
Abstract
The interest in the use of stem cells as a source for therapy has increased dramatically over the last decades. Different stem cell types have been tested in both in vitro and in vivo models, because of their properties such as differentiation potential, trophic effects and immune modulatory properties. To further optimize the use of different stem cell types for the treatment of disease in a clinical setting, it is necessary to know more about the in vivo behavior of these cells following engraftment. Until now, the golden standard to preclinically evaluate cell therapy was histology, which is an invasive method as the animals need to be sacrificed. This hampers the generation of dynamic information and results in only one single point in time available for analysis per animal. For more information regarding cell migration, in situ persistence, viability, proliferation and differentiation, molecular imaging can be used for imaging cells after transplantation dynamically and longitudinally, in a noninvasive way. With this technology, it becomes possible to track cells within the same subjects over a long period of time.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The concept of cellular therapy has existed for more than a century, and technologic advances from preclinical and early clinical studies promise to revolutionize the treatment of today’s health care problems, for example, autoimmune diseases, ischemic cardiovascular disease, and neurodegenerative disorders.
Cellular therapy can be defined as the transplantation of living cells for the treatment of medical conditions. Three major objectives of cellular therapy are regeneration of damaged tissue, replacement of function by secretion of biologically active molecules and redirection of aberrant processes. Stem cell therapy comprises a significant branch in the field of cellular therapy, as these primitive cells have the innate ability to differentiate towards different cell types, depending on their origin. In addition, it has also been shown that some stem cell types also secrete trophic factors that activate endogenous cell populations [1, 2].
A major hurdle blocking widespread clinical acceptance of cellular therapy is the fact that the mechanisms underlying success, or failure, are still poorly understood. The unraveling and optimization of these complex processes would benefit greatly from the development of dedicated metrics. These metrics can be long-term, such as improvement in tissue function or survival, or short-term, such as direct measurements of the transplanted cell numbers, their localization or functionality. Fast metrics that are noninvasive and allow longitudinal or kinetic data are ideal, since they improve the data generation of limited experiments and allow early treatment modification.
In vivo imaging is considered to be of pivotal importance in designing optimal treatment strategies. The clinically relevant in vivo imaging modalities comprise planar scintigraphy, SPECT, PET, and MRI. However, optical techniques such as FLI and BLI are very sensitive and are of great importance in the preclinical research field.
Optical imaging
Optical imaging is the imaging of visible light, with two different well-validated strategies: bioluminescence imaging (BLI) and fluorescence imaging (FLI). Both techniques have been used for in vitro and ex vivo research, e.g. fluorescence microscopy and luminescence assays, but these concepts have now also been extended to the noninvasive, in vivo imaging field. While both techniques detect levels of photons of visible light wavelengths, the key difference between these techniques is the way how these photons are generated (Fig. 1).

Basic principles of fluorescence and bioluminescence. With fluorescence, a fluorophore is excited by light at a specific wavelength, and emits light from another wavelength. For bioluminescence, D-luciferin is oxidized by luciferase in the presence of ATP and oxygen, generating visible light, pyrophosphate (PPi), adenosine monophosphate (AMP) and carbon dioxide (CO2)
Fluorescence occurs when an excitation light at a specific wavelength triggers the emission of light at another specific wavelength in a fluorophore such as green fluorescent protein (GFP).
Bioluminescence is the production of visible light resulting from an enzymatic reaction, catalyzed by enzymes called luciferases. Unlike for FLI, there is no need for excitation light in BLI. However, the exogenous luciferase gene has to be introduced into the host cell, tissue or organism. Two main luciferases are used for BLI: Firefly luciferase (Fluc) and Renilla luciferase (Rluc). Fluc oxidizes its substrate D-luciferin in the presence of oxygen and ATP, generating a detectable visible light signal. For Rluc, no ATP is needed and its substrate coelenterazin is oxidized to visible light photons in presence of only oxygen. An advantage of using Fluc is the possibility of detecting viable cells or tissues, due to the need for ATP in the catalyzation reaction. In most species, there is a very low background light production, leading to a very high signal to noise ratio. This implies that BLI is a very sensitive technique, with a relatively low resolution due to photon scatter. Another problem is the attenuation of the generated light photons within the tissue – mainly caused by absorption of the light by the hemoglobin present in the body. The main advantage of optical imaging is that it is very rapid, allows high throughput and is straightforward to implement, together with a relatively low cost, and the possibility to acquire different molecular signals at the same time (multiplexing). Its main limitation is the poor tissue penetration and signal attenuation, limiting their translational potential for clinical use. However, as a rapid and inexpensive tool, they have shown to be of great value in a wide range of research domains.
BLI is a very sensitive preclinical imaging modality, and it has been widely used for the visualization of stem cell engraftment in animals [3] but also for the imaging of endogenous stem cells in the mouse brain [4].
Magnetic resonance imaging
The fundamental principle underlying magnetic resonance imaging (MRI) is that unpaired nuclear spins, called magnetic dipoles (mainly hydrogen atoms in water), align themselves when placed into an externally applied magnetic field. In an MRI scanner, there is a strong magnet that produces a magnetic field surrounding the subject under investigation, creating a basic aligned state of the hydrogen atoms. After a transient perturbation by radio frequency pulses, the magnetic dipoles in the subject return to the basic aligned state, and emit electromagnetical waves that are detected by the scanner. The frequency and temporal characteristics of these radio waves are dependent on the molecular and tissue composition of the depicted object, based on the hydrogen atoms within water molecules [5]. Different magnetic contrasts can be generated for different purposes; this can be done by adjusting the pulse excitation and timing parameters. Well known and commonly used parameters for MRI are the so-called T1 and T2 weighting.
MRI is routinely used for anatomical imaging, because of its good soft tissue contrast and spatial resolution. On the other hand, the contrast between certain tissues can sometimes be too low, and contrast agents can be used. Furthermore, the sensitivity of MRI is typically low in the milli- to micromolar range. Moreover, quantification is sometimes limited because of the negative contrast generated by certain categories of contrast agents (e.g. paramagnetic iron-based contrast agents).
Nuclear imaging
Nuclear imaging techniques are based on the injection of a radiopharmaceutical (tracer) consisting of a radionuclide (a radioisotope) that is chemically linked to a molecule with a well-characterized biological behavior. Photons are emitted when the radionuclide decays and their detection by the imaging device allows determination of the three dimensional distribution of the tracer within the body. Nuclear imaging gives detailed information at the molecular level and therefore it is also being referred to as molecular imaging.
For nuclear imaging, there are two main modalities: positron emission tomography (PET) and single photon emission computed tomography (SPECT). The sensitivity is in the picomolar range, so sub-pharmacological amounts of tracer can be detected. Another strength of this technique is the translational aspect, since the same tracer molecule can be used over a range of species, going from mice over large animal models up to humans. Since in general little anatomical information is obtained with PET or SPECT, it is therefore often combined with other imaging modalities that provide detailed anatomical information such as X-ray based computed tomography (CT) or MRI. The development of PET/CT, SPECT/CT and even PET/MRI devices, which are now available at both the preclinical and clinical level, ensures optimal localization of the molecular information. In Table 1, the main differences between PET and SPECT for stem cell labeling are given.
Positron Emission Tomography (PET)
PET is based on the utilization of radioisotopes that emit a positron upon radioactive decay. An excess of protons in an unstable nuclide leads to the conversion of a proton to a neutron, and emission of a positron (β+) and a neutrino. The emitted positron scatters in the tissue while losing its kinetic energy, and then annihilates with an electron (its antiparticle), resulting in the emission of two high-energy photons of 511 keV at an angle of ~180° from each other (Fig. 2). These photons can be detected with a PET-camera. A PET-camera consists of a series of crystal detector rings that are coupled to a coincidence detection system [6]. The nearly simultaneous detection of two 511 keV photons by two opposite detectors within the scanner indicates an annihilation event has taken place on the line between these detectors. The line on which the annihilation event occurs (response line) enables to determine the three dimensional distribution of the isotope in the body after an applied series of reconstruction algorithms (Fig. 3). The spatial resolution of state of the art clinical whole body scanners, measured as the full-width-of-half maximum (FWHM), is typically around 4 to 5 mm, whereas preclinical scanners for animal research achieve a resolution of 0.8 to 1.5 mm.

Annihilation of a positron (β+) with an electron (e−), resulting in the emission of two photons at an angle of 180°, with an energy of 511 keV each

Coincidence detection principle. Near-simultaneous detection of two annihilation photons by two detectors situated approximately 180° from each other, allows one to draw an imaginary line between the two detectors (a). Following an array of such events, it is possible to locate a positron-emitting source in three dimensions (b and c)
Advantages of PET are its intrinsic tomographic nature allowing the generation of three dimensional images that reflect the distribution of the tracer. As PET studies can be performed dynamically with frames as short as a few seconds, yielding a series of tomographic images over time, it allows kinetic modeling of the data. These images, instead of reflecting radioactivity concentration, allow assessment of relevant biological features such as perfusion, metabolism, tissue receptor density or enzymatic activity.
Modern clinical and preclinical cameras allow obtaining images of a relevant part of the body, including so-called whole body scans that typically range from the skull to the thighs, within a few to tens of minutes. PET radionuclides can be readily incorporated into biological molecules with no or minimal influence on their biochemical properties (e.g. carbon-11 replacing a carbon-12 atom or fluorine-18 replacing a hydroxyl group). This is of great interest when one wants to image molecular processes without biologically perturbing the process itself (i.e. the tracer principle).
The major shortcoming of PET is the short half-life of the radionuclides used (e.g. carbon-11: 20.334 min, fluorine-18: 109.771 min) and the resulting necessity for most of these isotopes to be generated on-site by a dedicated cyclotron after which the radionuclide needs to be incorporated in the final radiopharmaceutical followed by purification, formulation and quality control. Not every institute or hospital is equipped with a cyclotron and an equipped radiopharmacy lab, which can be a barrier to perform PET imaging. Some PET radioisotopes, such as gallium-68 or rubidium-82, can be extracted from a mother/daughter generator which precludes the need for a cyclotron. The widespread adoption of the clinical use of PET has created an incentive to have radiopharmaceutical distribution available in many areas.
In addition, the short half-life of some of the most often used radionuclides can impede long-term follow-up of biological processes in vivo, although longer lived positron emitters such as iodine-124 (half-life 4.2 days) and zirconium-89 (half-life 3.2 days) are emerging in some clinical applications.
Single Photon Emission Computed Tomography (SPECT)
The principle of SPECT is based on isotopes that emit single gamma rays that have often a lower energy than the 511 keV photons detected by a PET scanner. The imaging is performed using a gamma camera that acquires multiple two dimensional projection images from multiple angles.
A gamma camera consists of a lead collimator used to define the direction of the detected gamma rays, a NaI (TI) scintillation crystal to convert the incident gamma rays to visible light, a light guide and a series of photomultiplier tubes that convert the visible light to a measurable current. This electrical current will be proportional to the energy of the detected gamma rays and will be used to generate an image. In clinical cameras, one or more detectors circulate around the subject while acquiring the information needed for reconstructing three dimensional images. In preclinical devices, a similar design can be used, or the animal can also be surrounded by detectors, allowing dynamic imaging. Generally, a computer is used to apply a tomographic reconstruction algorithm to the separate projection data obtained, generating a three dimensional image on the distribution of the radioisotope.
SPECT requires mechanical collimation of gamma rays emitted from the object to obtain information on the direction of the photons that hit the detector. The most common type of collimator for clinical imaging uses parallel holes to allow only photons that are perpendicular to the detector to reach the detector. For small-animal imaging, pinhole collimators are often used as they allow achieving a very high, submillimeter resolution (Fig. 4). A large fraction of the emitted photons will be absorbed by the collimator (this can lead to collimator absorption of more than 99 %) and thus the information from these photons is lost, which implies that the photon detection sensitivity of SPECT decreases accordingly and is typically one to two orders of magnitude lower than that of PET.

Basic components of a gamma camera: single photons are emitted from a subject, and due to the collimation, only photons that run perpendicular to the detector hit the scintillation crystal that will convert the photons to visible light. The generated light photons are guided and converted to an electric current in the photomultiplier tubes. This signal is then converted to an image (a). The most widely used types of collimators are the pinhole collimator for small-animal imaging, and the parallel hole collimator for clinical use (b)
The resolution in clinical applications is between 7 and 15 mm depending on camera and collimator characteristics but in current state of the art preclinical scanners resolution can be as low as 0.6 mm [7]. Another disadvantage of SPECT is the limited temporal resolution, as the detector circulates around the subject while acquiring planar projections which typically takes between 10 and 30 min per complete acquisition. Some dedicated solid state SPECT cameras can perform clinical grade images in 2 to 4 min. Modern preclinical devices have acquisition times that are in the 1 to 10 min range, depending on radionuclide and activity.
A marked advantage of SPECT is the possibility to image two different radioisotopes at the same time, so-called dual isotope imaging. This is only feasible if the gamma ray energies of these isotopes are sufficiently different, allowing multiplexing of two different molecular signals. Furthermore, the most common radioisotopes used in SPECT have half-lives ranging from ~6 h to ~8 days and so they allow the visualization of processes taking place on a moderately long timescale (tens of hours up to weeks).
Radiolabeling strategies for stem cells
Stem cells can be labeled with radioactive probes and visualized by nuclear imaging techniques. This radiolabeling procedure can be performed in two ways: in direct labeling, the tracer molecule is (stably) incorporated in, or attached to the cells by incubation in vitro; in indirect labeling, imaging reporter genes are introduced into host cells that encode proteins or molecules that will lead to the accumulation of a specific radioligand within cells in which the reporter is expressed. Currently, research in stem cell labeling is being conducted mainly in the preclinical field. This has changed the field of stem cell tracking in animals, with a great translational potential because many of the radioligands used for labeling stem cells have already been approved for other clinical indications by the American food and drug administration (FDA) and the European medicinal agency (EMA), and therefore are used routinely in clinical applications.
Direct labeling methods
Direct labeling of stem cells requires the incubation of the cells with the tracer in vitro prior to administration to the subject. These cells will thus be injected while containing the radionuclide after a labeling protocol in vitro (Fig. 5). Directly labeling stem cells generally involves the use of relatively long-lived isotopes, and gives information on the initial biodistribution and accumulation of cells in target organs. Direct stem cell labeling can yield very specific images of low numbers of cells because of the lack of background signal. The radiation dose to the stem cell recipient is also typically low as the activities used for cell labeling are relatively low. Also, direct labeling protocols can be implemented without the need of genetic cell manipulation. Therefore, translating direct labeling methods to the clinical setting is more straightforward than indirect strategies.

Schematic overview of the processes involved in direct cell labeling. Cells are first labeled with radionuclides in vitro through incubation, harvested and injected into a subject. This allows for the short-term visualization of these injected cells
Direct labeling has some disadvantages such as the dilution of the label after cell division, elution of the tracer from the cells over time, and the lack of cell tracking after tracer decay. Also, direct labeling strategies do not provide information on cell viability and cell death.
Three major pathways can be distinguished to directly label the cells. Firstly, tracer molecules can be taken up via a transporter mechanism (active pump or a passive channel) on the cell membrane; radioactive probes can also diffuse passively through the cell membrane, and thirdly they can bind on the outer side of the cell membrane (Fig. 6).

Direct cell labeling methods. Uptake of tracer molecules can occur via a pump or a channel, such as with 18 F-FDG. Afterwards, the tracer taken up by the cells will be phosphorylated leading to metabolic entrapment inside the cell. Tracers such as 111In-oxine are taken up via passive diffusion through the cell membrane, and once inside of the cell, these molecules will bind intracellular proteins, also leading to trapping of these molecules. Another direct labeling strategy involves the binding of molecules with a lipophilic tail to the outside of the cell membrane. This is the case for 18F-HFB. P phosphate group, i intracellular protein
Uptake via a pump or a channel
Labeling cells directly with radionuclides can be achieved through the uptake via pumps or channels located on the cell membrane. Hence, the tracer molecules are transported into the cytoplasm of the host cells. This implies that such molecules are slightly adapted forms of endogenous molecules present in the body. An advantage of such technique is the fact that one pump or channel can transport several molecules leading to signal amplification. This might allow the detection of lower cell numbers in the body. A prototypical example of this method is using 2-deoxy-2-[18F] fluoro-D-glucose, or 18F-FDG, which is taken up via the GLUT transporter family. FDG is routinely used in the clinic for the imaging of glucose metabolism of tumors, as well as brain and inflammation imaging [8–10].
-
a.
18F-FDG
18F-FDG is a glucose analog labeled with a positron-emitting fluorine-18 atom (half-life: 109 min) on the hydroxyl on the 2 carbon position. It is taken up by metabolically active cells and once inside the cytoplasm, it will enter the glycolytic pathway and will be phosphorylated by hexokinase to 18F-FDG-6-phosphate. No further metabolization will occur since the latter is not a substrate for phosphohexose isomerase. The phosphorylated molecule will not be able to diffuse back out of the cell, resulting in metabolic entrapment [11]. However, a substantial elution of the 18F-FDG from the cells after initial uptake has been reported by several groups [12–14]. This efflux is possibly caused by the high load of 18F-FDG within the cells, which cannot be entirely phosphorylated by hexokinase, and hence, the non-metabolized tracer molecules can diffuse back out of the cell. Furthermore, glucose phosphatase can actively dephosphorylate the phosphorylated 18F-FDG within the cells, and free 18F-FDG molecules can diffuse back to the extracellular space.
As PET images can be acquired up to some 5 to 6 tracer half-lives, monitoring of cells prelabeled with 18F-FDG is limited to a time frame of the first approximately 10 h after delivery. This limits the application of this labeling technique to the imaging of early biodistribution of injected cells and controlling the injection tract of labeled cells. Nevertheless, this can address important key questions, such as early sites of residence and in situ behavior in the next phase of engraftment in the tissue. Furthermore, labeling stem cells with 18F-FDG is a technique causing minimal or no radiotoxic effects on the cells. This is of utmost importance, since the labeling procedure as such is desired not to cause any side effects on the differentiation capacity and other biological properties possessed by stem cells intended for later transplantation [13, 15].
We have studied mesenchymal stem cells (MSCs) and multipotent adult progenitor cells (MAPCs) where different parameters were examined after incubation with 18F-FDG in vitro, such as proliferation, metabolic activity, differentiation capacity and ultrastructural characteristics. No significant changes in cell biology and function were observed (Fig. 7) [16].
Fig. 7 
Mouse MSCs and rat MAPCs were labeled with 18F-FDG, and showed an early tracer washout, with approximately 30–40 % of the tracer being retained inside the cells 3 h after labeling (panel A). Cell differentiation capacity of MSCs was not affected by 18F-FDG labeling (panel B). Small-animal PET-experiments with radiolabeled MAPCs and MSCs injected IV in mice showed a predominant accumulation in the lungs, and a substantial elution of 18F-FDG from both MSCs and MAPCs (panel C). This research was originally published in JNM. Wolfs E et al. 18F-FDG Labeling of Mesenchymal Stem Cells and Multipotent Adult Progenitor Cells for PET Imaging: Effects on Ultrastructure and Differentiation Capacity. JNM. 2013;54:447–454. © by the Society of Nuclear Medicine and Molecular Imaging, Inc. With permission of [16]
Until now, most of the experiments in the preclinical field using 18F-FDG were conducted for the assessment of early biodistribution in models of ischemia and myocardial infarction [12, 14]. Similar experiments were also already performed in patients, where hematopoietic stem cells (HSCs) were injected after myocardial infarction [17, 18].
In a landmark study, Hofmann et al. labeled autologous human bone marrow stem cells with 18F-FDG to monitor homing in patients with myocardial infarction [17]. Cells were either injected intracoronary or intravenously with or without first selecting the CD34+ population, since CD34 is a known marker of HSCs. The short-term kinetics of the biodistribution of the labeled cells was visualized with PET. With the intracoronary artery injection, 1.5 to 2.6 % of the total injected unselected HSCs were detected in the infarct zone. After injection of the 18F-FDG labeled CD34+ population, a more pronounced fraction of the injected cells homed to the infarcted zone, with approximately 25 % of the injected cells being detected in the zone of interest. With intravenous injection, no signal was detected in the infarct zone, and cells were trapped in the pulmonary capillary network. This study was the first to show the feasibility of 18F-FDG labeling of stem cells for tracking purposes in patients, and demonstrates large differences in cell behavior depending on the precise cell type and administration route (Fig. 8).
Fig. 8 
Myocardial homing and biodistribution of 18F-FDG labeled BMCs by Hofmann et al. Left posterior oblique (a) and left anterior oblique (b) views of chest and upper abdomen of a myocardial infarction patient after transfer of 18F-FDG–labeled, unselected BMCs into left circumflex coronary artery. BMC homing is detectable in the lateral wall of the heart (infarct center and border zone), liver, and spleen. Left posterior oblique (c) and left anterior oblique (d) views of chest and upper abdomen of a myocardial infarction patient after transfer of 18F-FDG–labeled, CD34+ BMCs into left anterior descending coronary artery. Homing of CD34 + cells is detectable in the anteroseptal wall of the heart, liver, and spleen. CD34 cell homing is most prominent in infarct border zone (arrowheads) but not infarct center (asterisk). With permission of [17]
In a study by Kang et al. [18] using 18F-FDG labeling of autologous HSCs, 17 patients with myocardial infarction were injected intracoronarily or intravenously. An accumulation of the injected cell population was detected in the infarct zone after intracoronary injection, whereas no cells were detected after intravenous injection, hereby confirming the results obtained by Hofmann et al.


Diffusion through the cell membrane
Another way of direct cell labeling is via the diffusion of radiolabeled molecules through the cell membrane. Here, the labeling procedure is not dependent on the presence of transporters or channels on the cells’ surface. For diffusion of tracers through the cell membrane, the radioisotopes of interest are typically bound to lipophilic molecules enabling their passive diffusion over the membrane. Once inside of the cell, these complexes dissociate releasing the isotopes that will bind to intracellular proteins, preventing the elution of the radioisotope from the cell. With this approach, several tracer molecules can pass the cell membrane and bind intracellular proteins. Hence, signal amplification can also occur with this labeling technique. Mainly two tracers for SPECT have been used for this approach: 111In-oxine and 99mTc-HMPAO.
-
a.
111In-oxine
111In-oxine is a molecule in which the radioisotope indium-111 is bound to the lipophilic chelator oxine, or 8-hydroxyquinoline. Due to the lipophilic nature of the 111In-oxine molecule, it is able to passively diffuse through the cell membrane. Once inside the cell, the complex dissociates, and the indium-111 nuclide binds to intracellular compounds acting as strong chelators, trapping the isotope stably inside of the cell [19]. 111In-oxine has been approved for SPECT imaging in humans by the FDA already in 1995, and is mainly used for labeling of leukocytes for the diagnosis of infectious diseases [19, 20]. 111In-oxine has a half-life of 2.8 days, and emits gamma rays of 171 and 245 keV. Therefore, tracking of labeled cells is possible up to 14 days after injection. However, binding of 111In-oxine has been shown to be somewhat reversible, and tracer leakage from the cells has been reported in different stem cell types such as MSCs and HSCs [21, 22]. Another drawback of 111In-oxine is the increased possibility of causing radiotoxic effects, with an impairment of the cellular viability and functionality, due to the emission of not only high-energy gamma rays (171 and 245 keV), but also due to damaging Auger electrons. Several groups have reported impaired cell function caused by 111In-oxine labeling [22, 23].
Nevertheless, 111In-oxine has been successfully used to image the biodistribution of stem cells in models of stroke and myocardial infarction [24, 25]. In a clinical setting, autologous MSCs have been labeled with 111In-oxine, as well as CD133+ peripheral blood progenitor cells [26–28]. Cells were successfully imaged using SPECT in patients with liver cirrhosis, chronic post infarction heart failure and chronic ischemic heart disease.
CD133+ peripheral blood progenitor cells were labeled with 111In-oxine in a clinical model of chronic post infarction heart failure [27]. Peripheral blood progenitor cells were sorted, and the CD133+ fraction was labeled in vitro using 111In-oxine. Cells were labeled with a high dose (111 MBq) or with a low dose (37 MBq), with a respective labeling efficiency of 28 and 15 %. An average viability in both conditions after labeling of 88 % was obtained. Labeled cells were injected intracoronarily in patients with chronic post infarction heart failure. A detailed analysis in two patients showed 6.9 to 8.0 % (after 2 h) and 2.3 to 3.2 % (after 12 h) residual radioactivity in the region of interest. Furthermore, no adverse events were observed during the procedure and up to 3 months follow-up. Therefore, this study demonstrated that CD133+ progenitor cells are capable of homing to the post-infarction remodeling myocardium after intracoronary injection (Fig. 9).
Fig. 9 
Anterior whole body scans acquired at 2, 12, 32, and 60 h after intracoronary injection of 111In-oxine-labeled CD133+ progenitor cells in a patient with chronic post infarction heart failure. Residual activity at the level of the heart is indicated by rectangles and clearly detectable for up to 36 h after injection. With permission of [27]
-
b.
99mTc-HMPAO
In order to circumvent the issue of radiation damage caused by 111In-oxine, another radiolabel has been used to label stem cells directly via diffusion through the cell membrane. 99mTc-HMPAO (hexamethylpropylene amine oxime) is a technetium-labeled molecule for SPECT with a half-life of 6 h, which is also able to diffuse through the cell membrane passively, after which a redox reaction with intracellular glutathione occurs, trapping the radionuclide in the cell. 99mTc-HMPAO has been used mainly for the detection of inflammation by labeling leukocytes showing low toxicity [29] and is in clinical routine use at the moment. Moreover, 99mTc-HMPAO by itself is used as a tracer for brain perfusion in routine clinical studies and is commercially available for these purposes.
In contrast with 111In-oxine, cell viability of both rat and human MSCs was not reduced after labeling with 99mTc-HMPAO [30, 31]. Nevertheless, a reduced colony forming ability has been observed, as well as a decreased proliferation caused by 99mTc-HMPAO labeling [30, 31] .
Clinical studies using 99mTc-HMPAO mainly involve the use of CD133+ and CD133- CD34+ stem cells in patients with chronic ischemic cardiomyopathy and myocardial infarction [32, 33]. Both groups were able to detect a population of injected stem cells in the infarcted area of the heart using SPECT.
-
c.
Other tracer molecules
Other molecules have been developed, in particular for PET imaging, such as 64Cu-pyruvaldehyde-bis (N4-methylthiosemicarbazone) (64Cu-PTSM). The lipophilicity of the this complex enables passive diffusion across the cell membrane, and after reduction within the cytoplasm the 64Cu-ion dissociates from the complex and binds to intracellular macromolecules, trapping it inside the cell. 64Cu has a half-life of 12.7 h and is one of the longer-lived PET radionuclides, enabling cell tracking up to 5 days. Likewise, different stem cell types have been labeled with this tracer such as HSCs, MSCs and ESCs [15, 34].

Binding to the cell membrane
A third way for directly labeling (stem) cells with a radioactive tracer molecule is via direct binding to the cell membrane.
One of the molecules that are being explored for these purposes is the radioactive tracer molecule hexadecyl-4-[18F] fluorobenzoate (18F-HFB), which consists of a lipophilic long-chain ester and a fluor-18 atom. It is efficiently and quickly absorbed into the cell membrane, without entering the cytoplasm. This molecule was first described by Ma et al. [35], who labeled rat MSCs with 18F-HFB, resulting in a yield of 25 %. As a proof of principle, labeled cells were injected in the tail vein of rats, and a clear signal in the lungs was shown.
The use of 18F-HFB was also investigated in cardiology, and compared with 18F-FDG [36]. 18F-HFB was shown to be a better tracer molecule for stem cell labeling and tracking in the setting of myocardial infarction, as there is less efflux of the 18F-HFB molecules from the cells compared to 18F-FDG, and the eluted molecules are not taken up by the surrounding cells, in contrast to 18F-FDG that can be taken up by surrounding myocytes and thus creating a nonspecific signal.
Indirect labeling methods
Indirect imaging of stem cells is based on the introduction of so-called “imaging reporter genes” into target cells. These reporter genes are transcribed into proteins which have a high affinity for a probe that can easily be detected through non-invasive imaging techniques. The main advantage of reporter gene imaging is that only viable cells with intact intracellular machinery can translate the gene into a protein that can be visualized. Furthermore, its transcription can be placed under the control of specific promoters or another gene, allowing transcription only in specific predefined conditions. In this approach, grafted cells are labeled in situ and assessed repetitively, typically using nuclear imaging methods, with a high sensitivity and short probe lifetimes (Fig. 10).

Schematic overview of the steps involved in indirect cell labeling. First, imaging reporter gene expression is induced in vitro in host cells. Reporter gene expressing cells are harvested, and injected into a subject. In a next step, the respective radiotracer is injected and imaging can be performed to determine the localization of the cells. Repetitive injection of the radiotracer allows for repetitive imaging and thus long-term visualization of the engrafted cells
Reporter genes
Reporter genes have been used for decades and have been developed for different imaging modalities such as optical imaging, nuclear imaging techniques and magnetic resonance imaging. The classic reporter genes with a widespread use in molecular biology are LacZ (encoding the enzyme β-galactosidase) and the ubiquitously used fluorescent proteins (such as green fluorescent protein, GFP, derived from the jellyfish Aequorea Victoria or other sources). The use of imaging reporter genes for nuclear imaging has some advantages over optical or magnetic resonance imaging-based approaches. Firstly, radionuclide-based methods offer a high sensitivity, making it possible to image relatively low levels of reporter gene expression. Furthermore, the use of radionuclide imaging for the detection of imaging reporter gene expression is (semi-) quantitative. The main advantage is the translational aspect, as the imaging modality and the tracers can be applied to humans [37]. The principal barrier for the latter is the induced overexpression of the reporter gene, which typically requires genetical modification of the administered cells.
Stem cells expressing nuclear imaging reporter genes can be visualized longitudinally in vivo using nuclear imaging techniques. These proteins have an affinity for a radioactive probe or tracer that can be injected repeatedly into the subject after its decay. This enables one to track and quantify the cells over a long time span without being limited to the half-life of the tracer used, like in the case of direct imaging. Furthermore, this labeling method does not only give information on cell trafficking, but it is also a measure of the in vivo viability and survival of grafted cells, as only viable cells will be expressing the imaging reporter genes. This is in contrast with the direct labeling method, where dead cells will pass on their label to neighboring cells or to the bloodstream.
Reporter gene delivery
Reporter genes are introduced into host cells using stable transfection or transduction with plasmids or viral vector systems, respectively. For stem cell imaging, in most applications a viral vector system is used that integrates the genes of interest into the DNA of the host cell and with a constitutive promoter driving the expression cassette. Until now, mostly lentiviral vectors have been used to transduce stem cells for indirect imaging [38]. With lentiviral vectors, derived from the lentivirus HIV-1, both dividing and non-dividing cells are transduced to a similar extent, and the genes of interest are integrated into the host cells’ DNA. This implies a stable and long-term transgene expression which will also be passed on to the daughter cells after cell division, and an in vivo expansion of the cell population will consequently result in an increased presence of the reporter proteins and thus increased signal [39]. Moreover, imaging reporter genes can be coupled to a therapeutic gene for the assessment of therapy in disease models.
The use of viral vectors (mostly lentiviral and retroviral vectors) to introduce reporter genes in a clinical setting has been questioned, as the introduction of genes into the host cell genome with this approach can cause insertional mutagenesis [40]. Since these genes are being introduced into specific sites such as transcriptionally active genes, around promoters and CpG islands in the host cells’ genome, certain vital genes can be activated or inactivated leading to mutations and/or the development of cancerous clones [41, 42]. Furthermore, reporter gene silencing is another aspect one has to consider when using viral vectors for long-term follow-up of grafted stem cells [43]. Reporter gene silencing refers to a gradual decrease of expression levels of the reporter gene due to epigenetic changes and gene repression during the differentiation processes in stem cells.
Several attempts have been done to overcome this issue of random integration by using gene targeting methods, such as the use of zinc finger nucleases (ZFN) to target one specific site in the genome [44]. ZFN have been designed and used to target the AAVS1 locus, encoding the ubiquitously expressed protein phosphatase 1, regulatory subunit 12C (PPP1R12C) gene, located on chromosome 19 [45, 46]. The AAVS1 locus is designated as a “safe harbor” locus, because the integration of target genes into this locus does not evoke pathological events [47], nor perturb the proliferation, karyotype or the expression of pluripotency genes in ESCs and iPSCs [44, 48] (Fig. 11). This approach was recently implemented for stem cell imaging, where the AAVS1 locus was targeted using ZFN to introduce a cassette of imaging reporter genes for multimodal imaging with fluorescence imaging or histology (mRFP; monomeric red fluorescent protein), BLI (Fluc; firefly luciferase) and PET (HSV-tk; herpes simplex virus thymidine kinase) [49].

Genome editing using ZFN. Two zinc finger nucleases consisting of a DNA binding domain (BD) and an endonuclease domain (scissors) dimerize, and generate a double stranded DNA break. A plasmid construct is presented containing a construct of interest flanked by two regions homologous to the regions flanking the double stranded DNA break, so-called homology regions (HR). Intrinsic homologous recombination will lead to the insertion of the construct of interest into the target location
Another challenge when using imaging reporter genes is that genes from a non-human source are prone to be recognized as ‘non-self’ and cause an immune reaction. Therefore, the search for human imaging reporter genes is now being intensified, as these proteins will be detected as ‘self’ with consequently less chances of evoking an immune response. The search for human imaging reporter genes is challenging, because these proteins are endogenous, and therefore are likely to result in a significant background signal. Additionally, cells expressing the imaging reporter genes will only take up a small fraction of the injected tracer. The major fraction of the injected tracer will be circulating in the bloodstream, and will either be taken up by other cells possibly expressing the same gene, and has to be cleared via its specific excretory tract.
Imaging reporter genes
For imaging and cell tracking, the introduction of imaging reporter genes can serve as a method for better understanding cell behavior in vitro, the molecular processes involved and the optimization of cell guidance in vivo. Moreover, two or more imaging reporter genes can be linked to each other into the same cassette driven by the same promoter by means of introducing linker sequences such as IRES (internal ribosomal entry site [50]) and peptide 2A sequences [51]. As a consequence, by coupling selected imaging reporter genes, multimodal imaging can be performed to corroborate data obtained by one modality by data from another one. Furthermore, imaging reporter genes also have the possibility to be used in a setting together with a therapeutic or suicide gene. Some imaging reporter genes used in nuclear medicine can also serve as a suicide or therapeutic gene. Both the sodium iodide symporter (NIS) [52] and the herpes simplex virus thymidine kinase (HSV-tk) enzyme [53] have shown to not only serve as an imaging reporter gene, but also as a suicide/therapy gene.
Overall, imaging reporter genes can be divided into three main groups: enzymes that will cause metabolic trapping of the reporter probe inside the cell, receptors to which the receptor probe will bind and finally transporters that will actively pump the probe from the extracellular space into the cell (Fig. 12).

Indirect labeling pathways involving imaging reporter genes. Imaging reporter genes are encoded to mRNA, which in turn, are translated to proteins. These reporter proteins can be enzymes that are able to metabolize molecules to a trapped form (eg HSV1-tk); receptors that are able to bind tracer molecules; or transporters that transport tracer molecules into the cell
-
a.
Enzymes
Enzymes as imaging reporter genes will metabolize specific radionuclides leading to their entrapment in the cells’ cytoplasm. Hence, only cells expressing the enzymatic imaging reporter gene, will contain the tracer molecules. One major advantage of enzymes as imaging reporter genes, is the fact that several probes will be modified by the same enzyme, and thus an accumulation of a higher number of modified probes and thus an amplification of the signal will occur [54–56]. The most extensively studied reporter gene for radionuclide imaging is the herpes simplex virus type 1 thymidine kinase (HSV1-tk).
-
1
Herpes simplex virus type 1 thymidine kinase
The viral HSV1-TK (which denotes the protein derived from the HSV1-tk gene) phosphorylates nucleoside analogs and has reduced substrate specificity, unlike its human variant. HSV1-TK thereby phosphorylates thymidine, its natural substrate, but also pyrimidine and acycloguanoside substrates. As a result of phosphorylation, substrates become negatively charged, preventing them to exit the cells, thus trapping them inside the cell. HSV1-TK is able to convert acycloguanosine pro-drugs (e.g. ganciclovir) to their cytotoxic effector metabolite, and therefore this enzyme is used mainly in cancer gene therapy protocols as a so-called “suicide” gene. The main idea is to deliver the HSV1-tk gene to tumor cells, and then use acycloguanosine to kill the cells expressing HSV1-tk. Accordingly, this HSV1-tk system was used with modest success in patients, mainly for the treatment of glioblastoma multiforme using ganciclovir at pharmacological levels [57, 58].
The HSV1-TK enzyme can also serve as a safeguard for cell and gene therapy, because cells expressing the HSV1-tk gene can be killed in a selective way, with a prodrug that is not harmful for non-expressing cells. In this way, ganciclovir will be converted to its cytotoxic metabolite after phosphorylation by the HSV1-tk expressing cells only.
Substrates of the HSV1-TK enzyme have been labeled with short- and long-living isotopes for both PET and SPECT imaging: 1) pyrimidine nucleoside analogs of which the uracil-based iodine-131 or iodine-124-labeled 2′-fluoro-2′-deoxy-5-iodo-1-β-D-arabinofuranosyl-5-iodouracil (124/131I-FIAU) has been used the most, and 2) purine nucleoside analogs of which the side chain fluorine-18-radiolabeled penciclovir analogue 9-(4-[18F]-fluoro-3-[hydroxymethyl]butyl)guanine (18F-FHBG) was mainly used for imaging. Unlike the use of HSV1-TK substrates for therapy, imaging based on these radiotracers will not cause a detrimental effect to the TK-expressing cells because the radiolabeled substrate is injected in subpharmacological doses [54, 59, 60].
HSV1-tk can thus be used as an imaging reporter gene for the imaging of gene transfer and (stem) cell therapy, as well as a therapeutic or “suicide” gene. These two features together in one single gene, renders the HSV1-tk to be an appealing gene for studies combining therapy and imaging approaches.
The shortcomings of HSV1-tk are its viral origin that can cause potential immune responses to the cells expressing the gene. Also, imaging in the brain is hampered by the fact that none of the tracers designed for HSV1-TK are able to cross the intact blood–brain barrier [60, 61]. Furthermore, therapeutic cells expressing HSV1-tk can be eradicated when antiviral therapy using HSV1-TK substrates is given. This can be problematic, especially in immunodeficient patients. Despite these shortcomings, HSV1-TK has been used for the imaging of stem cells in both clinical and preclinical settings. In preclinical studies involving HSV1-tk expression, stem cells were generally used as vehicles in suicide gene anti-cancer therapy [62], where gap-junction formation between the stem cells and the tumor cells occurs and intracellular molecules can be passed on from one cell to another, including phosphorylated - and thus toxic - ganciclovir leading to tumor cell death [63, 64].
Because HSV1-tk is not only a suicide gene, but also serves as an imaging reporter gene, several studies have used small-animal PET to determine cell fate and survival, predominantly in glioma models [65, 66]. Furthermore, also for murine ESCs expressing both Fluc and HSV1-tk it was possible to monitor their survival and proliferation after engraftment with both BLI and small-animal PET. Ganciclovir treatment resulted in a decrease in signal in tk-expressing tumors [67].
Recently, Perin et al. [68] have used HSV1-tk expressing MSCs for injections in a large animal porcine model of acute myocardial infarction. Long-term PET/CT was performed up to 5 months after injection, and it was shown that it was still feasible to in vivo monitor the fate of these intramyocardially injected MSCs (Fig. 13).
Fig. 13 
18F-FEAU PET/CT images observed in a pig 35 days after intramyocardial injection of sr39HSV1tk-expressing MSCs. PET/CT images of 18F-FEAU accumulation in three different locations of the heart (a–c). Images depict the possibility of imaging engrafted MSCs using PET/CT for at least 35 days, cells are situated at the red cross in the images. The location of aorta (Ao), left atrium (LA), right atrium (RA), left ventricle (LV), and right ventricle (RV), interventricular septum (SEP), and cervical vertebral body (VRT) are annotated on the images. With permission of [68]
Clinical studies regarding HSV1-TK have mainly focused on imaging of transgene expression in a setting of tumor therapy, mostly in glioma. Vectors for the expression of HSV1-tk were injected mostly intratumorally, and cell survival and treatment effect were monitored. In a first clinical study concerning HSV1-TK, retroviral vector producing murine cells were injected intratumorally in 15 patients with glioma, followed by ganciclovir treatment. This first study aimed to assess the safety, cell survival, vector release, possible immune response and antitumor effect of this treatment. Here, for the first time an indication for the bystander killing effect in patients was postulated because an antitumor effect was observed in 4 patients with a survival of the injected cells with, however, a limited gene transfer to the tumor cells [69]. Subsequent clinical studies have also included the use of HSV1-tk as an imaging reporter gene, with a clear focus on transgene expression [70, 71]. One case study published by Yaghoubi et al. [72], demonstrated the feasibility of tracking grafted transfected autologous cytolytic CD8+ T-cells expressing interleukin-13 and HSV1-tk with 18F-FHBG-PET in a patient with recurring glioblastoma multiforme (Fig. 14).
Fig. 14 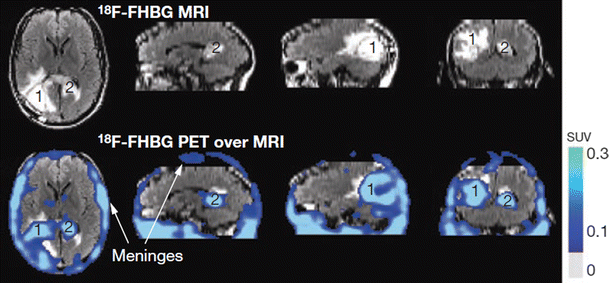
MRI and 18F-FHBG PET over MRI superimposed brain images, of a glioma patient who received infusions of autologous cytolytic CD8+ T cells expressing interleukin-13 and HSV1-tk. A surgically resected tumor [1] is evident in the left corner of his brain and a new, non-resected tumor in the center [2], near the corpus callosum. The infused cells had localized at the site of tumor 1 and trafficked to tumor 2. With permission of [72]
-
2
Other enzymatic reporter genes
HSV1-tk has been used as a therapeutic gene, as well as a reporter gene in both preclinical and clinical settings. It was shown to be essentially nontoxic, however, due to its viral nature, there are concerns about its potential for initiating an immunological response in the host [73, 74]. Therefore, human nucleoside kinase genes have been investigated such as the human mitochondrial thymidine kinase 2 gene (hTK2). hTK2 is one of four human deoxyribonucleoside kinases that are expressed in mitochondria [75], and it phosphorylates deoxythymidine, deoxycytidine, deoxyuridine and several antiviral and anticancer nucleoside analogs, such as 124I-FIAU [76–78].
Some other enzymatic systems different from the thymidine kinase genes have been proposed as imaging reporter genes, such as the varicella zoster virus thymidine kinase (VZV-tk) that can be imaged using fluorine-18 or carbon-11 radiolabeled bicyclic nucleoside analogues [79, 80]. Another enzymatic reporter gene is the aromatic L-amino acid decarboxylase that can be imaged with 6-[18F] fluoro-L-m-tyrosine [81]. Also, human tyrosinase has been used as a stand-alone multimodality imaging reporter gene in a human breast cancer cell line for in vitro and in vivo photoacoustic imaging (PAI), magnetic resonance imaging (MRI) and positron emission tomography (PET) [82]. However, to date these imaging reporter genes have not been used for stem cell tracking.
-
1
-
b.
Receptors
Receptors can also be used for imaging purposes with the advantage that the tracers designed do not need to cross the cell membrane, and bind on the outside of the cell. However, the proteins resulting from the expression of the imaging reporter genes will need to be located to the cell membrane and as only one ligand can bind to each receptor molecule, no signal amplification can be obtained, which implies that the ligand needs a high affinity for its receptor.
-
3
The dopamine 2 receptor
The dopamine 2 receptor (D2R) is primarily expressed in the brain striatum and pituitary gland. It is a 415 amino acid protein with seven transmembrane domains [83], and upon ligand binding, a G-protein is activated that leads to the inhibition of adenylyl cyclase and a subsequent decrease in cellular cyclic AMP levels [84]. The D2R can be imaged with BBB crossing tracers, for example, 3-(2′-[18F]-fluoroethyl)-spiperone ([18F]FESP) and [11C]raclopride. In the brain, a high endogenous expression of D2R will lead to a high background signal, particularly in the striatum, which may prevent the detection of subtle changes in D2R expression [54]. Another disadvantage of using the D2R as an imaging reporter gene is the possibility of endogenous dopamine to bind to the receptor, creating a competition between radioligands and endogenous dopamine levels. Furthermore, binding of a ligand to the D2R can ultimately lead to a modulation of cyclic AMP levels and thus activate its signaling cascade [54]. Therefore, a mutant version of the D2R was designed, in which binding of a ligand is uncoupled from activation of the signaling cascade. Still, this D2R80A has a preserved ligand binding capacity, with no physiological effect [85].
Nevertheless, the D2R is a human gene and will not be prone to an immune response against expressing cells. Therefore, the system has been used in some applications in which the D2R was mainly used as a second reporter gene in conjunction with another imaging reporter gene such as HSV1-tk or the human sodium iodide symporter (hNIS) [85–87]. Until now, the use of the D2R in stem cell trackings has not been applied further.
-
4
The human somatostatin receptor subtype 2
Another receptor that was validated as an imaging reporter gene for nuclear imaging is the human somatostatin receptor subtype 2 (hSSTr2). The hSSTr2 mediates the binding of somatostatin, which acts mainly as an inhibitory hormone in several organs in which it suppresses growth hormone release and inhibits pancreatic and gastrointestinal hormone release. This inhibitory signal also proved to have an antiproliferative effect in cancer cells [88].
The hSSTr2 is a G-protein coupled receptor situated on the plasma membrane, with seven transmembrane domains and belongs to the family of six somatostatin receptors (1, 2A, 2B, 2, 4 and 5). The hSSTr2 exists in two forms, subtype 2A and subtype 2B, due to alternative splicing of the SSTr2 mRNA. It is predominantly expressed in the gastrointestinal tract: pancreas, spleen and kidneys; as well as in the pituitary gland in the brain. It is also highly expressed in a number of neuroendocrine tumors [89]. In this setting, the hSSTr2 has been imaged for years in the clinic, as imaging its overexpression is a standard procedure in nuclear medicine for the detection of neuroendocrine tumors. Therefore, a number of tracer molecules were developed and used clinically, which is a major advantage when using the hSSTr2 as an imaging reporter gene [55]. Mainly, somatostatin analogs are used to develop tracers because the half-life of somatostatin in the circulation is limited to 2 or 3 min and rapidly gets broken down [89]. These analogs are biologically more stable, and they are developed for imaging and therapeutic purposes. Clinically, both 111In-pentreotide (which is commercially available (OctreoscanTM)) for SPECT and 68Ga-DOTATOC as well as 68Ga-DOTATATE for PET are the most used tracers. These molecules can also be labeled with yttrium-90 or lutetium-177 for peptide receptor radionuclide therapy and thus the hSSTr2 could be used as a suicide gene [90, 91]. Another advantage of hSSTr2 is the fact that it is a human gene, so the probability of evoking an immune response is very low. However, there are concerns regarding the activation of signal cascades upon binding of a receptor ligand. Therefore, the uncoupling of ligand binding to the activation of the signal cascade would be an appealing option, similar to the D2R80A mutation in the dopamine 2 receptor. Some work has already been done in this field, but further advances are still necessary [92]. Furthermore, a hemagglutinin (HA) sequence has been added on the extracellular N-terminus of the hSSTr2 gene, and imaging could be performed using a 99mTc-anti-HA antibody [93].
The hSSTr2 gene has been incorporated in viral vectors, and its gene expression has been monitored using radionuclides in a number of preclinical studies of tumor engraftment. Hence, it has been incorporated into an adenoviral vector that was used to transduce SKOV3 ovarian tumor cell lines. Noninvasive imaging of xenografts was performed using [111In]DTPA-D-Phe1-octreotide [94]. Also other somatostatin analogs were labeled with a radioactive compound, using Tc-99 m, In-111 and Re-188 [94–96].
In addition, adenoviral vectors encoding either hSSTr2 or GFP - as a control – were injected into subcutaneous pancreatic cell line tumors, followed by 111In-octreotide planar and SPECT imaging. The uptake of the tracer in hSSTr2 expressing tumors was significantly elevated compared to the controls expressing GFP. Also, mice with human breast cancer tumors were injected with an adenoviral vector encoding hSSTr2 or GFP, and imaged serially at 3 days and 2 weeks after injection of the vector. The signal decreased over time, with a low expression level of hSSTr2 after 3 weeks [97]. Nevertheless, a long term follow-up of hSSTr2 expression has been reported with adeno-associated vectors, up to 6 months [98]. This is in line with the fact that the choice of the vector delivery method has a significant influence on expression levels of reporter genes.
-
5
Other receptor-based reporter genes
An alternative to membrane receptors is the use of intracellular receptors such as the human estrogen receptor. Furukawa et al. have proposed to use the ligand binding domain of the estrogen receptor (hERL) together with 16 α-[F-18]-fluoro-17-β-estradiol (FES) ([18F]-FES) that has already been used in human studies [99]. Another reporter gene is the human type 2 cannabinoid receptor (CB2), which is the first receptor-based PET reporter system for imaging gene expression in the intact brain [100]. These reporter genes have not been used for stem cell tracking so far.
-
3
-
c.
Transporters
Another type of imaging reporter gene encodes transporter proteins that are translocated to the cell membrane after their expression. Transporter proteins will actively pump their specific probes from the extracellular space into the cell. One example of a transporter being used as a reporter gene for imaging is the human sodium iodide symporter or hNIS.
-
6
The human sodium iodide symporter
hNIS is expressed in high levels in the thyroid, and in lower levels in the gastric mucosa, salivary glands and lactating mammary gland. It belongs to the family of sodium/solute transporters, and it is an integral membrane glycoprotein with 13 transmembrane segments, situated on the basolateral membrane of thyroid follicular cells [101].
hNIS relies on the activity of the Na+/K+-pump which creates a Na+ electrochemical gradient over the basolateral membrane of the follicular cells. This is the driving force necessary for hNIS to transport two Na+ ions together with one I− ion. The I− ion is transported into the follicular cells against its electrochemical gradient. Following organification in the thyroid colloid, the transported I− ions will be used in the formation of thyroid hormones T3 and T4 [101]. hNIS transports I- ions, but is only partly selective. Other negatively charged ions are also transported such as ClO3 −, SCN−, SeCN−, NO3 −, Br−, BF4 −, IO4 − and BrO− 3 [102]. Furthermore, also all radioactive forms of I− are transported, as well as other isotopes such as technetium-99 m(99mTcO4 −) and rhenium-188/186 and these are in routine clinical use since decades for thyroid scintigraphy and radionuclide therapy. This is a major advantage, because these tracer molecules are readily available and their metabolism and clearance from the body are very well understood [103].
Besides the abovementioned properties that make hNIS an interesting imaging reporter gene, there is also little immunogenicity as it is a human protein. Furthermore, the low endogenous expression in a limited number of tissues will lead to a low background signal, allowing the use of NIS in a variety of imaging applications. Several groups have studied NIS gene transfer into a variety of tumor cell lines using either viral-vector or non-viral mediated gene delivery. All of these groups have reported a significantly increased radio-iodide uptake in NIS expressing tumor cells in vitro and in vivo [104–106].
There is the no organification of the I− ions taken up in non-thyroidal cells cells, and therefore a leakage of the tracer from the cells can occur. However, in masses such as tumors the leaked molecules can be taken up by surrounding cells, and the signal remains in the mass for a longer period of time [107, 108]. Despite this tracer leakage, hNIS remains an appealing reporter gene due to its many advantages listed above. Consequently, it has been used in various applications. hNIS expression was induced in rat cardiac-derived stem cells (rCDCs) which were injected intramyocardially, after the induction of myocardial infarction in rats. NIS-expressing rCDCs were visualized in the hypoperfused region using both 124I− PET and 99mTcO4 − SPECT up to 6 days post-injection. Here, 99mTcO4 − SPECT was more successful than 124I− PET, which might indicate the limitations of using I-124 as a radiotracer [109]. Likewise, adipose-derived stem cells expressing hNIS and GFP were injected into thigh muscles of mice, and animals underwent serial fluorescence imaging and 99mTcO4 − scintigraphy. Imaging was feasible up to 5 days following transplantation. Fluorescence imaging showed high within-group variability, whereas scintigraphic data provided more reliable information regarding cell tracking [110]. The expression of hNIS was also induced in MSCs using a multicistronic LV construct also encoding Fluc. Using these two imaging reporter genes, multimodal imaging was performed to visualize these cells in mice. 124I− PET was perfomed as well as Cherenkov luminescence imaging, where optical imaging devices are used to image radionuclides. Both modalities were successful in visualizing the engrafted MSCs, which was confirmed with BLI through the expression of Fluc [111].
Combined reporter gene imaging using hNIS with iron oxide labeling for MRI was performed by Higuchi et al. [112]. Human endothelial progenitor cells expressing hNIS and labeled with iron oxides were injected into the myocardium of nude rats. 124I− PET and MRI was performed on day 1, 3 and 7 after injection. Cells were successfully visualized with both PET and MRI on day 1, whereas the PET signal decreased from day 3 to day 7, and the MRI signal remained constant. Histological analysis depicted the presence of iron oxide-bearing macrophages, and an extensive apoptosis of cells at the transplantation site. This is a very important finding, showing that markers that establish cellular integrity (such as transporter reporter genes) outperform passive labels such as iron particles for the assessment of cell viability after transplantation.
Because hNIS can also be used for therapy, stem cells expressing hNIS have been used in tumor models where iodine-131 administration resulted in the inhibition of tumor growth [113, 114].
Recently, induced pluripotent stem cells (iPSCs) stably expressing hNIS were injected intramyocardially into a pig model of myocardial infarction. Cells could be visualized up to 15 weeks after injection, and co-injection of iPSCs and MSCs was likely to increase the engraftment of iPSCs at the target site [115] (Fig. 15).
Fig. 15 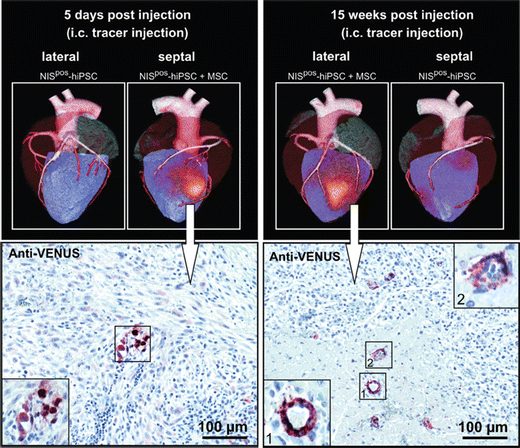
In vivo long-term SPECT/CT imaging of NIS-expressing iPSCs in pig hearts after 5 days and 15 weeks using I-123. Injection of iPSCs together with MSCs increased the iPSC engraftment in the heart. Injected iPS cells were also induced to express the fluorescent protein Venus, and therefore immunohistochemical anti-VENUS staining of tissue sections confirmed the presence of iPSCs. With permission of [115]
A phase 1 study applied hNIS in patients with localized prostate cancer. A replication-competent adenovirus encoding hNIS flanked by two suicide genes (Cytosine deaminase and thymidine kinase), was injected intraprostatically, and hNIS expression was imaged noninvasively using 99mTcO4 − SPECT up to 7 days. No extraprostatic signal was observed, demonstrating the safety of this approach for noninvasive imaging purposes in humans [116].
-
7
Other transporter-based imaging reporter genes
Another transporter system that has been validated for reporter gene imaging is the human norepinephrine transporter (hNET) [117]. It is known to be overexpressed in certain tumors (neuroblastoma, pheochromocytoma) and it plays an important role in cardiac innervation. Therefore, radioligands have been developed for the imaging of this transporter such as meta-iodobenzylguanidine (MIBG) labeled with iodine-123, iodine-124 and also iodine-131 [54]. hNET is a human protein, and therefore is not expected to cause an immune response. It has been used for the imaging of T cells, but has not been used for the tracking of grafted stem cells [118].
-
6



Future prospects of the field
Over the past decades, a variety of imaging techniques has been developed for non-invasive imaging of biological processes, ranging from small animals to man. Also, novel probes are being developed for the imaging of several molecules and biological interactions. The increasing availability of probes and imaging tools will therefore lead to a better understanding in biological concepts within living organisms, in a noninvasive and tomographic manner. Many clinical trials that are now ongoing are using cell therapy, and these would benefit from either direct or indirect labeling of these cells to enable tracking of cell migration following transplantation. Ultimately, this would lead to optimized cell delivery in therapeutic settings.
The use of reporter genes in humans still remains limited because of the genetic changes that are induced with relatively limited control over the exact genomic alteration. There is clearly a need to improve the genetic alteration of therapeutic cells with an emphasis on increased safety. Until now, introduction of imaging reporter genes into the host cell genome is uncontrolled, and random integrations may cause detrimental effects. This may be circumvented by the use of zinc finger nucleases [44], where integration of reporter genes can be targeted to specific genomic loci [49]. This will increase the safety in clinical applications of gene therapy and reporter gene imaging.
Data obtained from these nuclear modalities can be processed in a quantitative way, and represent the viable fraction of grafted cells repeatedly in a noninvasive setting.
The lack of anatomical information using nuclear imaging has been overcome by the use of hybrid cameras such as PET/CT cameras or the more recently emerging SPECT/CT and novel PET/MRI camera. These devices provide the exact anatomical correlate of the molecular signal. The establishment of core facilities for molecular imaging will also lead to better integrated worklflows and study protocols.
References
Le Blanc, K., Frassoni, F., Ball, L., Locatelli, F., Roelofs, H., Lewis, I., et al. (2008). Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet, 371(9624), 1579–86.
Sohni, A., & Verfaillie, C. M. (2011). Multipotent adult progenitor cells. Best Practice & Research Clinical Haematology, 24(1), 3–11.
Cao, Y. A., Wagers, A. J., Beilhack, A., Dusich, J., Bachmann, M. H., Negrin, R. S., et al. (2004). Shifting foci of hematopoiesis during reconstitution from single stem cells. Proceedings of the National Academy of Sciences of the United States of America, 101(1), 221–6.
Reumers, V., Deroose, C. M., Krylyshkina, O., Nuyts, J., Geraerts, M., Mortelmans, L., et al. (2008). Noninvasive and quantitative monitoring of adult neuronal stem cell migration in mouse brain using bioluminescence imaging. Stem Cells, 26(9), 2382–90.
Jacobs, R. E., & Cherry, S. R. (2001). Complementary emerging techniques: high-resolution PET and MRI. Current Opinion in Neurobiology, 11(5), 621–9.
Phelps, M. E., Hoffman, E. J., Mullani, N. A., & Ter-Pogossian, M. M. (1975). Application of annihilation coincidence detection to transaxial reconstruction tomography. Journal of Nuclear Medicine, 16(3), 210–24.
Vastenhouw, B., & Beekman, F. (2007). Submillimeter total-body murine imaging with U-SPECT-I. Journal of Nuclear Medicine, 48(3), 487–93.
Jones, T., & Rabiner, E. A. (2012). The development, past achievements, and future directions of brain PET. Journal of Cerebral Blood Flow and Metabolism, 32(7), 1426–54.
Krause, B. J., Schwarzenbock, S., & Souvatzoglou, M. (2013). FDG PET and PET/CT. Recent Results in Cancer Research, 187, 351–69.
Paterson, D. I., OMeara, E., Chow, B. J., Ukkonen, H., & Beanlands, R. S. (2011). Recent advances in cardiac imaging for patients with heart failure. Current Opinion in Cardiology, 26(2), 132–43.
Bhargava, K. K., Gupta, R. K., Nichols, K. J., & Palestro, C. J. (2009). In vitro human leukocyte labeling with (64) Cu: an intraindividual comparison with (111) in-oxine and (18) F-FDG. Nuclear Medicine and Biology, 36(5), 545–9.
Doyle, B., Kemp, B. J., Chareonthaitawee, P., Reed, C., Schmeckpeper, J., Sorajja, P., et al. (2007). Dynamic tracking during intracoronary injection of 18F-FDG-labeled progenitor cell therapy for acute myocardial infarction. Journal of Nuclear Medicine, 48(10), 1708–14.
Elhami, E., Goertzen, A. L., Xiang, B., Deng, J., Stillwell, C., Mzengeza, S., et al. (2011). Viability and proliferation potential of adipose-derived stem cells following labeling with a positron-emitting radiotracer. European Journal of Nuclear Medicine and Molecular Imaging, 38(7), 1323–34.
Zhang, Y., Thorn, S., DaSilva, J. N., Lamoureux, M., DeKemp, R. A., Beanlands, R. S., et al. (2008). Collagen-based matrices improve the delivery of transplanted circulating progenitor cells: development and demonstration by ex vivo radionuclide cell labeling and in vivo tracking with positron-emission tomography. Circulation. Cardiovascular Imaging, 1(3), 197–204.
Chen, M. F., Lin, C. T., Chen, W. C., Yang, C. T., Chen, C. C., Liao, S. K., et al. (2006). The sensitivity of human mesenchymal stem cells to ionizing radiation. International Journal of Radiation Oncology, Biology, and Physics, 66(1), 244–53.
Wolfs, E., Struys, T., Notelaers, T., Roberts, S. J., Sohni, A., Bormans, G., et al. (2013). 18F-FDG labeling of mesenchymal stem cells and multipotent adult progenitor cells for PET imaging: effects on ultrastructure and differentiation capacity. Journal of Nuclear Medicine, 54(3), 447–54.
Hofmann, M., Wollert, K. C., Meyer, G. P., Menke, A., Arseniev, L., Hertenstein, B., et al. (2005). Monitoring of bone marrow cell homing into the infarcted human myocardium. Circulation, 111(17), 2198–202.
Kang, W. J., Kang, H. J., Kim, H. S., Chung, J. K., Lee, M. C., & Lee, D. S. (2006). Tissue distribution of 18F-FDG-labeled peripheral hematopoietic stem cells after intracoronary administration in patients with myocardial infarction. Journal of Nuclear Medicine, 47(8), 1295–301.
Thakur, M. L., Segal, A. W., Louis, L., Welch, M. J., Hopkins, J., & Peters, T. J. (1977). Indium-111-labeled cellular blood components: mechanism of labeling and intracellular location in human neutrophils. Journal of Nuclear Medicine, 18(10), 1022–6.
Horcajada, J. P., Gutierrez-Cuadra, M., Martinez-Rodriguez, I., Salas, C., Parra, J. A., Benito, N., et al. (2012). High prevalence of upper urinary tract involvement detected by 111indium-oxine leukocyte scintigraphy in patients with candiduria. European Journal of Clinical Microbiology & Infectious Diseases, 31(3), 237–42.
Chin, B. B., Nakamoto, Y., Bulte, J. W., Pittenger, M. F., Wahl, R., & Kraitchman, D. L. (2003). 111In oxine labelled mesenchymal stem cell SPECT after intravenous administration in myocardial infarction. Nuclear Medicine Communications, 24(11), 1149–54.
Brenner, W., Aicher, A., Eckey, T., Massoudi, S., Zuhayra, M., Koehl, U., et al. (2004). 111In-labeled CD34+ hematopoietic progenitor cells in a rat myocardial infarction model. Journal of Nuclear Medicine, 45(3), 512–8.
Nowak, B., Weber, C., Schober, A., Zeiffer, U., Liehn, E. A., von Hundelshausen, P., et al. (2007). Indium-111 oxine labelling affects the cellular integrity of haematopoietic progenitor cells. European Journal of Nuclear Medicine and Molecular Imaging, 34(5), 715–21.
Mitkari, B., Kerkela, E., Nystedt, J., Korhonen, M., Mikkonen, V., Huhtala, T., et al. (2013). Intra-arterial infusion of human bone marrow-derived mesenchymal stem cells results in transient localization in the brain after cerebral ischemia in rats. Experimental Neurology, 239, 158–62.
Kraitchman, D. L., Tatsumi, M., Gilson, W. D., Ishimori, T., Kedziorek, D., Walczak, P., et al. (2005). Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation, 112(10), 1451–61.
Gholamrezanezhad, A., Mirpour, S., Bagheri, M., Mohamadnejad, M., Alimoghaddam, K., Abdolahzadeh, L., et al. (2011). In vivo tracking of 111In-oxine labeled mesenchymal stem cells following infusion in patients with advanced cirrhosis. Nuclear Medicine and Biology, 38(7), 961–7.
Schots, R., De Keulenaer, G., Schoors, D., Caveliers, V., Dujardin, M., Verheye, S., et al. (2007). Evidence that intracoronary-injected CD133+ peripheral blood progenitor cells home to the myocardium in chronic postinfarction heart failure. Experimental Hematology, 35(12), 1884–90.
Caveliers, V., De Keulenaer, G., Everaert, H., Van Riet, I., Van Camp, G., Verheye, S., et al. (2007). In vivo visualization of 111In labeled CD133+ peripheral blood stem cells after intracoronary administration in patients with chronic ischemic heart disease. The Quarterly Journal of Nuclear Medicine and Molecular Imaging, 51(1), 61–6.
Jorgensen, C., Couret, I., Bologna, C., Rossi, M., & Sany, J. (1995). Radiolabelled lymphocyte migration in rheumatoid synovitis. Annals of the Rheumatic Diseases, 54(1), 39–44.
Detante, O., Moisan, A., Dimastromatteo, J., Richard, M. J., Riou, L., Grillon, E., et al. (2009). Intravenous administration of 99mTc-HMPAO-labeled human mesenchymal stem cells after stroke: in vivo imaging and biodistribution. Cell Transplantation, 18(12), 1369–79.
Park, B. N., Shim, W., Lee, G., Bang, O. Y., An, Y. S., Yoon, J. K., et al. (2011). Early distribution of intravenously injected mesenchymal stem cells in rats with acute brain trauma evaluated by (99 m) Tc-HMPAO labeling. Nuclear Medicine and Biology, 38(8), 1175–82.
Goussetis, E., Manginas, A., Koutelou, M., Peristeri, I., Theodosaki, M., Kollaros, N., et al. (2006). Intracoronary infusion of CD133+ and CD133-CD34+ selected autologous bone marrow progenitor cells in patients with chronic ischemic cardiomyopathy: cell isolation, adherence to the infarcted area, and body distribution. Stem Cells, 24(10), 2279–83.
Kollaros, N., Theodorakos, A., Manginas, A., Kitziri, E., Katsikis, A., Cokkinos, D., et al. (2012). Bone marrow stem cell adherence into old anterior myocardial infarction: a scintigraphic study using Tl-201 and Tc-99 m-HMPAO. Annals of Nuclear Medicine, 26(3), 228–33.
Tarantal, A. F., Lee, C. C., Batchelder, C. A., Christensen, J. E., Prater, D., & Cherry, S. R. (2012). Radiolabeling and in vivo imaging of transplanted renal lineages differentiated from human embryonic stem cells in fetal rhesus monkeys. Molecular Imaging and Biology, 14(2), 197–204.
Ma, B., Hankenson, K. D., Dennis, J. E., Caplan, A. I., Goldstein, S. A., & Kilbourn, M. R. (2005). A simple method for stem cell labeling with fluorine 18. Nuclear Medicine and Biology, 32(7), 701–5.
Zhang, Y., Dasilva, J. N., Hadizad, T., Thorn, S., Kuraitis, D., Renaud, J. M., et al. (2012). (18) F-FDG cell labeling may underestimate transplanted cell homing: more accurate, efficient and stable cell labeling with hexadecyl-4-[(18) F] fluorobenzoate for in vivo tracking of transplanted human progenitor cells by positron emission tomography. Cell Transplantation, 21, 1821–35.
Gambhir, S. S., Herschman, H. R., Cherry, S. R., Barrio, J. R., Satyamurthy, N., Toyokuni, T., et al. (2000). Imaging transgene expression with radionuclide imaging technologies. Neoplasia, 2(1–2), 118–38.
De, A., Lewis, X. Z., & Gambhir, S. S. (2003). Noninvasive imaging of lentiviral-mediated reporter gene expression in living mice. Molecular Therapy, 7(5 Pt 1), 681–91.
Schipper, M. L., & Gambhir, S. S. (2000). Imaging Gene Expression: Concepts and Future Outlook. In C. Schiepers (Ed.), Diagnostic nuclear medicine (pp. p. 253–72). Berlin: Springer.
Li, Z., Dullmann, J., Schiedlmeier, B., Schmidt, M., von Kalle, C., Meyer, J., et al. (2002). Murine leukemia induced by retroviral gene marking. Science, 296(5567), 497.
Bushman, F., Lewinski, M., Ciuffi, A., Barr, S., Leipzig, J., Hannenhalli, S., et al. (2005). Genome-wide analysis of retroviral DNA integration. Nature Reviews Microbiology, 3(11), 848–58.
Schroder, A. R., Shinn, P., Chen, H., Berry, C., Ecker, J. R., & Bushman, F. (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell, 110(4), 521–9.
Ellis, J. (2005). Silencing and variegation of gammaretrovirus and lentivirus vectors. Human Gene Therapy, 16(11), 1241–6.
Hockemeyer, D., Soldner, F., Beard, C., Gao, Q., Mitalipova, M., DeKelver, R. C., et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nature Biotechnology, 27(9), 851–7.
Kotin, R. M., Linden, R. M., & Berns, K. I. (1992). Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO Journal, 11(13), 5071–8.
Tan, I., Ng, C. H., Lim, L., & Leung, T. (2001). Phosphorylation of a novel myosin binding subunit of protein phosphatase 1 reveals a conserved mechanism in the regulation of actin cytoskeleton. The Journal of Biological Chemistry, 276(24), 21209–16.
Smith, J. R., Maguire, S., Davis, L. A., Alexander, M., Yang, F., Chandran, S., et al. (2008). Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells, 26(2), 496–504.
DeKelver, R. C., Choi, V. M., Moehle, E. A., Paschon, D. E., Hockemeyer, D., Meijsing, S. H., et al. (2010). Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Research, 20(8), 1133–42.
Wang, Y., Zhang, W. Y., Hu, S., Lan, F., Lee, A. S., Huber, B., et al. (2012). Genome editing of human embryonic stem cells and induced pluripotent stem cells with zinc finger nucleases for cellular imaging. Circulation Research, 111(12), 1494–503.
Filbin, M. E., & Kieft, J. S. (2009). Toward a structural understanding of IRES RNA function. Current Opinion in Structural Biology, 19(3), 267–76.
Ibrahimi, A., Vande Velde, G., Reumers, V., Toelen, J., Thiry, I., Vandeputte, C., et al. (2009). Highly efficient multicistronic lentiviral vectors with peptide 2A sequences. Human Gene Therapy, 20(8), 845–60.
Ahn, B. C. (2012). Sodium iodide symporter for nuclear molecular imaging and gene therapy: from bedside to bench and back. Theranostics, 2(4), 392–402.
Rath, P., Shi, H., Maruniak, J. A., Litofsky, N. S., Maria, B. L., & Kirk, M. D. (2009). Stem cells as vectors to deliver HSV/tk gene therapy for malignant gliomas. Current Stem Cell Research & Therapy, 4(1), 44–9.
Deroose, C. M., Reumers, V., Debyser, Z., & Baekelandt, V. (2009). Seeing genes at work in the living brain with non-invasive molecular imaging. Current Gene Therapy, 9(3), 212–38.
Serganova, I., Ponomarev, V., & Blasberg, R. (2007). Human reporter genes: potential use in clinical studies. Nuclear Medicine and Biology, 34(7), 791–807.
Yaghoubi, S. S., Campbell, D. O., Radu, C. G., & Czernin, J. (2012). Positron emission tomography reporter genes and reporter probes: gene and cell therapy applications. Theranostics, 2(4), 374–91.
Immonen, A., Vapalahti, M., Tyynela, K., Hurskainen, H., Sandmair, A., Vanninen, R., et al. (2004). AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Molecular Therapy, 10(5), 967–72.
Valery, C. A., Seilhean, D., Boyer, O., Marro, B., Hauw, J. J., Kemeny, J. L., et al. (2002). Long-term survival after gene therapy for a recurrent glioblastoma. Neurology, 58(7), 1109–12.
Tjuvajev, J. G., Avril, N., Oku, T., Sasajima, T., Miyagawa, T., Joshi, R., et al. (1998). Imaging herpes virus thymidine kinase gene transfer and expression by positron emission tomography. Cancer Research, 58(19), 4333–41.
Yaghoubi, S., Barrio, J. R., Dahlbom, M., Iyer, M., Namavari, M., Satyamurthy, N., et al. (2001). Human pharmacokinetic and dosimetry studies of [(18)F]FHBG: a reporter probe for imaging herpes simplex virus type-1 thymidine kinase reporter gene expression. Journal of Nuclear Medicine, 42(8), 1225–34.
Jacobs, A., Braunlich, I., Graf, R., Lercher, M., Sakaki, T., Voges, J., et al. (2001). Quantitative kinetics of [124I] FIAU in cat and man. Journal of Nuclear Medicine, 42(3), 467–75.
Culver, K. W., Ram, Z., Wallbridge, S., Ishii, H., Oldfield, E. H., & Blaese, R. M. (1992). In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science, 256(5063), 1550–2.
Li, S., Tokuyama, T., Yamamoto, J., Koide, M., Yokota, N., & Namba, H. (2005). Bystander effect-mediated gene therapy of gliomas using genetically engineered neural stem cells. Cancer Gene Therapy, 12(7), 600–7.
Matuskova, M., Hlubinova, K., Pastorakova, A., Hunakova, L., Altanerova, V., Altaner, C., et al. (2010). HSV-tk expressing mesenchymal stem cells exert bystander effect on human glioblastoma cells. Cancer Letters, 290(1), 58–67.
Love, Z., Wang, F., Dennis, J., Awadallah, A., Salem, N., Lin, Y., et al. (2007). Imaging of mesenchymal stem cell transplant by bioluminescence and PET. Journal of Nuclear Medicine, 48(12), 2011–20.
Miletic, H., Fischer, Y., Litwak, S., Giroglou, T., Waerzeggers, Y., Winkeler, A., et al. (2007). Bystander killing of malignant glioma by bone marrow-derived tumor-infiltrating progenitor cells expressing a suicide gene. Molecular Therapy, 15(7), 1373–81.
Cao, F., Drukker, M., Lin, S., Sheikh, A. Y., Xie, X., Li, Z., et al. (2007). Molecular imaging of embryonic stem cell misbehavior and suicide gene ablation. Cloning and Stem Cells, 9(1), 107–17.
Perin, E. C., Tian, M., Marini, F. C., 3rd, Silva, G. V., Zheng, Y., Baimbridge, F., et al. (2011). Imaging long-term fate of intramyocardially implanted mesenchymal stem cells in a porcine myocardial infarction model. PloS One, 6(9), e22949.
Ram, Z., Culver, K. W., Oshiro, E. M., Viola, J. J., DeVroom, H. L., Otto, E., et al. (1997). Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nature Medicine, 3(12), 1354–61.
Jacobs, A., Voges, J., Reszka, R., Lercher, M., Gossmann, A., Kracht, L., et al. (2001). Positron-emission tomography of vector-mediated gene expression in gene therapy for gliomas. Lancet, 358(9283), 727–9.
Penuelas, I., Mazzolini, G., Boan, J. F., Sangro, B., Marti-Climent, J., Ruiz, M., et al. (2005). Positron emission tomography imaging of adenoviral-mediated transgene expression in liver cancer patients. Gastroenterology, 128(7), 1787–95.
Yaghoubi, S. S., Jensen, M. C., Satyamurthy, N., Budhiraja, S., Paik, D., Czernin, J., et al. (2009). Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nature Clinical Practice Oncology, 6(1), 53–8.
Riddell, S. R., Elliott, M., Lewinsohn, D. A., Gilbert, M. J., Wilson, L., Manley, S. A., et al. (1996). T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nature Medicine, 2(2), 216–23.
Verzeletti, S., Bonini, C., Marktel, S., Nobili, N., Ciceri, F., Traversari, C., et al. (1998). Herpes simplex virus thymidine kinase gene transfer for controlled graft-versus-host disease and graft-versus-leukemia: clinical follow-up and improved new vectors. Human Gene Therapy, 9(15), 2243–51.
Eriksson, S., Munch-Petersen, B., Johansson, K., & Eklund, H. (2002). Structure and function of cellular deoxyribonucleoside kinases. Cellular and Molecular Life Sciences, 59(8), 1327–46.
Al-Madhoun, A. S., Tjarks, W., & Eriksson, S. (2004). The role of thymidine kinases in the activation of pyrimidine nucleoside analogues. Mini Reviews in Medicinal Chemistry, 4(4), 341–50.
Wang, J., & Eriksson, S. (1996). Phosphorylation of the anti-hepatitis B nucleoside analog 1-(2′-deoxy-2′-fluoro-1-beta-D-arabinofuranosyl)-5-iodouracil (FIAU) by human cytosolic and mitochondrial thymidine kinase and implications for cytotoxicity. Antimicrobial Agents and Chemotherapy, 40(6), 1555–7.
Wang, J., Su, C., Neuhard, J., & Eriksson, S. (2000). Expression of human mitochondrial thymidine kinase in Escherichia coli: correlation between the enzymatic activity of pyrimidine nucleoside analogues and their inhibitory effect on bacterial growth. Biochemical Pharmacology, 59(12), 1583–8.
Deroose, C. M., Chitneni, S. K., Gijsbers, R., Vermaelen, P., Ibrahimi, A., Balzarini, J., et al. (2012). Preliminary validation of varicella zoster virus thymidine kinase as a novel reporter gene for PET. Nuclear Medicine and Biology, 39(8), 1266–74.
Chitneni, S. K., Deroose, C. M., Balzarini, J., Gijsbers, R., Celen, S., Debyser, Z., et al. (2007). A p-[18F] fluoroethoxyphenyl bicyclic nucleoside analogue as a potential positron emission tomography imaging agent for varicella-zoster virus thymidine kinase gene expression. Journal of Medicinal Chemistry, 50(26), 6627–37.
Eberling, J. L., Cunningham, J., Pivirotto, P., Bringas, J., Daadi, M. M., & Bankiewicz, K. S. (2003). In vivo PET imaging of gene expression in Parkinsonian monkeys. Molecular Therapy, 8(6), 873–5.
Qin, C., Cheng, K., Chen, K., Hu, X., Liu, Y., Lan, X., et al. (2013). Tyrosinase as a multifunctional reporter gene for Photoacoustic/MRI/PET triple modality molecular imaging. Scientific Reports, 3, 1490.
Bunzow, J. R., Van Tol, H. H., Grandy, D. K., Albert, P., Salon, J., Christie, M., et al. (1988). Cloning and expression of a rat D2 dopamine receptor cDNA. Nature, 336(6201), 783–7.
Strange, P. G. (1990). Aspects of the structure of the D2 dopamine receptor. Trends in Neurosciences, 13(9), 373–8.
Liang, Q., Satyamurthy, N., Barrio, J. R., Toyokuni, T., Phelps, M. P., Gambhir, S. S., et al. (2001). Noninvasive, quantitative imaging in living animals of a mutant dopamine D2 receptor reporter gene in which ligand binding is uncoupled from signal transduction. Gene Therapy, 8(19), 1490–8.
Chen, I. Y., Wu, J. C., Min, J. J., Sundaresan, G., Lewis, X., Liang, Q., et al. (2004). Micro-positron emission tomography imaging of cardiac gene expression in rats using bicistronic adenoviral vector-mediated gene delivery. Circulation, 109(11), 1415–20.
Hwang do, W., Kang, J. H., Chang, Y. S., Jeong, J. M., Chung, J. K., Lee, M. C., et al. (2007). Development of a dual membrane protein reporter system using sodium iodide symporter and mutant dopamine D2 receptor transgenes. Journal of Nuclear Medicine, 48(4), 588–95.
Bousquet, C., Puente, E., Buscail, L., Vaysse, N., & Susini, C. (2001). Antiproliferative effect of somatostatin and analogs. Chemotherapy, 47(Suppl 2), 30–9.
Reubi, J. C., Kvols, L., Krenning, E., & Lamberts, S. W. (1990). Distribution of somatostatin receptors in normal and tumor tissue. Metabolism, 39(9 Suppl 2), 78–81.
Forrer, F., Valkema, R., Kwekkeboom, D. J., de Jong, M., & Krenning, E. P. (2007). Neuroendocrine tumors. Peptide receptor radionuclide therapy. Best Practice & Research Clinical Endocrinology & Metabolism, 21(1), 111–29.
Ginj, M., Zhang, H., Waser, B., Cescato, R., Wild, D., Wang, X., et al. (2006). Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proceedings of the National Academy of Sciences of the United States of America, 103(44), 16436–41.
Parry, J. J., Chen, R., Andrews, R., Lears, K. A., & Rogers, B. E. (2012). Identification of critical residues involved in ligand binding and G protein signaling in human somatostatin receptor subtype 2. Endocrinology, 153(6), 2747–55.
Rogers, B. E., Chaudhuri, T. R., Reynolds, P. N., Della Manna, D., & Zinn, K. R. (2003). Non-invasive gamma camera imaging of gene transfer using an adenoviral vector encoding an epitope-tagged receptor as a reporter. Gene Therapy, 10(2), 105–14.
Rogers, B. E., McLean, S. F., Kirkman, R. L., Della Manna, D., Bright, S. J., Olsen, C. C., et al. (1999). In vivo localization of [(111)In]-DTPA-D-Phe1-octreotide to human ovarian tumor xenografts induced to express the somatostatin receptor subtype 2 using an adenoviral vector. Clinical Cancer Research, 5(2), 383–93.
Chaudhuri, T. R., Rogers, B. E., Buchsbaum, D. J., Mountz, J. M., & Zinn, K. R. (2001). A noninvasive reporter system to image adenoviral-mediated gene transfer to ovarian cancer xenografts. Gynecologic Oncology, 83(2), 432–8.
Zinn, K. R., Buchsbaum, D. J., Chaudhuri, T. R., Mountz, J. M., Grizzle, W. E., & Rogers, B. E. (2000). Noninvasive monitoring of gene transfer using a reporter receptor imaged with a high-affinity peptide radiolabeled with 99mTc or 188Re. Journal of Nuclear Medicine, 41(5), 887–95.
Singh, S. P., Yang, D., Ravoori, M., Han, L., & Kundra, V. (2009). In vivo functional and anatomic imaging for assessment of in vivo gene transfer. Radiology, 252(3), 763–71.
Cotugno, G., Aurilio, M., Annunziata, P., Capalbo, A., Faella, A., Rinaldi, V., et al. (2011). Noninvasive repetitive imaging of somatostatin receptor 2 gene transfer with positron emission tomography. Human Gene Therapy, 22(2), 189–96.
Furukawa, T., Lohith, T. G., Takamatsu, S., Mori, T., Tanaka, T., & Fujibayashi, Y. (2006). Potential of the FES-hERL PET reporter gene system – basic evaluation for gene therapy monitoring. Nuclear Medicine and Biology, 33(1), 145–51.
Vandeputte, C., Evens, N., Toelen, J., Deroose, C. M., Bosier, B., Ibrahimi, A., et al. (2011). A PET brain reporter gene system based on type 2 cannabinoid receptors. Journal of Nuclear Medicine, 52(7), 1102–9.
Dohan, O., De la Vieja, A., Paroder, V., Riedel, C., Artani, M., Reed, M., et al. (2003). The sodium/iodide Symporter (NIS): characterization, regulation, and medical significance. Endocrine Reviews, 24(1), 48–77.
Van Sande, J., Massart, C., Beauwens, R., Schoutens, A., Costagliola, S., Dumont, J. E., et al. (2003). Anion selectivity by the sodium iodide symporter. Endocrinology, 144(1), 247–52.
Chung, J. K. (2002). Sodium iodide symporter: its role in nuclear medicine. Journal of Nuclear Medicine, 43(9), 1188–200.
Mandell, R. B., Mandell, L. Z., & Link, C. J., Jr. (1999). Radioisotope concentrator gene therapy using the sodium/iodide symporter gene. Cancer Research, 59(3), 661–8.
Shimura, H., Haraguchi, K., Miyazaki, A., Endo, T., & Onaya, T. (1997). Iodide uptake and experimental 131I therapy in transplanted undifferentiated thyroid cancer cells expressing the Na+/I- symporter gene. Endocrinology, 138(10), 4493–6.
Spitzweg, C., O’Connor, M. K., Bergert, E. R., Tindall, D. J., Young, C. Y., & Morris, J. C. (2000). Treatment of prostate cancer by radioiodine therapy after tissue-specific expression of the sodium iodide symporter. Cancer Research, 60(22), 6526–30.
Schipper, M. L., Weber, A., Behe, M., Goke, R., Joba, W., Schmidt, H., et al. (2003). Radioiodide treatment after sodium iodide symporter gene transfer is a highly effective therapy in neuroendocrine tumor cells. Cancer Research, 63(6), 1333–8.
Faivre, J., Clerc, J., Gerolami, R., Herve, J., Longuet, M., Liu, B., et al. (2004). Long-term radioiodine retention and regression of liver cancer after sodium iodide symporter gene transfer in wistar rats. Cancer Research, 64(21), 8045–51.
Terrovitis, J., Kwok, K. F., Lautamaki, R., Engles, J. M., Barth, A. S., Kizana, E., et al. (2008). Ectopic expression of the sodium-iodide symporter enables imaging of transplanted cardiac stem cells in vivo by single-photon emission computed tomography or positron emission tomography. Journal of the American College of Cardiology, 52(20), 1652–60.
Quach, C. H., Jung, K. H., Paik, J. Y., Park, J. W., Lee, E. J., & Lee, K. H. (2012). Quantification of early adipose-derived stem cell survival: comparison between sodium iodide symporter and enhanced green fluorescence protein imaging. Nuclear Medicine and Biology, 39(8), 1251–60.
Wolfs, E., Holvoet, B., Gijsbers, R., Casteels, C., Roberts, S. J., Struys, T., et al. (2014). Optimization of multimodal imaging of mesenchymal stem cells using the human sodium iodide symporter for PET and Cerenkov luminescence imaging. PloS One, 9(4), e94833.
Higuchi, T., Anton, M., Dumler, K., Seidl, S., Pelisek, J., Saraste, A., et al. (2009). Combined reporter gene PET and iron oxide MRI for monitoring survival and localization of transplanted cells in the rat heart. Journal of Nuclear Medicine, 50(7), 1088–94.
Dwyer, R. M., Ryan, J., Havelin, R. J., Morris, J. C., Miller, B. W., Liu, Z., et al. (2011). Mesenchymal stem cell-mediated delivery of the sodium iodide symporter supports radionuclide imaging and treatment of breast cancer. Stem Cells, 29(7), 1149–57.
Knoop, K., Kolokythas, M., Klutz, K., Willhauck, M. J., Wunderlich, N., Draganovici, D., et al. (2011). Image-guided, tumor stroma-targeted 131I therapy of hepatocellular cancer after systemic mesenchymal stem cell-mediated NIS gene delivery. Molecular Therapy, 19(9), 1704–13.
Templin, C., Zweigerdt, R., Schwanke, K., Olmer, R., Ghadri, J. R., Emmert, M. Y., et al. (2012). Transplantation and tracking of human-induced pluripotent stem cells in a pig model of myocardial infarction: assessment of cell survival, engraftment, and distribution by hybrid single photon emission computed tomography/computed tomography of sodium iodide symporter transgene expression. Circulation, 126(4), 430–9.
Barton, K. N., Stricker, H., Brown, S. L., Elshaikh, M., Aref, I., Lu, M., et al. (2008). Phase I study of noninvasive imaging of adenovirus-mediated gene expression in the human prostate. Molecular Therapy, 16(10), 1761–9.
Moroz, M. A., Serganova, I., Zanzonico, P., Ageyeva, L., Beresten, T., Dyomina, E., et al. (2007). Imaging hNET reporter gene expression with 124I-MIBG. Journal of Nuclear Medicine, 48(5), 827–36.
Doubrovin, M. M., Doubrovina, E. S., Zanzonico, P., Sadelain, M., Larson, S. M., & O’Reilly, R. J. (2007). In vivo imaging and quantitation of adoptively transferred human antigen-specific T cells transduced to express a human norepinephrine transporter gene. Cancer Research, 67(24), 11959–69.
Conflicts of interest
The authors declare no potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wolfs, E., Verfaillie, C.M., Van Laere, K. et al. Radiolabeling Strategies for Radionuclide Imaging of Stem Cells. Stem Cell Rev and Rep 11, 254–274 (2015). https://doi.org/10.1007/s12015-014-9575-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-014-9575-3