
Abstract
Mesenchymal Stem Cells (MSCs), as an adult stem cell type, are used to treat various disorders in clinics. However, derivation of homogenous and adequate amount of MSCs limits the regenerative treatment potential. Although mesoderm is the main source of mesenchymal progenitors during embryonic development, neuromesodermal progenitors (NMPs), reside in the primitive streak during development, is known to differentiate into paraxial mesoderm. In the current study, we generated NMPs from human embryonic stem cells (hESC), subsequently derived MSCs and characterized this cell population in vitro and in vivo. Using a bFGF and CHIR induced NMP formation protocol followed by serum containing culture conditions; here we show that MSCs can be generated from NMPs identified by not only the expression of T/Bra and Sox 2 but also FLK-1/PDGFRα in our study. NMP-derived MSCs were plastic adherent fibroblast like cells with colony forming capacity and trilineage (osteo-, chondro- and adipo-genic) differentiation potential. In the present study, we demonstrate that NMP-derived MSCs have an endothelial tendency which might be related to their FLK-1+/PDGFRα + NMP origin. NMP-derived MSCs displayed a protein expression profile of characterized MSCs. Growth factor and angiogenesis related pathway proteins were similarly expressed in NMP-derived MSCs and characterized MSCs. NMP-derived MSCs keep characteristics after short-term and long-term freeze-thaw cycles and localized into bone marrow followed by tail vein injection into NOD/SCID mice. Together, these data showed that hESC-derived NMPs might be used as a precursor cell population for MSC derivation and could be used for in vitro and in vivo research.
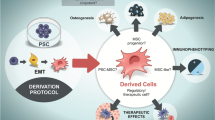
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cell therapy research and clinical approaches have been accelerated over the past 10 years [43]. Over 3000 cell transplantation clinical trials have been listed in the clinicaltrials.gov (https://clinicaltrials.gov/), and Mesencymal Stem Cell (MSC) based therapies comprised 44 % of all registered trials [43]. Cellular therapy has great importance due to the potential promising utilization in critical diseases such as cardiovascular diseases [4], neural diseases [50], and cancer [57]. Embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), and mesenchymal stem cells (MSCs) main cell sources for cell therapy research and applications, thanks to their differentiation capacity and self-renewal ability [1]. Choice of the appropriate cell source depends on many factors including ethical considerations, potency, optimized protocols and homogeneity [26].
MSCs among various cell types have been the aim of interest as a multipotent stromal cell source capable of differentiating into a variety mesoderm derived cell types including chondrocytes, osteoblasts, and adipocytes [54]. Because of immunomodulatory function, differentiation capacity, controlled cellular differentiation and long-term in vitro culturing capability [1], MSCs are used in the clinics for various disorders such as degenerative joint disease [38] and osteoarthiritis [18], cardiovascular diseases [4], aesthetic medicine [27], nervous and endocrine system diseases [20], and repair of damaged musculoskeletal tissues [29]. Although MSCs are important candidates in cellular therapies; functional heterogeneity because of different source of the donor, lack of adequate cell numbers, and harmful cryopreservation procedures limits the clinical treatments [35]. Inconsistent data from various trials of MSC-based cellular therapies might be explained unstandardized procedures and cultured cells [23] which could be controlled by establishment of culture conditions including derivation, expansion and characterization of the homogeneous MSC populations [36].
Human pluripotent stem cells (hPSC), including hESC and hiPSC, are candidates for cell therapy applications considering their typical features including pluripotency and self-renewal capacity [7]. More importantly, tissues derived from iPSCs have identical Human Leukocyte Antigen (HLA) profile with the donor cells, so that they are not rejected by the immune system [44]. Similar to iPSCs, human ESCs offer unprecedented regenerative potential to damaged tissues of the human body, as ESCs have unlimited proliferation potential and transform into various cell types [51]. ESC approaches support main investigation on the function and differentiation of human tissues and enhance the safety and effectiveness of human drugs [6, 39]. However, ES cell based therapy is restricted by various factors including tumorigenic properties, immunogenicity and lack of mature cell differentiation protocols [14].
Pluripotent stem cells can be used as unlimited sources to derive desired cell lineage for further research and therapy. Although PSC-derived MSC (PSC-MSC) developmental pathways are still unclear, identification of some of the differentiation protocols of PSCs in the cell culture enables homogeneous and high-quality MSC derivation from PSCs [1]. PSC-MSCs have significantly higher proliferation capacity and stronger immunomodulatory function compared to adult MSCs [34]. Remarkably, after the derivation of PSC-MSCs, cells still have osteogenic, chondrogenic, and adipogenic differentiation abilities, like the most distinguishing feature of adult MSCs [2]. However, in order to generate PSC-MSC, intermediate progenitors that belong to their differentiation pathways have to be clarified. For example, MSCs have multiple developmental origins which are containing neural crest and mesoderm [5, 42], and also, these pathways can be identified for determining intermediate progenitors [53].
Lineage-specific studies have identified that caudal epiblast (CE) contains a bipolar progenitor population, neural mesodermal progenitors (NMPs), located at the node streak border (NSB), spreading along with site into the caudal lateral epiblast (CLE), leading to neural and mesodermal precursors [45]. Recent grafting experiments in embryonic day 8.5 (E8.5) embryos have shown that the fate of NMP descendants is related to the relative levels of Sox2 or T protein expression [25] which caused by the potential to give rise to the spinal cord and paraxial mesoderm tissues such as cartilage and muscle cells [3, 47]. This advocates that NMP-derived MSC can exist during fetal development and also stay in our bodies during life [53]. Identification of NMPs as a potential intermediate cell source for homogenous MSC derivation might be useful in research and therapy. Wang and colleagues have shown the derivation of MSCs from NMPs [53] and we brought a new perspective to bank this cell population as a potential cell source.
Here, we generated and characterized MSCs from hESC-derived NMPs. We demonstrated the in vitro potential of these NMP-derived MSCs as a potential source for cell therapy and banking applications for regenerative medicine. Moreover, we elucidate fully characterization of NMP-derived MSCs population by in vivo homing capabilities to explore the clinical potential of this new alternative, safe, and promising cell population.
Materials and Methods
Cell Culture
Human ESC line H9 Cre-LoxP (Wicell, USA) which was labelled by GFP at the endogenous locus via Cre mediated recombination [19] was cultured in mTESR medium on Matrigel (Corning, USA) coated plates according to feeder free protocol [31]. Cells were incubated in a humidified incubator at 37 °C and 5 % CO2 atmosphere. The medium was replenished each day.
Human umbilical vein endothelial cells (HUVEC, ATCC-CRL 1730), mouse fibroblast cell line (L-929, ATCC-CCL-1) and SH-SY5Y neuroblastoma cells (ATCC-CRL-2266) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, USA) containing 10 % fetal bovine serum (FBS) (Invitrogen, UK), and 1 % of penicillin, streptomycin, and amphotericin B (PSA) (Invitrogen, UK).
ACS Telo cells (ASC52telo, ATCC-SCRC-4000) as hTERT immortalized adipose derived mesenchymal stem cells and dental pulp stem cells (DPSCs) which were previously isolated, characterized and cultured by our laboratory [12] were cultured in DMEM (low glucose, 1 g/L glucose) containing 10 % FBS, and 1 % PSA.
Differentiation of hESC into Neuromesodermal Progenitors (NMPs)
Human ES cells (H9 Cre-LoxP) were detached from coated wells with Versene (Gibco, USA) solution and plated at approximately 2 × 105 cells/well onto low attachment 24-well-tissue culture plates (Corning, USA) in N2B27 medium supplemented with 3 µM CHIR99021 (StemCell Technologies, USA), 12 ng/ml bFGF (Peprotech, UK) and 10 µM ROCK inhibitor Y-27,632 (StemCell Technologies, USA) as suspension culture for 72 h (Fig. 1a). N2B27 medium was used as a serum free culture medium for embryoid body generation which includes a mixture of 1:1 Neurobasal medium and DMEM-F12 medium (Gibco, USA), 0.5 % N2 supplement (Gibco, USA), 1 % B-27 supplement (Gibco, USA), 0.5 % of 10 % Bovine serum albumin (BSA) (Sigma-Aldrich, UK) in Phosphate Buffer Saline (PBS) (Gibco, USA), 1 % L-glutamine (Gibco, USA) and 1 % antibiotics including PSA.

Differentiation of hESCs into NMPs. (a) NMP derivation protocol. (b) NMP-derived MSC generation protocol. (c) T/Bra and Sox2 staining of cells at different time points (day 3, day 5 and day 24). (d) Number of T+/Sox2+ and T+/Sox2- colonies at D3 NMPs (e) Heat map representation of T/Bra and Sox2 immunostaining during MSC derivation. (f) T/Bra, Sox2, Wnt3, Nkx1-2 gene expression of day 3 NMPs. *P<0.05
NMP-derived MSC Generation in Cell Culture
NMPs (at day 3) were plated at approximately 106 cells on Matrigel (Gibco, USA)-0.1 % Gelatin (Sigma-Aldrich, UK) (1:200) solution coated 10 cm petri dishes (TPP, Switzerland) in N2B27 medium supplemented with 3 µM CHIR99021 and 10 µM ROCK inhibitor Y-27,632 for 48 h to induce mesoderm stage differentiation. ROCK inhibitor was removed from culture medium 24 h after plating. N2B27 culture medium was replenished with DMEM (1 g/L glucose) supplemented with 10 % FBS, 1 % PSA, 5 ng/ml bFGF for 72 h to induce MSC-like stage. MSC-like cells were re-plated at approximately 106 cells onto Matrigel-0.1 % Gelatin (1:200) coated 10 cm petri dishes in DMEM (1 g/L glucose) supplemented with 10 % FBS and 1 % PSA as shown in (Fig. 1b) and cultured until 24 days for further MSC stage differentiation. Two different stages were analyzed for MSC phenotype at day 10 (MSC I) and day 24 (MSC II). Cellular morphology was analyzed, and images were taken by a light microscope (Axio Vert.A1; Zeiss, Heidelberg, Germany).
Characterization of NMP-derived MSCs by Three Lineage Differentiation
NMP-derived MSCs were differentiated into chondrogenic, osteogenic, and adipogenic cells according to the literature [48] to confirm MSC characteristics. Differentiation medium contents used in the differentiation experiments were shown in Supplementary Table 1.
Cells were counted and plated onto 12-well-plates (TPP, Switzerland) at a cell density of 104 cells/ well in growth medium. After 24 h incubation, growth medium was replenished with osteogenic, chondrogenic and adipogenic medium. Cells were incubated in a humidified incubator at 37 °C and 5 % CO2 for 14 days and differentiation media were changed every other day. Differentiation of cells was confirmed by immunocytochemistry analysis, staining, and quantitative real time PCR Analysis (qPCR).
Staining of Differentiated MSC
Calcium deposition after osteogenic differentiation was visualized using Von Kossa staining (Abcam, UK). Cells were fixed with 4 % Paraformaldehyde (ThermoScientific, USA) at 4 ºC for 30 min. After fixation step, cells were stained with von Kossa kit according to the manufacturer’s instructions. Calcium depositions were visualized by using light microscope (Axio Vert.A1; Zeiss, Heidelberg, Germany).
Alcian blue staining (Sigma-Aldrich, UK) was performed to confirm chondrogenic differentiation. Cells were fixed with 4 % Paraformaldehyde and stained with Alcian blue solution according to the manufacturer’s instructions. Differentiated chondrocytes were observed under light microscope (Axio Vert.A1; Zeiss, Heidelberg, Germany).
Oil red staining (Sigma-Aldrich, UK) was performed to demonstrate lipid vesicles at the end of adipogenic differentiation. After 14 days of incubation, fixed cells were stained with Oil red according to the manufacturer’s instructions. Lipid particles were observed under light microscope (Axio Vert.A1; Zeiss, Heidelberg, Germany).
Quantitative Real Time PCR Analysis
Primers (Supplementary Table 2) were designed using Primer-BLAST software from the National Center for Biotechnology (Bethesda, MD, USA) and synthesized by Sentegen (Ankara, Turkey). \(\beta\)-Actin was used as a housekeeping gene. Total RNAs were isolated using Trizol reagent (Invitrogen, USA) according to the manufacturer’s instructions. Reverse transcription (RT) was performed using cDNA synthesis kit (Bio-Rad, USA). SYBR Green (Applied Biosystems, USA) method was used as described previously for qPCR analysis [15]. CFX96 RT-PCR system (Bio-Rad, USA) was used in all RT-PCR experiments.
Immunocytochemistry Analysis
Cells at different time points of the differentiation were stained with antibodies for NMP characterization, MSC characterization and confirmation of differentiation. Cells were fixed with 4 % PFA and permeabilized with 0.1 % Triton-X 100 solution (Sigma-Aldrich, UK). Afterwards, cells were washed with PBS and treated with 1 % BSA for blocking. Cells were incubated with the following primary antibodies including Sox2, T/Bra, CD45, CD73, Col1A, Col2A, PECAM1, VEGF, VE-cadherin and VCAM1 (SantaCruz Biotechnology, USA) and incubated overnight at 4 °C. Cells were rinsed and incubated with secondary antibodies including anti-mouse AlexaFluor 594 and anti-rabbit AlexaFluor 647 (Invitrogen, USA) for 30 min at room temperature. DAPI (1:1000) (ThermoScientific, USA) was used for nuclei staining. The images were taken using a confocal microscope (LSM 700; Zeiss, Heidelberg, Germany). Image J software was used for intensity measurements.
Self-Renewal Analysis of NMP-derived MSCs and Colony Forming Unit (CFU) Assay
Cells were subjected to self-renewal analysis by repeated colony formation assay according to the literature [41]. Cells were sub-cultured by four repeated colony forming unit assay (CFU) cycle for selected 10 different colonies as experimental replicates (Fig. 2c). Selected 10 colonies were dispersed and cultured at a cell density of 300 cells/well of 6-well plate (Corning, USA) in DMEM (1 g/L glucose) supplemented with 10 % FBS and 1 % PSA for 14 days (Fig. 2b) at each self-renewal cycle. Generated colonies were fixed at the end of 10 days for crystal violet staining (Sigma-Aldrich, UK). Images were taken by inverted microscope with equipped AxioCam ERc5s camera (ZEISS) and colonies were counted and analyzed by using Image J Software.

Characterization of NMP-derived MSC cells. (a) Cellular morphology during differentiation. Cells acquired MSC phenotype. (b) Colony formation capacity of MSC II cells. (c-i) Experimental design of sequential CFU assay for self-renewal analyses. (c-ii) Crystal violet stained colonies. (c-iii) Morphology of crystal violet stained MSC colonies. (d) Colony number and (e) colony diameter of MSC (MSC-II) colonies after 4 sequential CFU assays. (f) CD73 and CD45 immunostaining of MSC-II. (g) Cell doubling time and (h) cellular proliferation of NMP-derived MSC cells. (i) Differentiation of MSC-II cells into osteo-, chondro- and adipo-genic cells. Osteogenic differentiation was shown by vonKossa staining and Col I immunostaining. Chondrogenic differentiation was performed by Alcian Blue and Col II immunostaining. Adipogenic differentiation was shown by visualization of lipid droplets under microscope. *P<0.05
CFU assay was performed for confirm MSC characteristics after isolation, short-term and long-term freeze thaw cycles and self-renewal analysis as described above. Cells were seeded at a cell density of 300 cells/well of 6-well plate in DMEM (1 g/L glucose) supplemented with 10 % FBS and 1 % PSA. Cells were incubated for 14 days in a humidified cell culture incubator and medium was changed 3 times/week. Cells were fixed and stained with crystal violet. Images were taken by an inverted microscope with equipped AxioCam ERc5s camera (ZEISS) and colonies were counted and analyzed by using Image J Software.
Cell Proliferation Analysis
Cell counting analyses were done to check cell proliferation of NMP-derived MSCs at the end of 24 days. DPSCs and ASC Telo cells were used as characterized MSCs for comparison analysis. Cells were seeded onto 12-well plates at a cell density of 104 cells/well and cell proliferation was determined by cell counting.
Cell Doubling Analysis
Cell doubling of NMP-derived MSCs was calculated according to the protocol as described previously [9]. NMP-derived MSCs, DPSCs and ASC Telo cells were seeded onto 12-well plates at a cell density of 1 × 104 cells/well per well and counted every day to determine cell doubling time. Cell doubling time was calculated according to the formula [8]:
Where;
N(t) refers to the cell number at time t, N(0) is the cell number at time 0, gr is growth rate and t is time in hours. Thus, doubling time is calculated by fallowing formula:
Cell Cryopreservation Analysis of NMP-derived MSCs
Cell cryopreservation analyses were performed to explore banking capacity of NMP-derived MSCs. The effects of cryopreservation on NMP-derived MSCs were analyzed by repeated (short-term) and long-term freeze-thaw cycles according to previously described protocol by our group (Supplementary Fig. 5a) [11]. Different ten (C1- C10) colonies and 5 colonies of NMP-derived MSCs were subjected to long-term and short-term cryopreservation analysis.
Cells were seeded onto 150 mm tissue culture dishes (TPP, Switzerland) and incubated in humidified incubator until 80 % confluency. Cells were cryopreserved in 1.0 mL serum free freezing medium in 2 mL cryo-vials (CAPP, Germany) by using a controlled cell freezing container. Cells were kept at -80 °C freezer for 24 h and transferred to the -196 °C liquid nitrogen tank. This procedure was applied as four repeated freeze thaw cycles for short-term analysis and cells were kept at liquid nitrogen tank for 6 months for long-term storage. Trypan blue staining (Gibco, USA) was utilized to check cell viability after each cell thaw before plating [35]. The effect of cryopreservation on cell viability and MSC characteristics was investigated by morphology analysis and CFU assay. During each freezing process, cell images were taken with an invert microscope equipped with AxioCam camera (Axio Vert.A1; Zeiss, Heidelberg, Germany).
Cells were analyzed for cell surface markers and MSC characteristics after cryopreservation. Two colonies of cryopreserved NMP-derived MSCs were selected for further differentiation analysis to confirm MSC characteristics after short-term and long-term cryopreservation experiments. Cells were differentiated into osteogenic, chondrogenic and adipogenic lineages to confirm 3-lineage differentiation capacity of NMP-derived MSCs.
Tube Formation Assay
Tube formation analyses were performed to confirm endothelial differentiation capacity of NMP-derived MSCs. In vitro tube formation assay was performed on Matrigel-coated 24-well tissue culture plates as previously described by our group [10]. Briefly, pre-chilled plates were coated with Matrigel thawed at 4 °C overnight and incubated at 37 °C for 1 h to provide polymerization. 5 × 104 cells were re-suspended in serum-containing medium and 100 µl of cell suspension was put into the Matrigel-coated wells. Plate was incubated at 37 °C and 5 % CO2 in the incubator. The tube-like structures were observed after 24 h, and branches were counted in five randomly chosen areas for quantification.
Co-Culture Assay
Co-culture assay was conducted as previously described by our group [15]. HUVEC cells were stained by PKH 26 (red) cell labelling kit according to the manufacturer’s instructions. After staining, HUVEC and NMP-derived MSCs were co-cultured at 1:1, 1:3 and 3:1 ratio as 2 × 105 cells/well onto each well of 6-well plate. Cells were incubated in a humidified chamber at 37 °C and 5 % CO2 for 24 h. Pictures were taken using an inverted fluorescent microscope (Axio Vert.A1; Zeiss, Germany). Then, cells were trypsinized (Gibco, USA) and collected for flow cytometry analysis. Flow cytometry analyses were completed by Becton Dickinson FACS Calibur flow cytometry system (Becton Dickinson, CA).
Collection of Conditioned Medium
NMP-derived MSCs [(MSC I (D10)- MSC II (D24)], ASC Telo and DPSC were seeded onto T25 cell culture flasks (CAPP, Germany) and conditioned medium was collected when cells reached to 80 % confluency. Condition medium was filtered through the 0.22 µM syringe filter (GVS, Italy), aliquoted and stored at -20 °C until use.
Scratch Assay
Condition medium of NMP-derived MSCs were used for in vitro scratch analysis of fibroblast cells. Scratch assay was conducted according to the protocol as described previously [10]. L929 mouse fibroblast cells were seeded at 8 × 104 cells/well density onto the 12-well plates and incubated overnight in humidified incubator.
Next day, adherent cells were scratched with a sterile 200 µl pipette tip (CAPP, Germany), and the medium was immediately replaced with fresh media containing 20 % condition media. Wound healing experiments were performed using 20 % condition media collected from NMP-derived MSCs (D10 and D24), ASC Telo and DPSCs. Cells were observed under an inverted microscope (Zeiss PrimoVert) equipped with AxioCam ERc5s camera and Zen 2011 software (Carl Zeiss Microscopy, NY, USA) and images were taken at 0, 6 and 24 h.
Neuroprotective Activity
H2O2- induced toxicity model was established on SH-SY5Y neuroblastoma cell line to check neuroprotective activity. SH-SY5Y cells were plated onto 12-well plates with a cell density of 8 × 104 cells/well and incubated overnight in a humidified incubator. Next day, 300 µM H2O2 (Sigma-Aldrich, UK) and 20 % of condition media collected from NMP-derived MSCs (D10 and D24), ASC Telo and DPSCs were applied to the cells for 24 h. At the end of 24 h incubation, cell morphology was controlled, and images were taken by light microscope (Axio Vert.A1; Zeiss, Germany). Cell proliferation was determined by trypan blue staining and cells were counted using a hemocytometer.
Human Cytokine, Angiogenesis and Growth Factor Array
Cytokine, Angiogenesis and Growth Factor profile of NMP-derived MSCs were determined by protein membrane array analysis. Human Cytokine Array C1 (#AAH-CYT-1-8, RayBiotech, USA), Human Angiogenesis Antibody Array (#AAH-ANG-1-8, RayBiotech, USA), and Human Growth Factor Antibody Array C1 (#AAH-GF-1-8, RayBiotech, USA) were used to determine protein expression profile of cell at different time points (D0, D3, D5, D8, D10, D24). Protein panel of Cytokine, Growth Factor and Angiogenesis arrays were shown in Supplementary Table 3. Protein isolation was conducted for all experimental samples collected during differentiation of embryonic stem cells (D0, D3, D5, D8, D10, D24) and DPSCs as characterized MSCs. Experiments were conducted according to the manufacturer’s instructions. Briefly, proteins were isolated by Lysis Buffer of the array kit and protein concentrations were measured by BCA (ThermoScientific, USA) assay. Membranes were incubated in blocking buffer for 30 min and then treated with protein samples overnight at 4 °C on a rocking platform. Protein samples were removed, and membranes were washed 3 times with wash buffer. Biotinylated antibody cocktail diluted in blocking buffer were administered to the cells for 2 h at room temperature. Membranes were washed and treated with HRP for 2 h. Membranes were washed, protein expression was detected and visualized by a chemiluminescent system (Bio-Rad Biotechnology Inc., USA) [17]. Protein spot intensity measurements were conducted by Image Lab software Bio-Rad Laboratories Inc., Hercules, CA, USA) and results were analyzed by GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA).
In vivo Localization and Tissue Distribution analysis of NMP-derived MSCs
In vivo tissue and organ distribution analysis were performed for NMP-derived MSCs to perform in vivo characterization. The following procedure has been approved by the Yeditepe University Animal Experiments Ethics Committee (Approval number/date: 2019/12-4, 24.12.2019). Animals were housed at a constant temperature (23 ± 1 °C) and humidity (60 ± 10 %), maintained at a 12 h light/dark cycle and fed with food and water ad libitum. Experiments were conducted as described previously [46].
Experimental strategy was shown in Fig. 5a. 6 animals/experimental group were used to characterize NMP-derived MSCs in vivo. DPSCs and cells at different time points (days: D0, D3, D5, D8, D10, D24) were collected and 1 × 106 cells were transplanted into the NOD/SCID mice via tail vein injection. After 24 h, animals were sacrificed and samples including blood, spleen, liver, lung, heart, bone marrow and spinal cord were collected. Blood, spleen, liver, lung, heart, bone marrow and spinal cord samples were subjected to flow cytometry analysis.

Analysis of endothelial cells generated from NMP-derived MSCs. (a) Tube formation capacity of cells at different time points of differentiation protocol (Magnification: 4x). (b) Branch number of DPSCs, ASC-Telo, MSC I and MSC II cultured on Matrigel coated surfaces. (c) Tube-like structure formation of MSC II cells cultured on Matrigel:gelatin (1:200) coated surfaces. Cells generated branches between cells and tube-like structures between cell extensions. (d) PECAM1, VE-Cadherin, VCAM-1 and VEGF immunostaining and (e) expression profile of D24 MSC II cells. (f) Co-culture of MSC II and HUVEC cells at various ratios (g) Flow cytometry analysis of co-cultured MSC II and HUVEC cells and (h) percentage of cell number after one day culture. (i) Branch number of co-cultured MSC II and HUVEC cells after 24 h co-culture. *P<0.05
Erythrocytes were removed from whole blood by lysis buffer including 2.075 % ammonium chloride (NH4Cl) and 0.25 % sodium bicarbonate (NaHCO3) and washed twice with Flow cytometer (FCM) buffer prepared with PBS containing 1 % BSA and 0.5 % sodium azide (NaN3). Other tissues were divided into small pieces in FCM buffer and homogenized. The homogenates were filtered through a 40 μm nylon cell strainer (Greiner Bio-One, Germany) to discard debris. Then tissues were washed twice with FCM buffer and subjected to flow cytometry analysis to detect GFP-positive cells [22]. The flow cytometry analysis was conducted using Becton Dickinson FACS Calibur (Becton Dickinson, San Jose, CA, USA, model no: 342,975) flow cytometry system.
Statistical Analyses
One-way analysis of variance (ANOVA) with Tukey’s post-hoc test was utilized for statistical analysis. All statistical analysis was performed by GraphPad Prism version 8.0.0 for Windows (GraphPad Software, San Diego, California USA, www.graphpad.com). The p values less than 0.05 were accepted as statistically significant. Correleation matrix and principal component analysis (PCA) were generated by using the same program GraphPad Prism version 8.0.0 for Windows. Scatter plots were generated by an online tool ClustVis (https://biit.cs.ut.ee/clustvis/) [33].
Results
H9 Cre-LoxP Cells Successfully Differentiate into Neuromesodermal Progenitors (NMPs)
H9 Cre-LoxP hES cells were subjected to a 24 days differentiation protocol (Fig. 1b) which starts with the generation of NMPs (Fig. 1a) at the initial stage by activation of FGF and WNT pathways using bFGF and CHIR. H9 Cre-LoxP cells successfully differentiate into NMPs which was demonstrated by simultaneous T/Bra and Sox2 staining (Fig. 1c). An approximately 57 % of the NMP colonies were T/Bra and Sox2 double positive at day 3 (Fig. 1d) and double positive staining of NMP colonies reduced during differentiation (Fig. 1c). Cells loose T/Bra and Sox2 expression in a time dependent manner during 24 days differentiation (Fig. 1e, Supplementary Fig. 1a). Although there is still a robust expression of T/Bra and Sox2 at day 5 mesoderm cells (Fig. 1c, Supplementary Fig. 1a), staining intensity gradually reduced during differentiation into MSCs (Figure 1e). NMP marker genes such as T/Bra, Sox2, Wnt3, Nkx1-2 were significantly upregulated at NMPs (Fig. 1f). T/Bra and Sox2 gene expression was quite detected until day 8 and then sharply reduced during differentiation. Although Nkx1-2 specifically enhanced at day 3 and almost disappear at the remaining tie period, Wnt3 expression gradually increased until day 8, generate a peak and reduced at day 10 and day 24 by keeping its expression at a detectable level compared to D0 undifferentiated ES cells ((Figure 1f, Supplementary Fig. 1b). In addition, cells loose pluripotency gene expression during differentiation (Supplementary Fig. 1c). Oct3/4, Nanog and c-MYC gene expression was significantly reduced at the end of 24 days. MSC II population which is the MSC cells derived from NMPs after 24 days protocol did not show c-MYC gene expression and very small amount of Oct3/4 and Nanog expression was observed (Supplementary Fig. 1c).
NMPs Give Rise to Mesenchymal Stem Cells (MSCs) In Vitro
MSCs were derived from H9 Cre-LoxP hES cells through an initial 3 days NMP differentiation (Fig. 1a) followed by a 24-day MSC differentiation (Fig. 1b). Two different MSC population at day 10 (MSC I) and day 24 (MSC II) were analyzed for MSC phenotype. MSC-II resembles the characterized MSC phenotype and used for the further experiments. Cells lost ESC phenotype and acquired a spindle shape-fibroblast cell like morphology during differentiation as characterized MSCs such as DPSCs and ASC Telo cells. NMP-derived MSCs became plastic adherent starting from day 10 which were able to attach to the plastic tissue culture surfaces without extracellular matrix coating (Fig. 2a). MSCII cells represented MSC phenotype and generated colonies after single cell seeding onto plastic tissue culture surfaces (Fig. 2b). Colony formation assay was performed as 4 consecutive cycles (Fig. 2c-i). MSC II cells generated colonies at each cycle (Fig. 2c-ii) and cellular morphology was not changed (Fig. 2c-iii) indicating the self-renewal capacity of generated MSC II population. Colony number increased (Fig. 2d) but colony diameter decrease (Fig. 2e) after each cycle. This might be explained by MSC phenotype of cells after differentiation which enhanced surface attachment of single cells but decreased cellular proliferation as a result of aging in cell culture. NMP-derived MSC II cell population express high CD73 and low CD45 indicating the MSC characteristics of surface marker expression (Fig. 2f). Cells gradually increase CD73 protein expression and reduced CD45 during differentiation (Supplementary Fig. 2a). CD73 expression was detectable from the initial stages of MSC derivation protocol and enhanced in a time dependent manner (Supplementary Fig. 2a-2b). CD73, CD90 and CD105 gene expression were upregulated in NMP-derived MSC cells. MSC II at the end of the 24 days of differentiation protocol expressed high levels of marker genes including CD73 and CD90 but not CD105 (Supplementary Fig. 2c). Cell doubling and cellular proliferation were similar to characterized MSC populations such as DPSCs and ASC Telo cells. There was a minor increase in doubling time of NMP-derived MSC cells (Fig. 2g-h). MSC II cells were differentiated into mesoderm derived osteogenic, adipogenic and chondrogenic cell lineages. Osteogenic and chondrogenic differentiation were successfully performed, however cells were poorly differentiated into adipogenic cells (Fig. 2i). Differentiation capacity of MSC II cells was compared to MSC I cells, DPSCs and ASC Telo cells and successful differentiation was confirmed in vitro (Supplementary Fig. 3). MSC II cells successfully differentiated into 3 lineages which was similar to DPSCs and ASC Telo cells (Supplementary Fig. 3a-3b-3c-3d). MSC I cells showed a mild differentiation according to staining analysis (Supplementary Fig. 3b). However, differentiation related gene expression data demonstrated that MSC I and MSC II cell populations upregulated osteocalcin, adiponectin and Aggrecan genes as DPSCs and ASC Telo cells (Supplementary Fig. 4).
NMP-derived MSCs Keep Characteristics After Long-term Cryopreservation But Not Short-term Freeze-thaw Cycles
MSC II cell population was subjected to short-term freeze-thaw cycles and a long-term cryopreservation period (Supplementary Fig. 5a). Cellular morphology was not changed after repeated freeze-thaw cycles but cells were smaller after repeated freeze-thaw compared to regular MSC II population at day 24 (Supplementary Fig. 5b). CD45 expression was completely lost and CD73 expression was reduced at the end of fourth freeze-thaw cycle (Supplementary Fig. 5c) which might be explained by harmful effects of cryopreservation. Cellular proliferation was not significantly affected after each freeze-thaw cycle and cells adapted to cryopreservation process at the end of four short-term freeze-thaw cycles (Supplementary Fig. 5d). Cells were differentiated into osteogenic and chondrogenic lineages but lipid droplet generation as a marker of adipogenic differentiation was not observed (Supplementary Fig. 5e). Although adipogenic differentiation potential of NMP-derived MSCs were not superior before cryopreservation (Fig. 2i) compared to osteogenic and chondrogenic transformation, freeze-thaw cycles dramatically reduced adipogenic differentiation capacity (Supplementary Fig. 5e). In addition, osteogenic and chondrogenic differentiation was poor after four short-term freeze-thaw cycles. However, differentiation marker genes were expressed after three lineage differentiation of cells at the end of fourth freeze-thaw cycle (Supplementary Fig. 6).
Colony formation capacity of cells still existed after long-term cryopreservation (Supplementary Fig. 7a). Although colony diameter (Supplementary Fig. 7b) and number (Supplementary Fig. 7c) were variable between colonies, viable cell number showed the same trend for 10 different MSC II colonies (Supplementary Fig. 7d). CD73 expression reduced significantly after long-term cryopreservation (Supplementary Fig. 7e) indicating the reduction of MSC characteristics after long-term cryopreservation. Cells were able to differentiate into three mesoderm derived cell lineages which was confirmed by staining analysis (Supplementary Fig. 7f) and differentiation marker gene expressions (Supplementary Fig. 6).
NMP-derived MSCs Tend to Differentiate into Endothelial Cells
NMP-derived MSC II cell population exerted an endothelial phenotype in culture. MSC II cells generated tube-like structure on Matrigel coated wells which is an indicator of endothelial differentiation (Fig. 3a). MSC-I cells at D10 gathered together and generated limited number of branch like structures which was not a real tube formation morphology (Fig. 3a).

Protein profile of NMP-derived. (a) Hierarchical clustering and heat map representation of cells during differentiation protocol for (a.i.) Cytokine (aii) Growth Factor and (aiii) Angiogenesis arrays. (b) Correlation coefficient calculations of (bi) Cytokine (bii) Growth Factor and (biii) Angiogenesis arrays. (c) Principal component Analyses (PCA) of cells during differentiation protocol for (ci) Cytokine (cii) Growth Factor and (ciii) Angiogenesis arrays. (d) Scatter plot representation of (bi) Cytokine (bii) Growth Factor and (biii) Angiogenesis arrays
Branch number of MSC II cells was significantly increased on Matrigel coated wells compared to characterized MSCs and MSC-I (Fig. 3b). Moreover, CD73 expressing MSC-II cells were able to generate tube-like structures on Matrigel:gelatin (1:200) coated tissue culture plates in regular culture conditions which is an indicator of endothelial fate decision of NMP-derived MSC cells (Fig. 3c). This was shown by Flk-1 (Supplementary Fig. 8a) and PDGFRα (Supplementary Fig. 8b) immunocytochemistry analyses during differentiation of H9 Cre-LoxP hES cells. Cells demonstrated high Flk-1 and PDGFRα expression at the D3 NMP population and D24 MSC II during differentiation protocol (Supplementary Fig. 9a).
MSC II cells expressed endothelial markers such as PECAM1, VE-Cadherin, VCAM-1 and VEGF (Fig. 3d). PECAM1 and VE-Cadherin expression increased in a time dependent manner during differentiation. All endothelial markers except VCAM1 exerted an oscillatory phenotype during differentiation (Fig. 3e). VCAM-1 and VEGF expression is low compared to PECAM1 and VE-Cadherin and showed a mild increase in a time dependent manner (Supplementary Fig. 9b, Supplementary Figs. 10 and 11).
NMP-derived MSC II cell population was co-cultured with HUVEC cells to mimic endothelial behavior at culture conditions. MSC II cells generated tube-like structures and branches to surround the HUVEC cells at culture conditions (Fig. 3f). Increasing the MSC II cell ratio did not suppress HUVEC cell proliferation and vice versa at culture conditions after 24 h (Fig. 3g-h). However, increasing the MSC-II cell number significantly increased branch number at co-culture conditions (Fig. 3i) indicating the endothelial differentiation potential of NMP-derived MSC II cells.
NMP-derived MSCs demonstrated a MSC like protein expression profile
Protein membrane analyses were conducted to compare protein expression profile of proteins related to Cytokine, Growth Factor and Angiogenesis pathways (Fig. 4 and Supplementary Fig. 12). Hierarchical clustering and heat map representation of Growth Factor (Fig. 4aii) and Angiogenesis arrays (Fig. 4aiii) grouped MSC-II (day 24) and DPSCs together which was shown by correlation (Fig. 4b) and principal component analysis (Fig. 4c). D10 and MSC II (day 24) were clustered together for cytokine expression profile (Fig. 4a.i) more closely than DPSCs with a 0.43 correlation coefficient (Fig. 4bi) and 20.82 % PCA (Fig. 4ci). Correlation between the cells at different time points of differentiation protocol showed that there is a strong overall correlation between MSC II (day 24) and DPSCs for Growth Factor (Fig. 4bii) and Angiogenesis pathways (Fig. 4biii) which was supported by a 45.05 % (Fig. 4cii) and 44.17 PCA (Fig. 4ciii) scores respectively. The scatter-plot analysis of the protein expression profile of DPSCs versus MSC-II (day 24) demonstrated an even distribution for both Growth Factor (Fig. 4dii) and Angiogenesis arrays (Fig. 4diii) but not for Cytokine array (Fig. 4di) in the upper left and lower right areas.

In vivo characterization of NMP-derived MSCs. (a) Experimental design for in vivo tissue tissue distribution. (b) Flow cytometry analyses of GFP+ differentiated cells in various tissues and percentage of tissue distribution
Differentially expressed proteins including GM-CSF, GRO (α, β, γ), IL-6, IL-8, IL-10, PDGFR-β, EGFR, bFGF, IGF-1, MCP-1, TIMP1 and TIMP1 were detected in MSC II (D24) and DPSC groups compared to other differentiated cells (Supplementary Fig. 12).
NMP-derived MSCs are Localized into Bone Marrow
In vivo characterization of NMP-derived MSCs was performed by tissue distribution of differentiated cells by intravenous administration followed by Flow cytometry analysis (Fig. 5a). We compared the NMPs and NMP-derived MSCs at different time points for bone marrow localization as a marker of MSC phenotype. Both cell populations localized in spleen and bone marrow. Although 35 % of the MSC II population was localized into bone marrow, almost 20 % of the NMPs were migrated to the bone marrow region. Interestingly approximately 26 % NMPs were detected in spinal cord which is an NMP originated region during development (Fig. 5b).
We detected limited number of cells in lung, liver and heart for some of the cellular populations (Supplementary Fig. 13). Approximately 20 %, 6 % and 21 % cellular attachment were observed for day 3, day 10 and day 24 cells but not DPSCs in the spleen. In addition, an approximately 14 % day 10 and 20 % day 3 cells were detected in bone marrow, while only 5 % DPSCs migrate to the bone marrow region (Supplementary Fig. 13).
NMP-derived MSCs exert protective role
NMP-derived MSC cells (MSC I and MSC II) exerted wound healing properties and neuroprotective role in an oxidative stress induced toxicity model. Application of 20 % conditioned medium increased the scratch closure of fibroblast cells significantly compared to control group (Fig. 6a). We used conditioned medium of characterized ASC Telo and DPSCs cell to compare the wound healing activity of NMP-derived MSC I and MSC II cells. All experimental groups increased scratch closure of fibroblast cells compared to control group and no significant change was observed between characterized MSCs and NMP-derived MSC populations (Fig. 6b).

Analysis of protective properties of NMP-derived MSC. (a) Morphological images and (b) percentage of scratch closure analyses of conditioned medium on fibroblast cells. (c) Cell viability and (d) proliferation analysis of conditioned medium administrated SH-SY5Y cells under H2O2 induced toxicity
*P<0.05
In addition, application of 20 % conditioned medium increased cell viability and cell proliferation of SH-SY5Y after H2O2 induced oxidative stress (Fig. 6c and d). Although all MSC cells increased cell viability and proliferation, MSC I (day 10) increased the cell viability of neuroblastoma cell significantly.
Discussion
Success of cell therapy is depended on various factors including cell type and heterogeneity of cell source [32], cellular traits, in vitro/in vivo differentiation protocols and patient situation. Investigation of new cell sources is of interest among clinical therapies which were precisely understood in terms of progeny, fate decision, differentiation and safety.
NMP cells, as a progenitor cell population, give rise to the spinal cord neurons and paraxial mesoderm [24], which then generates somites prior to terminal differentiation into skeletal muscle, bone and dermis of late vertebrate development [37]. Identification of differentiation potential of NMPs will not only be an important step for understanding the developmental stages but also required for generation of clinically relevant cell populations. MSCs are preferred in clinical applications for treatment of various disorders including cardiovascular diseases, neurodegenerative diseases and orthopedic diseases [32] indicating their versatile applicability.
Here we show that human MSCs can also be isolated from embryonic stem cells via an intermediate NMP population. Our data support the derivation of MSCs from NMPs in vitro which was previously shown by Wang et al. [53]. We stablished an alternative differentiation protocol for derivation of MSCs from T/Bra and Sox2 double positive NMP population. Unlike Wang et al., we used a total 24 days differentiation protocol which was preceded by 3 days NMP generation followed by serum containing medium administration. This protocol allowed pluripotent stem cells to loose pluripotency gene expression profile and acquire an NMP phenotype at the initial stages which is very important for clinical applications to control tumorigenicty of pluripotent stem cells [14]. Although previous studies demonstrated an over 80 % T/Bra and Sox2 double positive NMP population [24, 53], we observed an approximately 57 % of double positive cells which might be related to the our differentiation protocol and concentration of bFGF and CHIR. Serum supplemented DMEM which was used by our group to derive adult stem cells previously [16] managed to transform NMP cell population into MSCs successfully. Two different time points were used to refer MSC population as MSC I and MSC II for 10 days and 24 days of in vitro differentiation protocol. NMP-derived MSCs were plastic adherent, had self-renewal capacity, and express MSC surface phenotype which were recorded as properties of pluripotent stem cell derived MSCs [55].
When compared to characterized MSC cell lines, we observe that there is a discrepancy in the three lineage differentiation potential. NMP-derived cells tend to differentiate into osteogenic cells which indicates the paraxial mesoderm fate of NMPs during vertebrate development. Notably, adipogenic differentiation was not successful in our experiments, suggesting that NMP-derived MSCs obtained from H9 Cre-LoxP cells were not capable of efficient fat cell differentiation. Although propensity of adipogenic differentiation for NMP-derived MSCs was observed in gene expression analysis of Adiponectin, cells failed to accumulate distinct and fine lipid droplets. This needs to be analyzed by application of different medium conditions on various pluripotent stem cell derived NMPs in further studies.
We observed FLK-1 expression at all stages during differentiation process but PDGFRα expression was detected notably at D3 NMP and D24 MSC II cell population. FLK-1+ and PDGFRα + cells represent lateral and paraxial mesoderm respectively during early mesoderm formation [13, 21, 56] and are associated with endothelial/hematopoietic cell fate decision [13]. We observed FLK-1 and PDGFRα expression in NMP cells which is not only related to their paraxial mesoderm fate but also might regulate the MSC differentiation of NMP cell population via an endothelial pathway. Vodyanik et al., demonstrated the derivation of MSCs from mesenchymoangioblast, a mesodermal progenitor with MSC and endothelial differentiation capacity, and suggested the progenitors with endothelial properties give rise to MSCs in their study [52]. Therefore, successful generation of NMP-derived MSCs with trilineage differentiation potential and endothelial characteristics observed in NMP-derived MSC cell population might be associated with FLK-1+ and PDGFRα + NMP cells at the initial stage of differentiation.
Moreover FLK-1+/PDGFRα + cells during early embryogenesis and ES cell differentiation is the part of primitive mesoderm which differentiate into MSCs [13, 21, 55]. These previous reports might explain the MSC differentiation potential of NMPs. High PDGFRα expression, as a marker of MSC [21], indicated the successful MSC generation at the end of 24 days protocol in our study.
With the identification of NMP-derived MSCs in the bone marrow and localization of NMPs in spinal cord, we enable to characterize in vivo cell behavior of MSCs and in vivo delineation of NMPs based on their progeny. Instead, we also confirmed the MSC phenotype after cryopreservation to characterize NMP-derived MSCs as potential candidates for clinical applications. Although cryopreservation did not affect the cellular proliferation, viability and growth, MSC characteristics reduced indicating the need for protocol optimization for cryopreservation. Differentiation of MSC II cells after long-term cryopreservation was better compared to cellular differentiation after four short-term freeze-thaw cycles.
The protein expression profile that segregate different cell populations during NMP-derived MSC differentiate demonstrated a high correlation for DPSCs and MSC cells at the end of 24 days. Although there is not a reported protein expression data, Wang et al. showed the similar gene expression patterns of NMP-derived MSCs and Bone Marrow Stem Cells (BMSCs) [53]. We preferred to use DPSCs because of their neural crest origin [58] during embryonic development which might resemble to NMP derived MSC population compared to other sources. Although cytokine array revealed a differential protein expression profile, some of the proteins such as GM-CSF, GROα and IL-10 which are secreted by or act on MSCs [28, 30, 40]; [49], were both upregulated in DPSCs and NMP-derived MSCs.
In the current study, we demonstrated the derivation of MSCs from a human ES cell line and characterized in vitro and in vivo. This NMP-derived MSC population has an endothelial background which regulates its MSC differentiation potential and behavior in culture after differentiation. Protein expression profile of NMP-derived MSCs indicates that these cells might pave the way for therapeutic purposes by regulating immune function, inflammation and cellular responses. In vivo therapeutic potential and molecular pathways underlying the cell fate decision during MSC differentiation of NMPs should be investigated by further studies. Identification of NMP-derived MSCs as a potential source for adult stem cells might be a promising candidate for future cell-based therapies.
Data Availability
Data is available upon request.
Code Availability
Not applicable.
References
Abdal Dayem, A., Lee, S. B., Kim, K., Lim, K. M., Jeon, T., Seok, J., & Cho, S. G. (2019). Production of mesenchymal stem cells through stem cell reprogramming. International journal of molecular sciences, 20(8), 1922
Ardeshirylajimi, A., Soleimani, M., Hosseinkhani, S., Parivar, K., & Yaghmaei, P. (2014). A comparative study of osteogenic differentiation human induced pluripotent stem cells and adipose tissue derived mesenchymal stem cells. Cell Journal (Yakhteh), 16(3), 235
Attardi, A., Fulton, T., Florescu, M., Shah, G., Muresan, L., Lenz, M. O., Lancaster, C., Huisken, J., van Oudenaarden, A., & Steventon, B. (2018). Neuromesodermal progenitors are a conserved source of spinal cord with divergent growth dynamics. Development, 145(21).
Augustin, M. (2012). Preconditioning methods in cell therapy of the heart, Doctoral dissertation, [online] Available at: https://helda.helsinki.fi/handle/10138/36906. Accessed 15 Oct 2021.
Bernitz, J. M., & Moore, K. A. (2014). Haematopoietic Stem Cells: Uncovering the origins of a niche. eLife, 3, e05041
Bremer, S., & Hartung, T. (2004). The use of embryonic stem cells for regulatory developmental toxicity testing in vitro-the current status of test development. Current Pharmaceutical Design, 10(22), 2733–2747
Chen, K. G., Mallon, B. S., McKay, R. D., & Robey, P. G. (2014). Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell, 14(1), 13–26
ATCC Animal Cell Culture Guide. [online] Available at: https://www.atcc.org/resources/culture-guides/animal-cell-culture-guide. Accessed 15 Oct 2021.
Demirci, S., Doğan, A., Apdik, H., Tuysuz, E. C., Gulluoglu, S., Bayrak, O. F., & Şahin, F. (2018). Cytoglobin inhibits migration through PI3K/AKT/mTOR pathway in fibroblast cells. Molecular and Cellular Biochemistry, 437(1), 133–142
Demirci, S., Doğan, A., Karakuş, E., Halıcı, Z., Topçu, A., Demirci, E., & Sahin, F. (2015). Boron and poloxamer (F68 and F127) containing hydrogel formulation for burn wound healing. Biological Trace Element Research, 168(1), 169–180
Demirci, S., Doğan, A., Şişli, B., & Sahin, F. (2014). Boron increases the cell viability of mesenchymal stem cells after long-term cryopreservation. Cryobiology, 68(1), 139–146
Demirci, S., Kaya, M. S., Doğan, A., Kalay, Å, ALTIN, N. Ã, & Şahin, Y. A. R. A. T. A. (2015). Antibacterial and cytotoxic properties of boron-containing dental composite. Turkish Journal of Biology, 39(3), 417–426
Ding, G., Tanaka, Y., Hayashi, M., Nishikawa, S. I., & Kataoka, H. (2013). PDGF receptor alpha+ mesoderm contributes to endothelial and hematopoietic cells in mice. Developmental Dynamics, 242(3), 254–268
Doğan, A. (2018). Embryonic stem cells in development and regenerative medicine. Cell Biology and Translational Medicine, 1, 1–15
Doğan, A., Demirci, S., Apdik, H., Apdik, E. A., & Şahin, F. (2017). Dental pulp stem cells (DPSCs) increase prostate cancer cell proliferation and migration under in vitro conditions. Tissue and Cell, 49(6), 711–718
Doğan, A., Yalvaç, M. E., Şahin, F., Kabanov, A. V., Palotás, A., & Rizvanov, A. A. (2012). Differentiation of human stem cells is promoted by amphiphilic pluronic block copolymers. International Journal of Nanomedicine, 7, 4849
Dou, Z., Ghosh, K., Vizioli, M. G., Zhu, J., Sen, P., Wangensteen, K. J., & Zhou, Z. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature, 550(7676), 402–406
Doyle, E. C., Wragg, N. M., & Wilson, S. L. (2020). Intraarticular injection of bone marrow-derived mesenchymal stem cells enhances regeneration in knee osteoarthritis. Knee Surgery, Sports Traumatology, Arthroscopy, 28, 3827–3842
Du, Z. W., Hu, B. Y., Ayala, M., Sauer, B., & Zhang, S. C. (2009). Cre recombination-mediated cassette exchange for building versatile transgenic human embryonic stem cells lines. Stem Cells, 27(5), 1032–1041
Eirin, A., & Lerman, L. O. (2014). Mesenchymal stem cell treatment for chronic renal failure. Stem Cell Research & Therapy, 5(4), 1–8
Farahani, R. M., & Xaymardan, M. (2015). Platelet-derived growth factor receptor alpha as a marker of mesenchymal stem cells in development and stem cell biology. Stem Cells International, 2015, 1–8
Fujiki, Y., Tao, K., Bianchi, D. W., Giel-Moloney, M., Leiter, A. B., & Johnson, K. L. (2008). Quantification of green fluorescent protein by in vivo imaging, PCR, and flow cytometry: comparison of transgenic strains and relevance for fetal cell microchimerism. Cytometry Part A: The Journal of the International Society for Analytical Cytology, 73(2), 11–118
Galipeau, J. (2013). The mesenchymal stromal cells dilemma—does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy, 15(1), 2–8
Gouti, M., Tsakiridis, A., Wymeersch, F. J., Huang, Y., Kleinjung, J., Wilson, V., & Briscoe, J. (2014). In vitro generation of neuromesodermal progenitors reveals distinct roles for wnt signalling in the specification of spinal cord and paraxial mesoderm identity. PLoS Biology, 12(8), e1001937
Henrique, D., Abranches, E., Verrier, L., & Storey, K. G. (2015). Neuromesodermal progenitors and the making of the spinal cord. Development, 142(17), 2864–2875
Herberts, C. A., Kwa, M. S., & Hermsen, H. P. (2011). Risk factors in the development of stem cell therapy. Journal of Translational Medicine, 9(1), 1–14
Hirvonen, T. (2014). Glycan binding proteins in therapeutic mesenchymal stem cell research. Doctoral dissertation, [online] Available at: https://helda.helsinki.fi/handle/10138/135978. Accessed 15 Oct 2021.
Hwang, J. H., Shim, S. S., Seok, O. S., Lee, H. Y., Woo, S. K., Kim, B. H., & Park, Y. K. (2009). Comparison of cytokine expression in mesenchymal stem cells from human placenta, cord blood, and bone marrow. Journal of Korean Medical Science, 24(4), 547
Kiani, A. A., Kazemi, A., Halabian, R., Mohammadipour, M., Jahanian-Najafabadi, A., & Roudkenar, M. H. (2013). HIF-1α confers resistance to induced stress in bone marrow-derived mesenchymal stem cells. Archives of Medical Research, 44(3), 185–193
Kyurkchiev, D., Bochev, I., Ivanova-Todorova, E., Mourdjeva, M., Oreshkova, T., Belemezova, K., & Kyurkchiev, S. (2014). Secretion of immunoregulatory cytokines by mesenchymal stem cells. World Journal of Stem Cells, 6(5), 552
Ludwig, T. E., Bergendahl, V., Levenstein, M. E., Yu, J., Probasco, M. D., & Thomson, J. A. (2006). Feeder-independent culture of human embryonic stem cells. Nature Methods, 3(8), 637–646. https://doi.org/10.1038/nmeth902
Lukomska, B., Stanaszek, L., Zuba-Surma, E., Legosz, P., Sarzynska, S., & Drela, K. (2019). Challenges and controversies in human mesenchymal stem cell therapy. Stem Cells International, 2019, 1–10.
Metsalu, T., & Vilo, J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Research, 43(W1), W566–W570
Peng, K. Y., Lee, Y. W., Hsu, P. J., Wang, H. H., Wang, Y., Liou, J. Y., & Yen, B. L. (2016). Human pluripotent stem cell (PSC)-derived mesenchymal stem cells (MSCs) show potent neurogenic capacity which is enhanced with cytoskeletal rearrangement. Oncotarget, 7(28), 43949
Phinney, D. G. (2012). Functional heterogeneity of mesenchymal stem cells: implications for cell therapy. Journal of Cellular Biochemistry, 113(9), 2806–2812
Pittenger, M. F., Discher, D. E., Péault, B. M., Phinney, D. G., Hare, J. M., & Caplan, A. I. (2019). Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regenerative Medicine, 4(1), 1–15
Pourquié, O. (2001). Vertebrate somitogenesis. Annual Review of Cell and Developmental Biology, 17(1), 311–350
Ripoll, C. B. (2010). Adult stem cell therapy in the twitcher mouse model of Krabbe’s disease utilizing mesenchymal lineage stem cells. Tulane University
Rolletschek, A., Blyszczuk, P., & Wobus, A. M. (2004). Embryonic stem cell-derived cardiac, neuronal and pancreatic cells as model systems to study toxicological effects. Toxicology Letters, 149(1–3), 361–369
Saldaña, L., Bensiamar, F., Vallés, G., Mancebo, F. J., García-Rey, E., & Vilaboa, N. (2019). Immunoregulatory potential of mesenchymal stem cells following activation by macrophage-derived soluble factors. Stem Cell Research & Therapy, 10(1), 1–15
Sarugaser, R., Hanoun, L., Keating, A., Stanford, W. L., & Davies, J. E. (2009). Human mesenchymal stem cells self-renew and differentiate according to a deterministic hierarchy. PloS One, 4(8), e6498
Sheng, G. (2015). The developmental basis of mesenchymal stem/stromal cells (MSCs). BMC Developmental Biology, 15(1), 1–8
Şişli, H. B., Hayal, T. B., Seçkin, S., Şenkal, S., Kıratlı, B., Şahin, F., & Doğan, A. (2019). Gene editing in human pluripotent stem cells: recent advances for clinical therapies. Cell Biology and Translational Medicine, 7, 17–28
Steinemann, D., Göhring, G., & Schlegelberger, B. (2013). Genetic instability of modified stem cells-a first step towards malignant transformation? American Journal of Stem Cells, 2(1), 39
Steventon, B., & Arias, A. M. (2017). Evo-engineering and the cellular and molecular origins of the vertebrate spinal cord. Developmental Biology, 432(1), 3–13
Su, W., Zhou, M., Zheng, Y., Fan, Y., Wang, L., Han, Z., & Xiang, R. (2011). Bioluminescence reporter gene imaging characterize human embryonic stem cell-derived teratoma formation. Journal of Cellular Biochemistry, 112(3), 840–848
Sweetman, D., Wagstaff, L., Cooper, O., Weijer, C., & Münsterberg, A. (2008). The migration of paraxial and lateral plate mesoderm cells emerging from the late primitive streak is controlled by different Wnt signals. BMC Developmental Biology, 8(1), 1–15
Taşlı, P. N., Doğan, A., Demirci, S., & Şahin, F. (2016). Myogenic and neurogenic differentiation of human tooth germ stem cells (hTGSCs) are regulated by pluronic block copolymers. Cytotechnology, 68(2), 319–329
Truong, M. D., Choi, B., Kim, Y., Kim, M., & Min, B. H. (2017). Granulocyte macrophage–colony stimulating factor (GM-CSF) significantly enhances articular cartilage repair potential by microfracture. Osteoarthritis and Cartilage, 25(8), 1345–1352
van Poll, D., Parekkadan, B., Rinkes, I. B., Tilles, A. W., & Yarmush, M. L. (2008). Mesenchymal stem cell therapy for protection and repair of injured vital organs. Cellular and Molecular Bioengineering, 1(1), 42–50
Vanderlaan, R. D., Oudit, G. Y., & Backx, P. H. (2003). Electrophysiological profiling of cardiomyocytes in embryonic bodies derived from human embryonic stem cells: Therapeutic implications. American Heart Association
Vodyanik, M. A., Yu, J., Zhang, X., Tian, S., Stewart, R., Thomson, J. A., & Slukvin, I. I. (2010). A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell, 7(6), 718–729
Wang, H., Li, D., Zhai, Z., Zhang, X., Huang, W., Chen, X., & Zou, Z. (2019). Characterization and therapeutic application of mesenchymal stem cells with neuromesodermal origin from human pluripotent stem cells. Theranostics, 9(6), 1683
Wei, X., Yang, X., Han, Z., Qu, F., Shao, L., & Shi, Y. (2013). Mesenchymal stem cells: a new trend for cell therapy. Acta Pharmacologica Sinica, 34(6), 747–754
Xu, M., Shaw, G., Murphy, M., & Barry, F. (2019). Induced pluripotent stem cell-derived mesenchymal stromal cells are functionally and genetically different from bone marrow‐derived mesenchymal stromal cells. Stem cells, 37(6), 754–765
Yamaguchi, T. P., Dumont, D. J., Conlon, R. A., Breitman, M. L., & Rossant, J. (1993). flk-1, an flt-related receptor tyrosine kinase is an early marker for endothelial cell precursors. Development, 118(2), 489–498
Yuan, Z., Lourenco, S. D. S., Sage, E. K., Kolluri, K. K., Lowdell, M. W., & Janes, S. M. (2016). Cryopreservation of human mesenchymal stromal cells expressing TRAIL for human anti-cancer therapy. Cytotherapy, 18(7), 860–869
Zhu, Y., Zhang, P., Gu, R. L., Liu, Y. S., & Zhou, Y. S. (2018). Origin and clinical applications of neural crest-derived dental stem cells. The Chinese Journal of Dental Research, 21(2), 89–100
Acknowledgements
This study was supported by Yeditepe University and Turkish Academy of Sciences Outstanding Young Scientists Award (TÜBA-GEBİP 2020). Albert A Rizvanov was supported by KFU state assignment 0671-2020-0058.
Funding
This study was supported by Yeditepe University and Outstanding Young Scientists Award (TÜBA-GEBİP 2020). Albert A Rizvanov was supported by KFU state assignment 0671-2020-0058.
Author information
Authors and Affiliations
Contributions
Ayşegül Doğan, Fikrettin Şahin, Albert A. Rizvanov and Selinay Şenkal contributed to the study conception and design. Material preparation, data collection and analysis were performed by Taha Bartu Hayal and Selinay Şenkal. In vitro experiments were conducted by Ayşegül Doğan, Taha Bartu Hayal, Selinay Şenkal and Derya Sağraç. In vivo experiments were conducted by Hatice Burcu Şişli, Engin Sümer and Fikrettin Şahin. Flow cytometry analysis and Immunocytochemistry experiments were performed by Ayla Burçin Asutay and Binnur Kıratlı. The first draft of the manuscript was written by Ayşegül Doğan, revised by Albert A. Rizvanov and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of Interest/Competing Interests
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 22 kb)

Supplementary Figure 1
Characterization of NMPs. (a) T/Bra and Sox2 staining of cells at different time points (D3, D5, D8, D10 and D24). (b) T/Bra, Sox2, Wnt3, Nkx1-2 gene expression at different time points (D3, D5, D8, D10 and D24) of differentiation protocol. (c) OCT3/4, Nanog, c-MYC gene expression at different time points. *P<0.05. (PNG 5829 kb)

Supplementary Figure 2
Characterization of NMP-derived MSC population. (a) CD73 and CD45 immunostaining of cells during differentiation (b) Heat map representation of CD73 and CD45 immunostaining during MSC derivation (c) CD73, CD90 and CD105 gene expression analysis during differentiation protocol. *P<0.05. (PNG 6445 kb)

Supplementary Figure 3
Three lineage differentiation analyses of NMP-derived MSCs. Osteo-, chondro- and adipo-genic differentiation of (a) MSC-II, (b) MSC-I, (c) ASC telo s and (d) DPSCs. (PNG 15537 kb)

Supplementary Figure 4
Gene expression analyses after three lineage differentiation of NMP-derived MSCs. Osteo-, chondro- and adipo-genic differentiation related gene expression analyses of MSC II, MSC I, ASC telo and DPSCs. Osteocalcin, Aggrecan and Adiponectin gene expression were calculated. *P<0.05. (PNG 138 kb)

Supplementary Figure 5
Repeated short-term freeze-thaw cycles of MSC II cell population. (a) Experimental design of four repeated short-term freeze-thaw cycles. (b) Morphological images of selected MSC II colonies after each freeze-thaw. (c) CD73 and CD45 immunostaining of cells short-term freeze-thaw cycles (d) Viable cell number of MSC II cells after four repeated short-term freeze-thaw cycles. (e) Three lineage differentiation analyses of NMP-derived MSCs (MSC II) after short-term freeze-thaw cycles. (PNG 1153 kb)

Supplementary Figure 6
Gene expression analyses after three lineage differentiation of cryopreserved MSC II cells. Osteo-, chondro- and adipo-genic differentiation related gene expression analyses of MSC II followed by short-term and long-term cryopreservation analyses. Osteocalcin, Aggrecan and Adiponectin gene expression were calculated. *P<0.05. (PNG 141 kb)

Supplementary Figure 7
Characterization of MSC II cells after long-term cryopreservation. (a) Crystal violet staining and morphological analyses. (b) Diameter and (c) Number of MSC II cell colonies after long-term cryopreservation. (d) Viable cell number of MSC II cells after long-term cryopreservation (e) CD73 and CD45 immunostaining of cells after long-term cryopreservation (f) Three lineage differentiation analyses of NMP-derived MSCs (MSC II) after long-term cryopreservation. (PNG 22023 kb)

Supplementary Figure 8
Immunocytochemistry analyses of (a) FLK-1 and (b) PDGFRα during differentiation protocol. (PNG 1714 kb)

Supplementary Figure 9
Heat map representation of (a) FLK-1 and PDGFRα, (b) PECAM1, VE-Cadherin, VCAM-1 and VEGF immunostaining during NMP-derived MSC derivation. (PNG 59 kb)

Supplementary Figure. 10
Immunocytochemistry analyses of VE-Cadherin and VCAM-1 during NMP-derived MSC derivation. (PNG 6186 kb)

Supplementary Figure 11
Immunocytochemistry analyses of PECAM1 (CD31) and VEGF immunostaining during MSC derivation. (PNG 5033 kb)

Supplementary Figure 12
Protein membrane array analysis of differentiated cells (a) Cytokine, Growth Factor and Angiogenesis arrays of cells at different time points of the differentiation protocol. (b) Selected differentially expressed proteins Cytokine, Growth Factor and Angiogenesis arrays. (PNG 3566 kb)

Supplementary Figure 13
Flow cytometry analyses of GFP+ differentiated cells in various tissues and percentage of tissue distribution during differentiation. (PNG 2558 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Şenkal, S., Hayal, T.B., Sağraç, D. et al. Human ESC-derived Neuromesodermal Progenitors (NMPs) Successfully Differentiate into Mesenchymal Stem Cells (MSCs). Stem Cell Rev and Rep 18, 278–293 (2022). https://doi.org/10.1007/s12015-021-10281-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-021-10281-0