
Abstract
Platelets (PLTs) are small anucleate blood cells that release from polyploidy megakaryocytes(MKs). PLT transfusion is standard therapy to prevent hemorrhage. PLT transfusion is donor‐dependent way which have limitations including the inadequate donor blood supply, poor quality, and issues related to infection and immunity. Overcoming these obstacles is possible with in vitro production of human PLTs. Currently several cells have been considered as source to in vitro production of PLTs such as hematopoietic stem cells (HSCs), embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). However, HSCs are a limited source for PLT production and large-scale expansion of HSC-derived PLT remains difficult. Alternative sources can be ESCs which have unlimited expansion capacity. But ESCs have ethical issues related to destroying human embryos. iPSCs are considered as an ideal unlimited source for PLT production. They are able to differentiate into any cells and have the capacity of self-renewal. Moreover, iPSCs can be acquired from any donor and easily manipulated. Due to new advances in development of MK cell lines, bioreactors, feeder cell-free production and the ability of large scale generation, iPSC-based PLTs are moving toward clinical applicability and considering the minimal risk of alloimmunization and tumorigenesis of these products, there is great hopefulness they will become the standard source for blood transfusions in the future. This review will focus on how to progress of in vitro generation of PLT from stem cell especially iPSCs and some of the successful strategies that can be easily used in clinic will be described.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
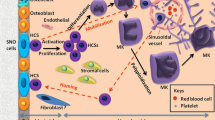
Platelets (PLTs) which were constantly derived from the polyploidy megakaryocytes (MKs) in the bone marrow (BM) are small non-nucleated blood cells of about 2-4 µm in diameter [1]. PLTs that circulate in the bloodstream are generally implicated in maintaining hemostasis, thrombosis, inflammatory responses and vascular integrity through a blood clotting mechanism [2]. In human, the normal PLT count is approximately 1.5 to 4 × 1011 in each µL of blood and almost 1 × 1011 PLTs are produced each day [3]. Although typically, the healthy human body is programmed to preserve an adequate PLT count, clinically many severe disorders may be developed through abnormal change in PLT level. Thrombocytopenia (decreased PLT production less than 150,000/μL or impaired PLT function) may occur because of BM failure, drug treatment, repeated-chemotherapy, autoimmune disease, and invasive surgery that result in risk of uncontrolled bleeding, thrombocythemia (increased PLT production more than 450,000/μL) and stroke [4].
Since the 1950s, following the progress in methods of PLT isolation from blood [5], allotransfusion of PLTs has been standard therapy in patients with low PLT in order to prevent or treat hemorrhages [6]. On the other hand, platelet-rich plasma (PRP) which is obtained after centrifugal concentration of whole blood has received widespread attention in the medical and cosmetic fields. Overall, in recent years PLT-based treatment has gradually become an important treatment approach in hemostasis management, regenerative medicine and tissue engineering [7, 8].
Nevertheless, there is a serious shortage in PLT supply in clinic. PLT availability is completely dependent on donating blood [4]. PLT products cannot be frozen because of once transfused they are rapidly cleared from the circulation [9], storage at room temperature significantly preserved the viability but due to PLT function instability and susceptibility to bacterial flourish, PLT lifespan is restricted to just 4 or 5 days [9, 10], consequently, stocks must be constantly replaced [4]. Even if there is enough available supply, PLT transfusion cannot be appropriate strategy because PLT transfusion refractoriness (PTR) can result in an allograft rejection through human leukocyte antigen (HLA) or human PLT antigen (HPA) mismatching [2, 11]. Also, PTR can be developed due to cytokines and other bioactive ingredients of PLT product that can result in shortened PLTs survival and causing complications such as fever, allergic symptoms, and transfusion-related acute lung injury (TRALI) [12].
A considerable potential strategy that may help to overcome restriction of PLT sources is the in vitro PLT production. To establish a successful protocol for PLT generation, the key point is choosing the best cell as a starting source.
MKs as unipotent progenitors of PLT are expandable cells, but they are a rare population in BM [4, 12], furthermore, using MKs as the starting source is simply not feasible [4]. The upstream multipotent progenitor of MKs, hematopoietic stem cells (HSCs), can self-renew and differentiation to MK lineage. HSCs can be collected from umbilical cord blood, BM, and mobilized peripheral blood (PB) but their amounts are low (one unit of UCB include 1 million HSCs) and HSC expansion is not efficient to produce enough PLTs for clinical use [13].
Human pluripotent stem cells (hPSCs) including human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs) are able to self-renew and have the capacity to differentiate to all human cell types including MK lineage. Since the both hESCs and hiPSCs can be proliferated and be conserved for longtime in vitro condition so they can be considered as controllable sources for PLT production [12]. Due to ethical challenges regarding hESC about the destruction of human embryos there are constraints for hESC use.
hiPSCs are freed from the ethical concepts and can be produced from various types of somatic cells. Moreover, patient-specific iPSCs can represent a readily available source for autologous transplantation. Even iPSCs derived from HLA-incompatible donors would enable escape from the alloimmune response by manipulation of the HLA or HPA expression [12, 14]. Due to potential tumorigenicity risk of hPSC, residual of iPSCs should be eliminated by irradiation before transfusion, that this strategy is efficient for enucleated cells such as PLTs [13]. Nevertheless, iPSCs can be considered as an ideal resource for in vitro PLT production.
Generation of PLT Using HSC (CD34+cell)
HSCs, also known as CD34+ cells, have the potential self-renew ability and can differentiate into all hematopoietic lineage cells. Hence CD34+ cells provide a good approach for in vitro PLT production. The CD34+ cells represent a rare population in the BM, accordingly they can be isolated from BM. Additionally, CD34+ cells can be isolated noninvasively from peripheral blood (PB) after the administration of granulocyte-colony stimulating factor (G-CSF) or from umbilical cord blood (UCB) [4]. The different reports have been represented production of functional MKs and PLTs from CD34+cells. In the first report, it was shown that PLTs could produce by culturing PB0-derived CD34+ cells in aplastic canine serum [15]. In order to production of MK and PLT from CD34+ cells, various cytokines have been examined. Thrombopoietin (TPO) a cytokine that also known as MK growth and development factor has an important role in MK proliferation and differentiation and also preserving the CD34+ cell population so in the past, it was used in standard protocols for CD34+ cell differentiation into PLTs, but TPO is not useful for large scale generation because of low mitotic activity [16]. In 2001, it was shown that addition of the chemokine stromal cell-derived factor 1α (SDF-1α) into a serum-free culture supplemented with TPO is able to generation of CD34+-derived MKs 12 folds more than culture supplemented with TPO alone. But this amount is not enough for clinical use [17]. Subsequent study reported that a combination of TPO with Flt3-ligand (FL), IL-6 and IL-11 in serum-free medium result in generation of a suitable number of immature MK progenitor cells expressing both CD34 and CD41 antigens that after transfusion they could recover PLTs [18]. In continue, to promote large scale PLT production under serum-free condition, three-phase culture was defined as a new strategy. Multiphase culture process can give rise to high expansion of CD34+ cell, high ploidy MK production, and promoting PLT production. However, maintenance of CD34+ cell activity in long-term culture is a key problem that co-culture of CD34+ cell with human telomerase catalytic subunit (hTERT)-transfected human stromal cells as an immortalized cell can solve the problem [19]. Matsunaga et al. proved that functional human PLT could be produced from UCB-derived CD34+ cells in three‐step serum‐free co-culture system consist of hTERT stroma in the presence of stem cell factor (SCF), Flt‐3/Flk‐2 ligand (FL), TPO and interleukin‐11 [20]. In first step, CD34+ cells were expanded on hTERT stroma with SCF/TPO/FL. In next step, expanded CD34+ cells were co-culture with hTERT stroma and SCF/TPO/FL/IL‐11 that leads to differentiation and expansion of MKs. In the last step, cells were transferred to liquid culture medium containing SCF,FL,TPO and IL-11 in order to MK differentiation into PLTs [20]. Results shown that culture in multiphase condition with diverse conditions in each phase with effect on in vitro megakaryopoiesis causes a considerable quantity of PLTs be produced. This method is suitable for clinical purposes [20].
Several groups have shown that gradients of pH and pO2 enhance Mk maturation in the BM niche with effect on megakaryopoiesis [21, 22]. According to this finding in order to large-scale CD34+-derived PLT production, Swapna and collaborators increased pH and pO2 in a 3-phase culture to mimic the BM niche gradient. Moreover, different cytokine cocktails were used for different stages of Mk differentiation. The study reported that switching from 5% O2 and pH 7.2 to 20% O2 and pH 7.4 along with cytokine cocktails can greatly increase production of mature Mk [23].
In a fascinated study, Yang et al. showed a rotary three-dimensional (3D) culture system could promote megakaryopoiesis and improve efficiency of CD34+-derived PLT generation compared with static condition [24]. Therefore, using this physical strategy along with other biochemical strategies can be helpful for large scale generation of PLTs.
There are a number of small molecules which can be used to control cell self-renewal, lineage differentiation, regeneration and reprogramming. It has been proved that these chemical molecules can be versatile tools for manipulating cell fates toward desired cells [25,26,27]. It can be inferred that chemical molecules could use also for PLT production from stem cells including CD34+ cells. Huang N et al. reported that a small chemical molecule inhibitor of transforming growth factor-beta (TGF-β) pathway (616,452 molecule) is able to promote polyploidization and shortened maturation of MKs through TPO-independent pathways. In this method, UCB or BM-derived CD34+ cells were cultured in StemSpan SFEM media containing SCF, TPO, Flt-3 ligand and IL-3 and were induced by 616,452 molecule in a dose-dependent fashion. The results showed that 616,452 molecule is a robust inducer of MK production and NSG mice transplanted with this small molecule treated cells were able to MK reconstitution in the BM and peripheral PLT production [28].
Bhatlekar et al. introduced anti-apoptotic BCL2L2 as a new candidate that can improve amount of produced PLT in culture system. In this study, UCB-derived CD34+ cells were cultured in serum free media in the presence of TPO and SCF. Results showed that Lentviral mediated BCL2L2 overexpression regulates apoptosis in cultured MKs and promotes pro- PLT formation that is associated with PLT number [29].
It should be noted, although the idea of producing PLTs from CD34+ stem cells was initially accepted there is the major drawback in large scale production. Existence of donor-specific antigens, low expansion power, low efficiency of megakaryopoiesis and low number of produced PLT are the other major problems to establishing CD34+-based PLT production [30].
Generation of PLT Using ESCs
ESCs are pluripotent cells derived from the inner cell mass of embryos that are able to proliferate and differentiate into any cell types. Moreover, ESCs can be potential source for the large‐scale PLT production [31]. Initially Gaur et al. indicated that ESCs can differentiate into MKs through two-dimensional “monolayer” system. In this method, undifferentiated hESCs were cultured on OP9 stromal cell monolayer to facilitate hematopoiesis. Although functional PLTs were produced in this strategy, yield of MKs was low (less than one MK per ESC) [32].
Takayama et al. promoted generation of MKs from hESCs through the formation of unique sac-like structures that were named embryonic stem cell-derived sacs (ES-sacs). In this study, to increase formation of ES-sacs and improve efficiency, hESCs were cultured on C3H10T1/2 or OP-9 stromal cells as a feeder in the presence of vascular endothelial growth factor (VEGF). “The ES-sacs were consisted of multiple cysts demarcated by cellular monolayers with some of the properties of endothelial cells. The spherical cells inside sacs expressed primarily CD34 that were able to form hematopoietic colonies in semi-solid culture to differentiate into mature MKs in the presence of TPO by day 24.” Since the ES-sacs can develop an appropriate microenvironment for creating hematopoietic progenitor cells (HPCs), this approach could lead to produce functional PLTs. However, number of produced PLT by this efficiency improvement strategy were far fewer than produced PLT from BM‐derived cells. In general, since this method requires a conventional stroma co-culture as feeder, it cannot appropriate for clinical use [33]. Therefore, it seems that feeder free condition is necessary for clinical-grade production.
Lu et al. reported an efficient approach for large-scale generation of MKs from hESCs under feeder-free condition through embryoid bodies (EBs) differentiation. However, generated PLTs by this condition had function in mice, only seven PLTs per MK were yielded [34]. As a result, the yield of PLT production in the feeder-free/serum-free conditions was lower than feeder-on/serum-on conditions.
In recent years, 3D culture systems have been proven as a feasible technic for hESC expansion [35, 36]. Based on that, in order to large‐scale generation of MKs, Zhang et al. developed a 3D suspension induction method with using special polystyrene CellSTACK culture chamber. In this method before culture, the hESC colonies were digested into single cells and transferred onto chamber. In the chamber, single hESCs began to aggregate to form EBs. EBs gradually produced many hematopoietic cells that mimicked embryonic megakaryopoiesis. This 3D suspension induction method could generate (3.4 ± 2.5) × 108 CD41a+ MKs which was more than previous reports. Moreover, it was xeno-free/feeder-free/transgene-free system that can be a good choice for high efficient and large-scale clinical–grad production of MKs and PLTs [37].
Basically, because of ethical concerns about the destruction of human embryos, using hESCs as a source is ineffective in clinic.
IPSC Technology
In 2006, it was reported that iPSCs were generated from murine embryonic fibroblasts (MEFs) by delivery of pluripotency transcription factors)TF) Oct4, Sox2, Klf4, and c-Myc (OKSM cocktail) that called Yamanaka factors (YFs) into the cells by retroviral vector [38]. In 2007, four TFs were successfully used to generate iPSCs from human fibroblasts [39]. Like to ESCs, iPSCs have self-renewal capacity and can differentiate into any cell type, also have potential to improve personalized medicine using patient-specific iPSCs [40]. Generation of iPSCs eliminated some technical and ethical limitations of using ESCs, and the potential of hiPSCs to generate functional tissues led to iPSCs technology consider as a powerful tool for therapeutic applications in regenerative medicine [40, 41]. Obviously, the advent of iPSC technology opened up a new window in stem cell biology and stem cell-based therapy, as well as unique opportunities in disease modelling, drug discovery, cell availability and immune rejection studies [40, 41]. Although clinical trials have not been accompanied using iPSCs derived hematopoietic cells so far, the possibility of substituting damaged tissues by iPSC-derived tissue has been represented in treatment macular degeneration, parkinson’s and heart disease which some of have successfully toward clinical trials [42,43,44]. It schematically shows the various strategies to produce iPSCs and its applications in Fig. 1. At the first, several integrative viral vectors (e.g. retroviruses and lentiviruses) were used for transduction of YFs. Although integrative approaches can strongly induce pluripotency in somatic cells, insertional mutagenesis in oncogenic genes such as Klf4 and c-Myc can occur by viral integration which can leads to malignant tumor formation [45]. Because of safety concerns, Yu et al. suggested substituting NANOG and LIN28 with KLF4 and c-Myc and Montserrat et al. suggested replacing of Oct4 with GATA3 to improve the safety of iPSCs generation [46, 47].Now days, in order to iPSC-based cell therapies, new strategies have been suggested for cell reprogramming without genetic and epigenetic alterations in the iPSCs. Besides, the use of non-integrative viruses such as adeno and sendai viruses, non-viral vectors such as episomal plasmids, microRNAs, metabolites, small molecules, synthesized RNAs and proteins have been established for iPSC generation for clinical approaches [48,49,50,51,52,53,54,55].Although non-integrative methods lead to produce integration-free iPSCs, the important disadvantage of some of them is low reprogramming efficiency compared to the integrative vectors. Among them, sendai viruses, episomal plasmids and synthetic mRNAs are the commonly used in basic and clinical researches to produce integration-free iPSCs because of their high efficiency and relative simplicity and the episomal plasmids and sendai viruses have been the preferred for clinical applications [56]. Interestingly, it was shown that the gene expression profiles of integration-free iPSCs were more similar to hESCs than the iPSCs generated by integrating methods [57].

iPSC production and applications. Reprograming somatic cells into iPSCs can be done by various strategies. Integrative methods such as retroviruses and lentiviruses have the high reprogramming efficiency with less safety. But non-integrative methods (e.g., sendai virus, mRNA, or episome) have low reprogramming efficiency with high safety. The generation of iPSCs can be used in drug screening, cell therapy, and disease modeling
In 2014, the first clinical trial to evaluate the safety of iPSC-derived cells was launched by Masayo Takahashi of the RIKEN in Japan [58]. In this study, human iPSC-derived retinal pigment epithelial (RPE) cells were used to treat macular degeneration that improvement of the vision was observed. The trial was subsequently suspended because of finding the gene abnormality in the iPSCs of second patient [58].
Generally, iPSC-based therapy is still limited due to its inherent tumorigenicity. Elimination of remaining of undifferentiated iPSCs with cell irradiation is one of the strategies to reduce tumorigenicity risk. However, cell irradiation is only useful for enucleate cells such as PLTs, that causes hiPSCs be a considerable source for unlimited production of this enucleate cell [59].
IPSC-derived MK and PLT Productions
Fundamentally, PLTs are produced from iPSCs in three steps. The first step is the generating iPSCs from the differentiated mature cells. The second step is differentiation of iPSCs into HSCs which can be done by two different approaches, and the final step is HSCs differentiation into MKs.
The first approach for differentiation of human iPSCs into HSCs is co-culture of iPSCs with feeder cells (e.g. C3H10T1/2 and OP-9 stromal cells) under two-dimensional (2D) culture. Feeder cells consist of important cellular component that are necessary for iPSCs growth and differentiation. Moreover, the feeder cells are able to promote hematopoiesis [60, 61]. The second approach for differentiation of iPSCs into HSCs is EB formation under feeder-free 3D culture system [62, 63].
For the first time Takayama et al. reported that functional PLTs can be produced from hiPSCs through reprogramming human dermal fibroblasts (HDFs) by OKSM cocktail. Briefly, co-culturing hiPSCs on C3H10T1/2 or OP-9 feeder cells in the presence of VEGF led to generation of a hematopoietic niche including hematopoietic progenitors, which was called iPSC-sac-like structure. iPSC-sac-like structures were cultured in the presence of human TPO and other cytokines (IL-6, IL-11, and SCF) to generate mature MKs and release of PLTs. The function of produced PLTs was confirmed in NOD-SCID- IL-2 γc−/− thrombocytopenia mouse model. Although in this method production efficiency of MKs from hiPSCs was low (about three MKs per hiPSCs), results showed that the initial up-regulation of c-Myc expression in the presence of other TFs effect on reprogramming efficiency of somatic cells into hiPSCs and number of produced MK. In the following, down-regulation of c-Myc expression effect on production efficiency of PLTs. In generally, results showed that c-Myc plays a key role in the formation of MKs and PLTs so that initial increase and then decline in c-Myc expression are critical for megakaryopoiesis and effective PLT production [60].
Nishimura et al. generated canine induced pluripotent stem cells (ciPSCs) from canine embryonic fibroblasts. In continue, ciPSCs differentiated into MKs and PLTs on OP9 stromal cells supplemented with VEGF. The produced ciPSCs-drived PLTs were morphologically and functionally close to human blood PLTs. Functional PLTs had been derived from hiPSCs so far and it was the first study about generating functional PLTs from large-animal models [61]. Result of this study can be helpful to resolve produce iPSCs derived PLTs in human.
The two above studies have been depended on using serum and xeno feeder cells. In order to prevent immunogenic reactions associated with them, feeder free substitutes were considered such as CELLstart (Life Technologies), Matrigel (BD),recombinant proteins [64], synthetic polymers [65] and synthetic materials [66].
Feng et al. defined a method for large scale production of functional PLTs from hiPSCs under serum/ xeno/feeder-free conditions through EB strategy. In this study matrigel-coated surface with mTeSR1 medium were used as feeder free condition and efficiency of MK differentiation into PLT was enhanced by the small molecule iBET151 as inhibitor of c-Myc expression. Results showed that produced PLTs were morphologically and functionally similar to peripheral PLTs. Moreover, knockout of the β2 microglobulin gene in iPSCs could provide a source for universal PLTs that are negative for HLA class I. Nevertheless, output approximately was six PLTs per MK that was still very low compared to number of PLTs produced from BM-derived MKs [62].
Liu et al. tried to make an efficient system for clinical-grade generation of MK progenitors from hiPSCs in essential 8 medium (a xeno-free and feeder-free medium) by replacing TPO and BSA by FDA-approved pharmacological reagents. These reagents were included romiplostim (Nplate, a TPO analog), oprelvekin (recombinant IL-11), and plasbumin (human albumin). In this system PLT production efficiency was low that may be due to lack of feeder cells [63].
The above studies that were based on using cytokine cocktails could not be successful to progress the efficiency production. Therefore, genetic manipulation strategy was considered. Nakamura et al. tried to produce immortalize MK progenitor cell line (imMKCL) for using as a stock in cell bank. imMKCL was produced by overexpression of c-MYC, BMI1, and BCL-XL in hiPSC-derived HPCs which could be stable for up to 5 months. Due to the proliferation capacity, it was estimated that imMKCL produce over 1011 PLTs, but it was obtained only 10 PLTs per MK and function of produced PLT was not equal to peripheral PLTs [67].
Moreau et al. reported a novel technology for large scale production of functional MKs from hiPSCs by direct sequential differentiation of hiPSCs into MKs. This ‘directed differentiation method was called MK forward programming (MK-FOP) and relied on exogenous expression of GATA1, FLI1, and TAL1 in a chemically-defined condition. Matured MKs-FOP produced by this method could be cryopreserved and banked for several months with purity over 90%. Although this approach was suitable for large-scale production, produced PLTs were less mature than peripheral PLTs and after transferring into mice had a short half-life [68].
Mostly, sufficient production of PLTs for clinical requirements was unproductive due to the low number of released PLTs from iPSC-derived MKs. The maturation of MKs, its differentiation into pro-PLTs and amount of released PLTs are all really rely on the microenvironment [69]. Therefore, an important step toward the industrial-scale generation of PLTs in vitro is the development of bioreactors that physically mimics the in vivo environment [12]. Initially, in several studies microfluidic system was developed to mimicking in vivo environment. But its laminar flow which was only based on shear stress and vorticity, was not very successful in effective thrombopoiesis and industrial-scale generation of PLT [70, 71]. Ito Y et al. showed turbulent flow with an optimal level of shear stress and turbulent energy is a crucial physical regulator for PLT release. According to they developed a turbulent flow-based bioreactor that was able to generation of 100 billion-order PLTs from hiPSC-MKs. The turbulent flow facilitates PLT shedding with promoting release of IGFBP2, MIF, and Nardilysin from MKs [72].
On the other hand, the discovery of effective substances that can strikingly improve the efficiency and yield of produced PLT is another important step toward the industrial-scale generation. Many studies have been done to finding these effective substances. The PLT function initiates with its adhesion to the extracellular matrix (ECM) by interaction between platelet glycoprotein Ibα (GPIbα) and von Willebrand factor (vWF). Matrix metalloproteinase (MMPs) are involved in shedding of GPIbα that lead to decline in quality of PLT production [73]. Moreover, loss of GPIbα results in rapid PLT loss after transfusion. So high expression of GPIbα is a critical factor in life span of circulating PLTs. It was demonstrated that GPIbα shedding can occur in iPSCs derived PLT too [60]. Hence, regulation of MMPs could contribute to maintain PLT function and efficient generation of hiPSCs-derived PLT. Feng et al. described that MMP8 inhibitor is able to prevent GPIb shedding during iPSCs differentiation into PLT [62]. Unfortunately, the safety of MMP8 inhibitor have never been confirmed which is a drawback for its clinical application. Hirata and colleagues showed GPIbα ectodomain shedding occurs by temperature-dependent activation of MMP17 (ADAM17) in 37◦C. This study indicated the selective inhibition of MMP17 using KP‐457 could preserve GPIbα and generate iPSC‐derived PLTs with improved hemostatic function in vivo in 37◦C. It has been emphasized that KP‐457 has low risk of genetic and systemic toxicities. This strategy can effectively enhance clinical–grad production of functional hiPSC-derived PLTs [74].
As an alternative to producing PLT from hiPSCs the generation of MK-like progenitors by direct conversion strategies (bypassing the pluripotent state) is a faster way to generating PLT. Directed differentiation of fibroblasts into MK-like progenitors has been done by two different studies using three factors (NF‐E2, MAFG, and MAFK) [75], and six factors (GATA2, RUNX1, GATA1, TAL‐1, LMO2, and c‐MYC) [76]. These MK-like progenitors enabled to release functional PLTs in mice. But directed differentiation strategies have low efficiency and need efficiency improvement.
Summary of different approaches for in vitro generation of PLT was shown in Fig. 2. In September 2018, Japanese Ministry of Health, Labor and Welfare approved a plan for the first world’s clinical trial to treat an aplastic anemia patient with thrombocytopenia by the infusion of autologous iPSC-derived PLTs. Center of iPSC research and application (CiRA) of kyoto university was responsible for the cell manufacturing, their non-clinical safety evaluation and their quality assessment. In this project, autologous iPSC-derived PLTs were generated from peripheral blood mononuclear cell (PBMCs) of patient using episomal plasmids. Collected PBMCs were reprogrammed into iPSCs. Then, iPSCs were differentiated into the imMKCLs by overexpression of c-MYC, BMI1 and BCL-XL under the doxycycline control promoter so that the transgenes switching off led to imMKCL maturation. The imMKCLs were stored as a master cell line in liquid nitrogen. The master cell line was proliferated and differentiated into PLT in turbulent flow-based bioreactors to improve thrombopoiesis. In order to maintain PLT function, TA-316, a thrombopoietin mimetic compound, KP-547, an ADAM17 inhibitor were used to inhibit of GPIbα shedding. Finally, the produced PLTs were purified, concentrated, washed, irradiated and finally transfused into the patient. The patient received PLT in three doses: at first,1 × 1010 PLTs, 3 months later 3 × 1010 PLTs and finally 5 months after the second dose 1 × 1011 PLTs. No significant adverse event was observed during one year full monitoring after the last dose [77].

Schematic of the different methods for in vitro PLT production. At the first, iPSC-derived MKs were produced using cytokines. In the following, iPSC-derived MKs were produced using transgenes and cytokines. Also. MK-like progenitors can be generated by direct differentiation of fibroblasts using several TFs. Industrial-scale generation of iPSCs-derived PLTs needs to expansion of MK lines together with the development of bioreactors
Advances in Producing Immune-compatible IPSCs-derived PLT
It is clearly evident that repeated PLT transfusions cause serious acute or delayed complications due to HLA or HPA mismatch [64]. Washing PLT products before transfusion helps to prevent acute reactions [12]. Moreover, induced graft-versus-host disease (GVHD) by contaminating lymphocytes in the PLT concentrate could be prevented by irradiation or leukodepletion of PLT products. But transfusion-related GVHD is not able to evoke by iPSCs derived PLT products because of absent of lymphocytes in these products [12].
A significant clinical problem that complicates the PLT transfusion is PTR which is caused by expression of alloantigens (e.g., ABO, HLA class I, and HPA). The most frequent immune reason for PTR is the existence of specific alloantibodies against HLA class I epitopes [78]. Of course, ABO‐compatible PLT transfusion reduces the frequency of PTR and guidelines often propose fresh and ABO compatible PLT products for transfusion [79, 80]. Overall, using HLA-matched PLTs or autologous PLTs is essential to prevent immune reactions induced by repeated allogeneic PLT transfusions, but they have some obstacles such as shortage of stock and extra cost. To address these concerns, the perfect products are autologous PLTs that are generated from patient-derived iPSCs. Although autologous PLTs do not induce alloimmune response, they have high costs and variable quality. To overcome the limitations of autologous PLTs, the derivation of allogeneic PLTs from allogeneic iPSCs line bank have considered. Since, allogenic products must be matched with one of the two class I HLA loci so include a large population of donors [81]. Currently, the advancement of genome editing technologies specially CRISPR/Cas has been created a new perspective for HLA and HPA manipulation of iPSC-derived PLTs Fig. 3. Silencing of HLA class I or deletion of β2 microglobulin which is required for HLA Class I assembly, could be completely dismissed PTR [82]. Borger et al. with silencing of HLA class I using shRNA established a stable HLA-universal iPSCs line under feeder/animal-free conditions without any effect on the differentiation capacity of the iPSCs line into MKs. These HLA-universal iPSC-derived MKs were enabled to release functional PLTs and as well escape alloimmune response related to antibody-dependent cell-mediated cytotoxicity(ADCC) and cellular-dependent cytotoxicity(CDC), which can be used as a source of poorly immunogenic cells. In this regard, since silencing with shRNA do not completely remove the β2-microglobulin protein so NK cell cytotoxic activity is suppressed by the residual HLA class I expression [83].

Summary of the strategies for overcoming alloimmune responses following PLT transfusion. Autologous iPSCs-derived PLTs do not induce alloimmune responses but they have high cost and variable quality. Allogeneic iPSC lines with homologous-type class I have less immune response because allogenic products must be matched with one of the two locus HLA class I that include a large population of donors. Currently, advances in genome editing technologies lead to engineering iPSCs derived PLT by manipulation of HLAs and HPAs
Feng et al. have generated universal PLTs that are negative for the HLA class I by knocking out the β2-microglobulin gene (B2M). These HLA Class I null PLTs can be released and functional. Although the alloreactivity of NK cells has not been proved after PLT transfusion, it may occur after HLA Class I null PLT transfusion due to the “missing self” response, since β2M–/– cells are sensitive to NK cells [62]. Thus, in knockout HLA-I dependent studies, fuse of recombinant non-polymorphic HLA-E or HLA-G into β2M, or deletion of the HLA-A and HLA-B couple with retaining HLA-C are required to suppress of NK cell [84]. However, it has been reported that retaining HLA-C leads to induce of alloimmune response by anti-HLAC [85].
It has recently been shown that knockout (KO) of HLA class I in iPSCs lines by CRISPR/Cas9 system reduces the immune response without NK cell activation. Suzuki et al. successfully produced iPSC-derived HLA class I knockout PLTs (HLA-KO iPLATs) from HLA-KO imMKCLs using the CRISPR/Cas9 system for KO of B2M. HLA-KO iPLATs were enabled to evade NK cell-mediated and anti-HLA I antibody-mediated immune responses in vitro and in vivo assay [86]. Of course, due to the risk of off-target in CRISPR/Cas9 evaluation of safety is essential before clinical trials. Paired CRISPR/Cas9 nickases can reduce concerns about the risk of off-target [87, 88]. Norbnop et al. produced HLA-KO functional PLTs from iPSCs using paired CRISPR/Cas9 nickase [89].
Beside, Although PTR associated to incompatible HPA is lower than PTR associated to HLA class I, antiHPA antibodies can induce critical clinical responses such as post-transfusion purpura (PTP) and fetal/neonatal alloimmune thrombocytopenia (FNAIT). Since single nucleotide polymorphisms (SNPs) of HPA molecules are responsible for antiHPA antibodies knockout cannot be useful for manipulating of HPA. In this way Zhang et al. modified HPA by converting SNPs using CRISPR/Cas9 to suppress the alloimmune response [90].
Conclusion
The increasing demand for PLT products in clinic and low rates of blood donation have led to remarkable efforts toward manufacturing of PLTs in vitro. It is currently possible to generate PLT from stem cells such as HSCs, ESCs, and iPSCs. HSCs are a source for in vitro PLT production which could make a steady source of blood donations. However, this source is still limited because HSCs cannot be indefinitely expanded and also have concern about existence of donor-specific antigen and donor dependency. hESC sources have barriers such as ethical concerns about the destruction of human embryos, and also differentiated hESCs express high levels of MHCI that lead to rejection in mismatched patients. Todays, iPSCs are regarded as an unlimited universal source for produce of many cells. Because not only iPSCs have pluripotent potential and self-renewal ability but also they can be generated from any donor, and easily manipulated. Moreover, iPSCs don’t have ethical issues. Thus, these cells have more advantageous than HSCs and ESCs. Many studies have been done for PLT production from iPSCs. Although numbers of them have been succeeded in producing functional PLTs, none of them could achieve to the high efficient and large scale production. So more studies are needed to find a new strategy to produce enough and efficient functional PLT for clinical purposes. Generally, modifications in culture systems such as using multiphase culture can improve cell expansion, production of high-ploidy MKs and promoting PLT production. Moreover, improvement of physical strategies along with other biochemical strategies can help large scale generation. Advances in culture chamber and using rotary 3D culture system instead of static condition could promote efficiency of PLT generation. Likewise, development of turbulent flow-based bioreactors that physically mimics the in vivo environment help thrombopoiesis that led to increase quality and quantity of industrial-scale generation. A Key step toward the industrial-scale generation of iPSC-derived PLTs is expansion of MK lines that solves the problem of storage. Moreover, EB formation method that can gradually produce many hematopoietic cells are a good choice as start point for iPSCs differentiation. The discovery of small molecules and effective substances which can control self-renewal, lineage differentiation and reprogramming could strikingly improve the efficiency.
To prevent complications of repeated PLT transfusions autologous PLT can be helpful, but it has some limitations that cause consider to allogenic PLT. Of course, using allogenic PLT has also the problem about allo-reactivity related to HLA Class I that producing β2M null iPSCs-universal PLT and HLA-matched PLT line bank can solve the problem. Although the alloreactivity of NK cells has not been proved after PLT transfusion, it may occur after HLA Class I null PLTs transfusion due to the “missing self” response. KO of HLA class I using CRISPR/ Cas9 system can reduce the immune response without NK cell activation and paired CRISPR/Cas9 nickases can decrease risk of off-targets related to CRISPR/CAS9 system. Although frequency of PTR associated to incompatible HPA is lower than HLA class I, converting SNPs of HPA by CRISPR/ Cas9 system can suppress alloimmune responses related to incompatible HPA.
Ultimately, iPSCs can be a safe and unlimited source to supply the required PLT for clinical applications but efforts should be continued for high efficient and large scale production. iPSC-based PLT products have yet to be confirmed in humans, but since PLTs are non-proliferating anucleate cells with minimal genetic aberrations, it is hoped that using in vitro produced PLTs in clinic will come true soon.
Availability of Data and Materials
Not applicable.
Abbreviations
- iPSCs:
-
Induced Pluripotent Stem Cells
- hiPSC:
-
Human Induced Pluripotent Stem Cells
- PLTs:
-
Platelets
- MKs:
-
Megakaryocytes
- PRP:
-
Platelet-Rich Plasma
- BM:
-
Bone marrow
- PB:
-
Peripheral Blood
- HLA:
-
Human Leukocyte Antigen
- HPA:
-
Human Platelet Antigen
- hPSCs:
-
Human pluripotent stem cells
- PTR:
-
Platelets Transfusion Refractoriness
- TRALI:
-
Transfusion-Related Acute Lung Injury
- UCB:
-
Umbilical Cord Blood
- HSCs:
-
Hematopoietic stem cells
- TPO:
-
Thrombopoietin
- SCF:
-
Stem Cell Factor
- ESCs:
-
Embryonic Stem Cells
- hESCs:
-
Human Embryonic Stem Cells
- VEGF:
-
Vascular Endothelial Growth Factor
- ES-sacs:
-
Embryonic Stem Cell–Derived Sacs
- HPCs:
-
Hematopoietic Progenitor Cells
- TF:
-
Transcription Factors
- YFs:
-
Yamanaka factors
- EB:
-
Embryoid Body
- MEF:
-
Murine Embryonic Fibroblast
- HDF:
-
Human Dermal Fibroblasts
- ciPSCs:
-
Canine Induced Pluripotent Stem Cells
- imMKCLs:
-
Immortalize MK Progenitor Cell Line
- MK-FOP:
-
MK Forward Programming
- MMPs:
-
Matrix Metalloproteinase
- vWF:
-
Von Willebrand Factor
- GPIbα:
-
Platelet Glycoprotein Ibα
- GVHD:
-
Graft-versus-host disease
- B2M:
-
β2-Microglobulin
- KO:
-
Knockout
- CDC:
-
Cellular-dependent Cytotoxicity
- ADCC:
-
Antibody-dependent Cell-mediated Cytotoxicity
- FNAIT:
-
Fetal/neonatal Alloimmune Thrombocytopenia
- PTP:
-
Post-Transfusion Purpura
References
van der Meijden, P. E. J., & Heemskerk, J. W. M. (2019). Platelet biology and functions: New concepts and clinical perspectives. Nature Reviews. Cardiology, 16(3), 166–179.
Stanworth, S. J., et al. (2015). Platelet refractoriness–practical approaches and ongoing dilemmas in patient management. British Journal of Haematology, 171(3), 297–305.
Semple, J. W., Italiano, J. E., & Freedman, J. (2011). Platelets and the immune continuum. Nature Reviews Immunology, 11(4), 264–274.
Karagiannis, P., Sugimoto, N., & Eto, K. (2019). 66 - Stem Cell-Derived Platelets, in Platelets (Fourth Edition), A.D. Michelson, Editor. Academic Press. 1173–1189.
Dillard, G. H. L., Brecher, G., & Cronkite, E. P. (1951). Separation, Concentration, and Transfusion of Platelets. Proceedings of the Society for Experimental Biology and Medicine, 78(3), 796–799.
Szczepiorkowski, Z. M., & Dunbar, N. M. (2013). Transfusion guidelines: When to transfuse. Hematology. American Society of Hematology. Education Program, 2013, 638–644.
Etulain, J. (2018). Platelets in wound healing and regenerative medicine. Platelets, 29(6), 556–568.
Garbin, L. C., & Olver, C. S. (2020). Platelet-Rich Products and Their Application to Osteoarthritis. Journal of Equine Veterinary Science, 86:102820.
Rumjantseva, V., & Hoffmeister, K. M. (2010). Novel and unexpected clearance mechanisms for cold platelets. Transfusion and Apheresis Science, 42(1), 63–70.
Jenkins, C., et al. (2011). Bacterial contamination in platelets: Incremental improvements drive down but do not eliminate risk. Transfusion, 51(12), 2555–2565.
Brown, C. J., & Navarrete, C. V. (2011). Clinical relevance of the HLA system in blood transfusion. Vox Sanguinis, 101(2), 93–105.
Sugimoto, N., & Eto, K. (2017). Platelet production from induced pluripotent stem cells. Journal of Thrombosis and Haemostasis, 15(9), 1717–1727.
Avanzi, M. P., & Mitchell, W. B. (2014). Ex Vivo production of platelets from stem cells. British Journal of Haematology, 165(2), 237–247.
Karagiannis, P., Endo, H., & Eto, K. (2016). Generating Blood from iPS Cells. In H. Schulze & J. Italiano (Eds.), Molecular and Cellular Biology of Platelet Formation: Implications in Health and Disease (pp. 399–420). Springer International Publishing.
Mazur, E., et al. (1990). Isolation of large numbers of enriched human megakaryocytes from liquid cultures of normal peripheral blood progenitor cells. Blood, 76, 1771–1782.
Pineault, N., & Boisjoli, G. J. (2015). Megakaryopoiesis and ex vivo differentiation of stem cells into megakaryocytes and platelets. ISBT Science Series, 10(S1), 154–162.
Guerriero, R., et al. (2001). Stromal cell–derived factor 1α increases polyploidization of megakaryocytes generated by human hematopoietic progenitor cells. Blood, 97(9), 2587–2595.
De Bruyn, C., et al. (2005). Ex vivo expansion of megakaryocyte progenitor cells: Cord blood versus mobilized peripheral blood. Stem Cells Development, 14(4), 415–424.
Kawano, Y., et al. (2003). Ex vivo expansion of human umbilical cord hematopoietic progenitor cells using a coculture system with human telomerase catalytic subunit (hTERT)–transfected human stromal cells. Blood, 101(2), 532–540.
Matsunaga, T., et al. (2006). Ex Vivo Large-Scale Generation of Human Platelets from Cord Blood CD34+ Cells. Stem Cells, 24(12), 2877–2887.
Mostafa, S. S., Miller, W. M., & Papoutsakis, E. T. (2000). Oxygen tension influences the differentiation, maturation and apoptosis of human megakaryocytes. British Journal of Haematology, 111(3), 879–889.
Yang, H., Miller, W. M., & Papoutsakis, E. T. (2002). Higher pH promotes megakaryocytic maturation and apoptosis. Stem Cells, 20(4), 320–328.
Panuganti, S., et al. (2013). Three-stage ex vivo expansion of high-ploidy megakaryocytic cells: Toward large-scale platelet production. Tissue engineering. Part A, 19(7–8), 998–1014.
Yang, Y., et al. (2016). Integrated Biophysical and Biochemical Signals Augment Megakaryopoiesis and Thrombopoiesis in a Three-Dimensional Rotary Culture System. Stem Cells Translational Medicine, 5(2), 175–185.
Zhang, Y., et al. (2012). Small molecules, big roles – the chemical manipulation of stem cell fate and somatic cell reprogramming. Journal of Cell Science, 125(Pt 23), 5609–5620.
Li, W., et al. (2013). Chemical approaches to studying stem cell biology. Cell Research, 23(1), 81–91.
Yu, C., et al. (2014). Chemical approaches to cell reprogramming. Current Opinion in Genetics & Development, 28, 50–56.
Huang, N., et al. (2016). Identification of a potent small molecule capable of regulating polyploidization, megakaryocyte maturation, and platelet production. Journal of hematology & oncology, 9(1), 136–136.
Bhatlekar, S., et al. (2019). Anti-apoptotic BCL2L2 increases megakaryocyte proplatelet formation in cultures of human cord blood. Haematologica, 104(10), 2075–2083.
Nurhayati, R. W., Ojima, Y., & Taya, M. (2016). Recent developments in ex vivo platelet production. Cytotechnology, 68(6), 2211–2221.
Keller, G. M. (1995). In vitro differentiation of embryonic stem cells. Current Opinion in Cell Biology, 7(6), 862–869.
Gaur, M., et al. (2006). Megakaryocytes derived from human embryonic stem cells: A genetically tractable system to study megakaryocytopoiesis and integrin function. Journal of Thrombosis and Haemostasis, 4(2), 436–442.
Takayama, N., et al. (2008). Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood, 111(11), 5298–5306.
Lu, S. J., et al. (2011). Platelets generated from human embryonic stem cells are functional in vitro and in the microcirculation of living mice. Cell Research, 21(3), 530–545.
Storm, M. P., et al. (2010). Three-dimensional culture systems for the expansion of pluripotent embryonic stem cells. Biotechnology and Bioengineering, 107(4), 683–695.
Lei, Y., & Schaffer, D. V. (2013). A fully defined and scalable 3D culture system for human pluripotent stem cell expansion and differentiation. Proc Natl Acad Sci U S A, 110(52), E5039–E5048.
Zhang, B., et al. (2021). Large-scale generation of megakaryocytes from human embryonic stem cells using transgene-free and stepwise defined suspension culture conditions. Cell Prolife, 54(4):e13002.
Takahashi, K., & Yamanaka, S. (2006). Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell, 126(4), 663–676.
Takahashi, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131(5), 861–872.
Shi, Y., et al. (2017). Induced pluripotent stem cell technology: A decade of progress. Nature reviews Drug discovery, 16(2), 115–130.
Moradi, S., et al. (2019). Research and therapy with induced pluripotent stem cells (iPSCs): Social, legal, and ethical considerations. Stem Cell Research & Therapy, 10(1), 341.
Li, F., Hu, J., & He, T. C. (2017). iPSC-based treatment of age-related macular degeneration (AMD): The path to success requires more than blind faith. Genes Dis, 4(2), 41–42.
Takahashi, J. (2020). iPS cell-based therapy for Parkinson’s disease: A Kyoto trial. Regen Ther, 13, 18–22.
Turner, D., et al. (2020). Clinical-based Cell Therapies for Heart Disease-Current and Future State. Rambam Maimonides Medical Journal, 11(2):e0015.
Yamanaka, S. (2012). Induced Pluripotent Stem Cells: Past, Present, and Future. Cell Stem Cell, 10(6), 678–684.
Yu, J., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science, 318(5858), 1917–1920.
Montserrat, N., et al. (2013). Reprogramming of Human Fibroblasts to Pluripotency with Lineage Specifiers. Cell Stem Cell, 13(3), 341–350.
Stadtfeld, M., et al. (2008). Induced pluripotent stem cells generated without viral integration. Science, 322(5903), 945–949.
Fusaki, N., et al. (2009). Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proceedings of the Japan Academy, Series B, 85(8), 348–362.
Woltjen, K., et al. (2009). piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature, 458(7239), 766–770.
Worringer, Kathleen A., et al. (2014). The let-7/LIN-41 Pathway Regulates Reprogramming to Human Induced Pluripotent Stem Cells by Controlling Expression of Prodifferentiation Genes. Cell Stem Cell, 14(1):40–52.
Esteban, M. A., et al. (2010). Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell, 6(1), 71–79.
Hou, P., et al. (2013). Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science, 341(6146), 651–654.
Warren, L., et al. (2010). Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell, 7(5), 618–630.
Kim, D., et al. (2009). Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell, 4(6), 472–476.
Musunuru, K., et al. (2018). Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circulation Genomic and Precison Medicine, 11(1):e000043.
Liu, Y., et al. (2012). The gene expression profiles of induced pluripotent stem cells (iPSCs) generated by a non-integrating method are more similar to embryonic stem cells than those of iPSCs generated by an integrating method. Genetics and molecular biology, 35(3), 693–700.
Mandai, M., et al. (2017). Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. New England Journal of Medicine, 376(11), 1038–1046.
Gekas, C., & Graf, T. (2010). Induced pluripotent stem cell-derived human platelets: One step closer to the clinic. The Journal of experimental medicine, 207(13), 2781–2784.
Takayama, N., et al. (2010). Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. The Journal of experimental medicine, 207(13), 2817–2830.
Nishimura, T., et al. (2013). Generation of functional platelets from canine induced pluripotent stem cells. Stem Cells Dev, 22(14), 2026–2035.
Feng, Q., et al. (2014). Scalable Generation of Universal Platelets from Human Induced Pluripotent Stem Cells. Stem Cell Reports, 3(5), 817–831.
Liu, Y., et al. (2015). Efficient generation of megakaryocytes from human induced pluripotent stem cells using food and drug administration-approved pharmacological reagents. Stem cells translational medicine, 4(4), 309–319.
Rodin, S., et al. (2010). Long-term self-renewal of human pluripotent stem cells on human recombinant laminin-511. Nature Biotechnology, 28(6), 611–615.
Mei, Y., et al. (2010). Combinatorial development of biomaterials for clonal growth of human pluripotent stem cells. Nature Materials, 9(9), 768–778.
Lee, H., et al. (2016). Establishment of feeder-free culture system for human induced pluripotent stem cell onDAS nanocrystalline graphene. Scientific Reports, 6(1), 20708.
Nakamura, S., et al. (2014). Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell, 14(4), 535–548.
Moreau, T., et al. (2016). Large-scale production of megakaryocytes from human pluripotent stem cells by chemically defined forward programming. Nature Communications, 7, 11208.
Ingavle, G., et al. (2019). Mimicking megakaryopoiesis in vitro using biomaterials: Recent advances and future opportunities. Acta Biomaterialia, 96, 99–110.
Thon, J. N., et al. (2014). Platelet bioreactor-on-a-chip. Blood, 124(12), 1857–1867.
Blin, A., et al. (2016). Microfluidic model of the platelet-generating organ: Beyond bone marrow biomimetics. Scientific Reports, 6(1), 21700.
Ito, Y., et al. (2018). Turbulence Activates Platelet Biogenesis to Enable Clinical Scale Ex Vivo Production. Cell, 174(3), 636-648.e18.
Hosseini, E., Mohtashami, M., & Ghasemzadeh, M. (2019). Down-regulation of platelet adhesion receptors is a controlling mechanism of thrombosis, while also affecting post-transfusion efficacy of stored platelets. Thrombosis Journal, 17(1), 20.
Hirata, S., et al. (2017). Selective Inhibition of ADAM17 Efficiently Mediates Glycoprotein Ibα Retention During Ex Vivo Generation of Human Induced Pluripotent Stem Cell-Derived Platelets. Stem cells translational medicine, 6(3), 720–730.
Ono, Y., et al. (2012). Induction of functional platelets from mouse and human fibroblasts by p45NF-E2/Maf. Blood, 120(18), 3812–3821.
Pulecio, J., et al. (2016). Direct Conversion of Fibroblasts to Megakaryocyte Progenitors. Cell Reports, 17(3), 671–683.
Madrid, M., et al. (2021). Autologous Induced Pluripotent Stem Cell–Based Cell Therapies: Promise, Progress, and Challenges. Current Protocols, 1(3):e88.
Basire, A., & Picard, C. (2014). Platelet allo-antibodies identification strategies for preventing and managing platelet refractoriness. Transfusion Clinique et Biologique, 21(4–5), 193–206.
Heal, J. M., et al. (1993). The role of ABO matching in platelet transfusion. European Journal of Haematology, 50(2), 110–117.
Estcourt, L. J., et al. (2017). Guidelines for the use of platelet transfusions. British Journal of Haematology, 176(3), 365–394.
Gourraud, P.-A., et al. (2012). The Role of Human Leukocyte Antigen Matching in the Development of Multiethnic “Haplobank” of Induced Pluripotent Stem Cell Lines. STEM CELLS, 30(2), 180–186.
Figueiredo, C., & Blasczyk, R. (2021). Generation of HLA Universal Megakaryocytes and Platelets by Genetic Engineering. Frontiers in Immunology, 12.
Börger, A. K., et al. (2016). Generation of HLA-Universal iPSC-Derived Megakaryocytes and Platelets for Survival Under Refractoriness Conditions. Molecular Medicine, 22, 274–285.
Xu, H., et al. (2019). Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell, 24(4), 566-578.e7.
Saito, S., et al. (2002). Platelet transfusion refractoriness caused by a mismatch in HLA-C antigens. Transfusion, 42(3), 302–308.
Suzuki, D., et al. (2019). iPSC-Derived Platelets Depleted of HLA Class I Are Inert to Anti-HLA Class I and Natural Killer Cell Immunity. Stem Cell Reports.
Gopalappa, R., et al. (2018). Paired D10A Cas9 nickases are sometimes more efficient than individual nucleases for gene disruption. Nucleic Acids Research, 46(12), e71–e71.
Ran, F. A., et al. (2013). Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell, 154(6), 1380–1389.
Norbnop, P., et al. (2020). Generation and characterization of HLA-universal platelets derived from induced pluripotent stem cells. Scientific reports, 10(1), 8472–8472.
Zhang, N., et al. (2016). CRISPR/Cas9-mediated conversion of human platelet alloantigen allotypes. Blood, 127(6), 675–680.
Acknowledgements
We acknowledge the staffs of Pediatric Cell and Gene Therapy Research Center, Gene, Cell & Tissue Research Institute, Tehran university of medical sciences, Tehran, Iran.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
EI and ZS were contributed in paper writing and generated figures. AAH were contributed in data discussion and revised the manuscript LPL and AA were contributed in revised the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent for Publication
Not applicable.
Competing Interest
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Izady, E., Saltanatpour, Z., Liu, LP. et al. Toward in Vitro Production of Platelet from Induced Pluripotent Stem Cells. Stem Cell Rev and Rep 18, 2376–2387 (2022). https://doi.org/10.1007/s12015-022-10366-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-022-10366-4