations, which generate dysfunctional proteins and numerous subsequent pathogenetic mechanisms, such as impaired TNF receptor shedding, defective intracellular TNF receptor trafficking to the cell surface, and subverted TNF-independent cell activation with increased production of IL-1 and reactive oxygen species [23, 24]. The molecular link between TRAPS and IL-1 is not yet clear: the pathogenesis of clinical pictures may vary with each mutation, but it is possible that IL-1 might act as a TNF-downstream proinflammatory cytokine, or that aggregates of misfolded TNF receptors stimulate intracellular signals resulting in enhanced production of IL-1 and other chemokines [25]. TRAPS phenotype is characterized by week-long febrile episodes, differently from the other HPFS, combined with skin, muscular, joint, abdominal, and ocular manifestations, which occur spontaneously or after trivial triggers [26]. Molecular analysis is required for diagnosis of TRAPS and prognosis is related to the risk of renal amyloidosis, which can be observed in 25 % of patients carrying specific TNFRSF1A mutations [27]. A positive family history for recurrent pericarditis and a poor response to colchicine may represent striking clues suggesting the presence of a TNFRSF1A mutation in patients with idiopathic recurrent acute pericarditis [28]. More than 140 TNFRSF1A variants have been recognized and associated with protean TRAPS clinical phenotypes, while low penetrance variants, as P46L and R92Q, are characterized by lower risk of amyloidosis and milder symptoms or shorter duration of attacks [29].
Cryopyrin-Associated Periodic Syndrome
The cryopyrin-associated periodic syndrome (CAPS) is a heterogeneous group of diseases characterized by excessive IL-1β release, resulting in severe systemic and organ inflammation, including familial cold autoinflammatory syndrome, Muckle-Wells syndrome, and neonatal onset multisystem inflammatory disorder (NOMID, also called chronic infantile neurologic cutaneous articular syndrome or CINCA syndrome), which are inherited as autosomal dominant conditions. These diseases usually start in childhood and were originally thought to be distinct clinical entities, but they are actually part of a spectrum of symptoms with increasing severity [30]. In general terms, CAPS is caused by at least 170 gain-of-function mutations in the NLRP3 gene, which encodes a key component of the inflammasome (see Fig. 1) that regulates the secretion of IL-1β, called cryopyrin (or NLRP3) [31]: all three conditions are caused by mutations in the same gene, though certain mutations result in a specific clinical syndrome, and others have been identified in more than one clinical picture, even within the same family, suggesting the existence of additional influences, either genetic or environmental, that might affect clinical presentations, probably through the loss of a regulatory step in the NLRP3 inflammasome activation with increased caspase-1 activation and excessive release of IL-1β. Moreover, somatic mutations, occurring during fetal development, can be found in a relevant number of apparently mutation-negative cases of CAPS patients [32]. The phenotype is clinically easy to recognize, including inflammation of the skin, joints, eyes, bones, and meninges, in combination with persistent elevation of acute phase reactants and increased white blood cell count. Common for the three conditions are recurrent outbreaks of systemic inflammation with fever, urticaria-like rashes a few hours after generalized cold exposure, joint pains, and fatigue. At the more severe point of the spectrum of CAPS, there are further symptoms and signs, such as hearing loss, hypertrophic arthropathy with typical bony overgrowth of the epiphysis of long bones and patella, aseptic chronic meningitis, elevated intracranial pressure with ventriculomegaly, mental and growth retardation [33–35]. The most evident findings in NOMID are cartilage abnormalities with epiphyseal modifications: synovial fluid in the joints of NOMID patients is sterile and contains predominantly polymorphonuclear cells. These patients also display dysmorphic features, such as prominent forehead, large head, saddleback nose, and midface hypoplasia, which cause a sibling-like resemblance among patients [36].
Mevalonate Kinase Deficiency
Mevalonate kinase deficiency (MKD) is a rare autosomal recessive metabolic disease caused by mutations in the MVK gene, leading to subverted enzymatic activity of mevalonate kinase, occurring worldwide, though most reported patients were born in Netherlands and Italy [37]: the disease originates from disrupted cholesterol biosynthesis and decreased production of unsaturated lipid chains, known as nonsterol isoprenoids, in particular geranylgeranyl groups, which have been shown to increase the release of IL-1β [38].
Most MVK mutations impair the stability of the mevalonate kinase enzyme, but not its catalytic activity, justifying the role that environmental factors might exert on the disease expression, which ranges from the so-called hyper-IgD syndrome, distinguished by monthly inflammatory attacks, to “mevalonic aciduria”, which in addition to inflammatory symptoms is characterized by psychomotor retardation, dysmorphic features, cerebellar ataxia, cataracts, hyperbilirubinaemia, and failure to thrive [39]. The classic MKD phenotype appears when the residual enzymatic activity is less than 10 % of normal, while neurological signs occur when enzymatic activity is nearly absent [40]. As many physicians are still not familiar with this disease, the diagnostic delay in MKD is currently more than 7 years [41]. The disease starts inevitably in the first months of life with febrile attacks every month, lasting 4–7 days, combined with heterogeneous skin signs, joint pain, and severe gastrointestinal complaint, sometimes provoked by childhood vaccinations or common infections: a typical febrile attack might also mimic many infectious and rheumatologic disorders of childhood [42]. Highly characteristic of MKD is the recurring course of febrile episodes which are associated with increased urinary excretion of mevalonic acid (only during fever spikes), while serum IgD elevation can be observed in any phase of the disease. However, the measurement of IgD is not a reliable method to diagnose MKD, as 28 % of patients do not have elevated IgD levels [43]. In most patients, the clinical expression of MKD tends to change with increasing age, and a decrease in attack frequency is often observed over time. Macrophage activation syndrome related to upregulated proinflammatory cytokine production and requiring intensive care support has been also reported during the disease course [44]. Due to the lack of clinical criteria, diagnosis of MKD is based on the identification of two pathogenic MVK mutations or by detection of decreased enzyme activity combined with increased excretion of mevalonic acid in urine collected during a febrile attack. In addition, the combination of early age of clinical onset, severity of abdominal complaint, presence of diarrhea, enlarged lymph nodes, and splenomegaly during febrile flares is openly contributive for a definite diagnosis [45].
Clues for the Management of Hereditary Periodic Fever Syndromes
Daily colchicine is the standard therapy for the prevention of acute FMF attacks and also amyloid A-associated amyloidosis: colchicine reduces FMF attack frequency, decreases attack severity, and shortens the overall duration of febrile attacks in most patients; it can also prevent, halt, and even reverse amyloidosis, which has been associated with the M694V MEFV mutation and SAA1.1/SAA1.1 genotype [46]. Treatment should not be aimed at the prevention of attacks, but to decrease chronic subclinical inflammation and its complications: therefore, annual physical examination with the periodic dosage of SAA is highly recommended in all FMF patients [47]. The optimal colchicine dosing regimens and time of introduction remain less clear: dosage ranges up from 0.05 mg/kg/body weight/day to a maximum of 3 mg/day and must be taken regularly on a life-long basis. A favorable response to colchicine is one of the Tel Hashomer major criteria that supports the diagnosis of FMF: nonresponders to colchicine are rare and should be distinguished from patients treated with insufficient dosages or those with poor compliance. Resistance to colchicine, defined by the lack of clinical and laboratory response, is observed in only 5–10 % of patients, being revealed by high SAA levels in attack-free periods [48]. Colchicine prophylactic role on the recurrence of FMF attacks was discovered serendipitously; however, alternative medications, such as IL-1 blockers, have now shown to be highly effective in colchicine-poor responders after the discovery that mutated pyrin leads to an increase in caspase-1 activity and subsequent increased IL-1 release [49]. Many reports have shown the brilliant efficacy in terms of reduction of attack frequency and SAA normalization of both competitive recombinant IL-1Ra anakinra (1–2 mg/kg/day for pediatric patients and 100 mg/day for adults) and the fully human anti-IL-1β monoclonal antibody canakinumab (150 mg in patients with a weight over 40 kg or 2 mg/kg in those less than 15–40 kg every 8 weeks) in patients with colchicine-resistant FMF. However, colchicine administration should be continued also during anti-IL-1 treatment to help in prevention of amyloidosis [50].
Management of TRAPS is more challenging than in other HPFS due to the considerable genetic heterogeneity and variable clinical spectrum of the disease. Inflammatory attacks are often responsive to the administration of corticosteroids, but patients may need increasing doses if frequent relapses occur or even long-time administration of steroids in order to prevent flares, with subsequent risk of important side effects. The anti-TNF inhibitor etanercept has been shown to prevent inflammatory attacks in a dose-dependent manner, allow the reduction of steroid dosage, and even work on TRAPS-related reactive amyloidosis as well [51]. Other TNF-neutralizing agents, such as infliximab and adalimumab, may cause paradoxical inflammatory attacks in TRAPS patients for different mechanisms, and caution should be used if administered [52]. IL-1 inhibitors, such as anakinra and canakinumab, have recently been shown to induce a stable and longer-lasting effect in controlling TRAPS clinical manifestations, and anakinra has been also successfully used as on-demand treatment [53].
The use of TNF-inhibitors is non-effective in determining the resolution of the whole clinical scenery of CAPS [54], differently from IL-1 blockade which is brilliantly successful in nearly all patients: anakinra has been the first biologic designed for the selective blockade of IL-1 in CAPS, given at a starting dose of 1 mg/kg per day by subcutaneous injection, and proving a sustained efficacy also on the stabilization of NOMID neurological manifestations [55, 56]. The efficacy of IL-1 blockade with anakinra is countered by its short half-life and day-by-day variations in activity; therefore, the development of IL-1 antagonists with higher affinity and longer half-life has facilitated patients’ compliance and management. Canakinumab targets IL-1β and is approved at standard doses for children and adults with all CAPS phenotypes [57]. While canakinumab is known to neutralize IL-1β by competing for binding to IL-1 receptor, which blocks signaling by the antigen-antibody complex, gevokizumab, another recombinant humanized anti-IL-1β monoclonal antibody, should act as a regulatory antibody by altering the electrostatic surface of IL-1β itself [58]. Canakinumab displays the advantage of bimonthly administration and is also approved for children, while gevokizumab has not yet been used in CAPS, but only in other autoinflammatory conditions, such as type 2 diabetes mellitus, Behçet’s disease-related uveitis, and generalized pustular psoriasis. Among HPFS, CAPS is the only condition for which there are different randomized controlled trials to support therapeutic suggestions: patients receiving canakinumab (at a dose of 2 mg/kg every 8 weeks) in a 48-week double-blind placebo-controlled randomized withdrawal study remained in remission, and this result was maintained in a 24-month continuation phase III study, although with a modified treatment schedule for NOMID patients (i.e., increased dosage until 5 mg/kg and/or increased frequency of administration) [59, 60]. Different clinical experiences have also proved that canakinumab given every 8 weeks (at a dose of 150 mg in adults and 2 mg/kg in children) provides rapid remission of most clinical and biochemical abnormalities of CAPS, though with limited efficacy in the structural bone lesions, and that dose adjustments are probably necessary for those patients with the most severe phenotype, without any substantial risk of adverse effects [61].
Therapeutic options in MKD encompass nonspecific anti-inflammatory approaches, such as non-steroidal anti-inflammatory drugs or corticosteroids, and biological agents that target specific cytokine pathways. Indeed, consistent with the involvement of IL-1β in MKD, anakinra administration has shown to relieve symptoms in various patients, even when administered on-demand [62]. Canakinumab injections every 4–8 weeks have been associated with both complete and partial remission of disease [63], while hematopoietic stem cell transplantation has been used in the very severely affected patients with MKD or in those with an established diagnosis of mevalonic aciduria [64].
Conclusive Thoughts
In conclusion, we made an overview on four classic hereditary causes of recurrent fever in children and adults, understanding how different misrupted innate immunity pathways can give heterogeneous patterns of disease. As we have seen, in HPFS we find the recurrence of episodic sterile inflammation, which is followed by symptom-free periods of variable duration, though recurrent inflammation over time might also lead to potentially irreversible chronic damages. Therefore, HPFS need to be considered and attentively managed to avoid the long-term complications of an overlooked systemic disorder: the chance to use biologics requires that diagnostic times of HPFS should be anticipated in order to suppress many complex clinical phenotypes and avoid the occurrence of secondary amyloidosis. In these last years, we have also witnessed a reconsideration of many multifactorial diseases, which have been linked to the inflammasome and IL-1β overactivity, as demonstrated by the dramatic response to specific IL-1 blockade. Obviously, the more we know about the molecular mechanisms of a disease, the better therapeutic approaches we can design to treat and potentially cure it: many IL-1-targeting agents have been developed, and findings regarding the regulation of IL-1 production in HPFS have paved the way to different clinical trials, which have been performed and are ongoing. In the future, empirical algorithms for diagnosis and treatment will be developed, and until then we hope that this comprehensive description might serve as a template for a proper evaluation of patients with HPFS.
References
Cooper EL (2010) Evolution of immune systems from self/not self to danger to artificial immune systems (AIS). Phys Life Rev 7(1):55–78. doi:10.1016/j.plrev.2009.12.001
Dzik JM (2010) The ancestry and cumulative evolution of immune reactions. Acta Biochim Pol 57(4):443–466
Buchmann K (2014) Evolution of innate immunity: clues from invertebrates via fish to mammals. Front Immunol 23(5):459. doi:10.3389/fimmu.2014.00459
Rigante D, Lopalco G, Vitale A et al (2014) Untangling the web of systemic autoinflammatory diseases. Mediat Inflamm 2014:948154. doi:10.1155/2014/948154
Aksentijevich I, Kastner DL (2011) Genetics of monogenic autoinflammatory diseases: past successes, future challenges. Nat Rev Rheumatol 7(8):469–478. doi:10.1038/nrrheum.2011.94
Lamkanfi M, Dixit VM (2012) Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28:137–161. doi:10.1146/annurev-cellbio-101011-155745
Cantarini L, Lopalco G, Cattalini M, Vitale A, Galeazzi M, Rigante D (2015) Interleukin-1: ariadne’s thread in autoinflammatory and autoimmune disorders. Israel Med Assoc J 17(2):93–97
Auron PE, Webb AC, Rosenwasser LJ, Mucci SF, Rich A, Wolff SM, Dinarello CA (1984) Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc Natl Acad Sci U S A 81(24):7907–7911
Mariathasan S, Monack DM (2007) Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 7(1):31–40
Vitale A, Rigante D, Lucherini OM et al (2013) Biological treatments: new weapons in the management of monogenic autoinflammatory disorders. Mediat Inflamm 2013:939847. doi:10.1155/2013/939847
Ben-Chetrit E, Levy M (1998) Familial Mediterranean fever. Lancet 351(9103):659–664
Centola M, Wood G, Frucht DM et al (2000) The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood 95(10):3223–3231
Touitou I, Lesage S, McDermott M et al (2004) Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat 24(3):194–198. doi:10.1002/humu.20080
Rigante D, Vitale A, Lucherini OM, Cantarini L (2014) The hereditary autoinflammatory disorders uncovered. Autoimmun Rev 13(9):892–900. doi:10.1016/j.autrev.2014.08.001
Balci B, Tinaztepe K, Yilmaz E et al (2002) MEFV gene mutations in familial Mediterranean fever phenotype II patients with renal amyloidosis in childhood: a retrospective clinicopathological and molecular study. Nephrol Dial Transplant 17(11):1921–1923
Bilginer Y, Akpolat T, Ozen S (2011) Renal amyloidosis in children. Pediatr Nephrol 26(8):1215–1227. doi:10.1007/s00467-011-1797-x
Livneh A, Langevitz P, Zemer D et al (1997) Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 40(10):1879–1885
Jéru I, Hentgen V, Cochet E et al (2013) The risk of familial Mediterranean fever in MEFV heterozygotes: a statistical approach. PLoS One 8(7):e68431. doi:10.1371/journal.pone.0068431
Marek-Yagel D, Berkun Y, Padeh S et al (2009) Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 60(6):1862–1866. doi:10.1002/art.24570
Rigante D, La Torraca I, Avallone L, Pugliese AL, Gaspari S, Stabile A (2006) The pharmacological basis of treatment with colchicine in children with familial Mediterranean fever. Eur Rev Med Pharmacol Sci 10(4):173–178
Rigante D, Frediani B, Galeazzi M, Cantarini L (2013) From the Mediterranean to the sea of Japan: the transcontinental odyssey of autoinflammatory diseases. Biomed Res Int 2013:485103. doi:10.1155/2013/485103
Aksentijevich I, Galon J, Soares M et al (2001) The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet 69(2):301–314
Caso F, Cantarini L, Lucherini OM et al (2014) Working the endless puzzle of hereditary autoinflammatory disorders. Mod Rheumatol 24(3):381–389. doi:10.3109/14397595.2013.843755
Cantarini L, Rigante D, Merlini G et al (2014) The expanding spectrum of low-penetrance TNFRSF1A gene variants in adults presenting with recurrent inflammatory attacks: clinical manifestations and long-term follow-up. Semin Arthritis Rheum 43(6):818–823. doi:10.1016/j.semarthrit.2013.12.002
Savic S, Dickie LJ, Wittmann M, McDermott MF (2012) Autoinflammatory syndromes and cellular responses to stress: pathophysiology, diagnosis and new treatment perspectives. Best Pract Res Clin Rheumatol 26(4):505–533. doi:10.1016/j.berh.2012.07.009
Rigante D, Lopalco G, Vitale A et al (2014) Key facts and hot spots on tumor necrosis factor receptor-associated periodic syndrome. Clin Rheumatol 33(9):1197–1207. doi:10.1007/s10067-014-2722-z
Stojanov S, McDermott MF (2005) The tumour necrosis factor receptor-associated periodic syndrome: current concepts. Expert Rev Mol Med 7(22):1–18
Rigante D, Cantarini L, Imazio M et al (2011) Autoinflammatory diseases and cardiovascular manifestations. Ann Med 43(5):341–346. doi:10.3109/07853890.2010.547212
Ravet N, Rouaghe S, Dodé C et al (2006) Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann Rheum Dis 65(9):1158–1162
Aksentijevich I, D Putnam C, Remmers EF et al (2007) The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum 56(4):1273–1285
Cantarini L, Lucherini OM, Frediani B et al (2011) Bridging the gap between the clinician and the patient with cryopyrin-associated periodic syndromes. Int J Immunopathol Pharmacol 24(4):827–836
Tanaka N, Izawa K, Saito MK et al (2011) High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum 63(11):3625–3632. doi:10.1002/art.30512
Hoffman HM, Wanderer AA, Broide DH (2001) Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol 108(4):615–620
Aganna E, Martinon F, Hawkins PN et al (2002) Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 46(9):2445–2452
Prieur AM, Griscelli C, Lampert F et al (1987) A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol 66:57–68
Rigante D (2010) The protean visage of systemic autoinflammatory syndromes: a challenge for inter-professional collaboration. Eur Rev Med Pharmacol Sci 14(1):1–18
Esposito S, Ascolese B, Senatore L et al (2014) Current advances in the understanding and treatment of mevalonate kinase deficiency. Int J Immunopathol Pharmacol 27(4):491–498
Mendey SH, Kuijk LM, Frenkel J, Waterham HR (2006) A role for geranylgeranylation in interleukin-1β secretion. Arthritis Rheum 54(11):3690–3695
Mendey SH, Schneiders MS, Koster J, Waterham HR (2006) Mutational spectrum and genotype-phenotype correlations in mevalonate kinase deficiency. Hum Mutat 27(8):796–802
van der Burgh R, ter Haar NM, Boes ML, Frenkel J (2013) Mevalonate kinase deficiency, a metabolic autoinflammatory disease. Clin Immunol 147(3):197–206. doi:10.1016/j.clim.2012.09.011
Berody S, Galeotti C, Koné-Paut I, Piram M (2015) A restrospective survey of patients journey before the diagnosis of mevalonate kinase deficiency. Joint Bone Spine 82(4):240–244. doi:10.1016/j.jbspin.2014.12.011
Bader-Meunier B, Florkin B, Sibilia J et al (2011) Mevalonate kinase deficiency: a survey of 50 patients. Pediatrics 128(1):e152–159. doi:10.1542/peds.2010-3639
Ammouri W, Cuisset L, Rouaghe S et al (2007) Diagnostic value of serum immunoglobulinaemia D level in patients with a clinical suspicion of hyper IgD syndrome. Rheumatology (Oxford) 46(10):1597–1600
Rigante D, Emmi G, Fastiggi M, Silvestri E, Cantarini L (2015) Macrophage activation syndrome in the course of monogenic autoinflammatory disorders. Clin Rheumatol 34(8):1333–1339. doi:10.1007/s10067-015-2923-0
Gershoni-Baruch R, Brik R, Zacks N, Shinawi M, Lidar M, Livneh A (2003) The contribution of genotypes at the MEFV and SSA1 loci to amyloidosis and disease severity in patients with familial Mediterranean fever. Arthritis Rheum 48(4):1149–1155
Ben-Zvi I, Livneh A (2011) Chronic inflammation in FMF: markers, risk factors, outcomes and therapy. Nat Rev Rheumatol 7(2):105–112. doi:10.1038/nrrheum.2010.181
Hentgen V, Grateau G, Kone-Paut I et al (2013) Evidence-based recommendations for the practical management of familial Mediterranean fever. Semin Arthritis Rheum 43(3):387–391. doi:10.1016/j.semarthrit.2013.04.011
Roldan R, Ruiz AM, Miranda MD, Collantes E (2008) Anakinra: new therapeutic approach in children with familial Mediterranean fever resistant to colchicine. Joint Bone Spine 75(4):504–505. doi:10.1016/j.jbspin.2008.04.001
Meinzer U, Quartier P, Alexandra JF, Hentgen V, Retornaz F, Koné-Paut I (2011) Interleukin-1 targeting drugs in familial Mediterranean fever: a case series and a review of the literature. Semin Arthritis Rheum 41(2):265–271. doi:10.1016/j.semarthrit.2010.11.003
Bulua AC, Mogul DB, Aksentijevich I et al (2012) Efficacy of etanercept in the tumor necrosis factor receptor-associated periodic syndrome: a prospective, open-label, dose-escalation study. Arthritis Rheum 64(3):908–913. doi:10.1002/art.33416
Nedjai B, Hitman GA, Quillinan N et al (2009) Proinflammatory action of the antiinflammatory drug infliximab in tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum 60(2):619–625. doi:10.1002/art.24294
Grimwood C, Despert V, Jeru I, Hentgen V (2015) On-demand treatment with anakinra: a treatment option for selected TRAPS patients. Rheumatology (Oxford) 54(9):1749–1751. doi:10.1093/rheumatology/kev111
Federico G, Rigante D, Pugliese A, Ranno O, Catania S, Stabile A (2003) Etanercept induces improvement of arthropathy in chronic infantile neurological cutaneous articular (CINCA) syndrome. Scand J Rheumatol 32(5):312–314
Koné-Paut I, Galeotti C (2014) Anakinra for cryopyrin-associated periodic syndrome. Expert Rev Clin Immunol 10(1):7–18. doi:10.1586/1744666X.2014.861325
Rigante D, Ansuini V, Caldarelli M, Bertoni B, La Torraca I, Stabile A (2006) Hydrocephalus in CINCA syndrome treated with anakinra. Childs Nerv Syst 22(4):334–337
Kuemmerle-Deschner JB, Hachulla E, Cartwright R et al (2011) Two-years results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann Rheum Dis 70(12):2095–2102. doi:10.1136/ard.2011.152728
Blech M, Peter D, Fischer P et al (2013) One target-two different binding modes: structural insights into gevokizumab and canakinumab interactions to interleukin-1β. J Mol Biol 425(1):94–111. doi:10.1016/j.jmb.2012.09.021
Lachmann HJ, Koné-Paut I, Kuemmerle-Deschner JB et al (2009) Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 360(23):2416–2425. doi:10.1056/NEJMoa0810787
Koné-Paut I, Lachmann HJ, Kuemmerle-Deschner JB et al (2011) Sustained remission of symptoms and improved health-related quality of life in patients with cryopyrin-associated periodic syndrome treated with canakinumab: results of a double-blind placebo-controlled randomized withdrawal study. Arthritis Res Ther 13:R202. doi:10.1186/ar3535
Anton J, Calvo I, Fernández-Martin J et al (2015) Efficacy and safety of canakinumab in cryopyrin-associated periodic syndromes: results from a Spanish cohort. Clin Exp Rheumatol 33(6 Suppl 94):S67–71
Steichen O, van der Hilst J, Simon A, Cuisset L, Grateau G (2009) A clinical criterion to exclude the hyperimmunoglobulin D syndrome (mild mevalonate kinase deficiency) in patients with recurrent fever. J Rheumatol 36(8):1677–1681. doi:10.3899/jrheum.081313
Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J (2011) On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis 70(12):2155–2158. doi:10.1136/ard.2011.149922
Galeotti C, Meinzer U, Quartier P et al (2012) Efficacy of interleukin-1-targeting drugs in mevalonate kinase deficiency. Rheumatology (Oxford) 51(10):1855–1859
Neven B, Valayannopoulos V, Quartier P et al (2007) Allogeneic bone marrow transplantation in mevalonic aciduria. N Engl J Med 356(26):2700–2703
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rigante, D., Frediani, B. & Cantarini, L. A Comprehensive Overview of the Hereditary Periodic Fever Syndromes. Clinic Rev Allerg Immunol 54, 446–453 (2018). https://doi.org/10.1007/s12016-016-8537-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-016-8537-8