
Abstract
Heart failure (HF) represents a main global healthy and economic burden with unacceptably high morbidity and mortality rates. In the current study, we evaluated the potential effect of mesenchymal stem cell (MSC)-derived exosomes (MSC-Exos) on oxygen–glucose deprivation (OGD)-induced damages to HL-1 cells and HF mice and searched for the possible mechanism. MSC-Exos ameliorated oxidative stress and reduced apoptosis in OGD-treated HL-1 cells. By microarray analysis, we found that MSC-Exos treatment significantly increased the microRNA (miR)-129-5p expression in HL-1 cells. miR-129-5p inhibitor attenuated the protective effect of MSC-Exos on OGD-treated HL-1 cells. miR-129-5p targeted tumor necrosis factor receptor–associated factor 3 (TRAF3), and TRAF3 loss reversed the effect of miR-129-5p inhibitor by blunting the NF-κB signaling. MSC-Exos injection alleviated ventricular dysfunction and suppressed oxidative stress, apoptosis, inflammation, and fibrosis in cardiomyocytes in mice with HF by inhibiting NF-κB signaling pathway through miR-129-5p/TRAF3. Our findings suggest that exosomal miR-129-5p from MSCs protects the heart from failure by targeting TRAF3 and the following NF-κB signaling. This regulatory axis may be a possible therapeutic target for HF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) is a major and expensive public health problem, requiring numerous office visits and hospitalizations in all countries of the world [1]. The most common trigger for HF is reduced left ventricular (LV) myocardial function; however, dysfunction of the myocardium, heart valves, or great vessels alone or in combination is also related to the onset and development of HF [2]. It presents as a multifactorial and systemic disease, during which structural, cellular, and molecular mechanisms are activated and function as a network after cardiac injury to maintain physiological functioning [3]. For one of them, when reactive oxygen species generation exceeds the ability of the antioxidant defense mechanisms to buffer them, oxidative stress develops, eventually resulting in HF [4]. Therefore, effectively combating oxidative stress is an important task for overcoming HF.
Mesenchymal stem cells (MSCs), multipotent stem cells that exist in multiple organs, such as bone marrow, adipose tissue, skeletal muscles, and dental pulp, have the capacity to differentiate into different cell types [5]. Due to several unique properties, MSCs may be more effective than other cell systems for diseases that are difficult to treat or untreatable [6]. Exosomes, spherical bilayer membrane vesicles, perform a wide range of biological activities, including inflammation induction, oxidative stress, and apoptosis, due to their cargos, including proteins, nucleic acids, lipids, and microRNAs (miRNAs) [7]. MSC-derived exosomes (MSC-Exos) have been proposed as an attractive therapeutic approach to improve cardiac function, and optimized exosomes can be developed via various engineering methods such as miRNA loading to improve the therapeutic effects of exosomes [8]. Here, we identified miR-129-5p as the most significantly upregulated miRNA in oxygen–glucose deprivation (OGD)-challenged HL-1 cells co-cultured with extracted MSC-Exos versus OGD-induced cells treated with phosphate-buffered saline (PBS). It was thus suggested that miR-129-5p was delivered by MSC-Exos to HL-1 cells. Similarly, exosomes containing miR-129-5p were found to be significantly more easily absorbed by colon cancer cells [9]. Interestingly, Kaplan–Meier and multivariate Cox regression analyses in a recent study suggested that high miR-129-5p expression was able to predict overall survival of patients with chronic HF, indicating its potential as a novel non-invasive biomarker for the diagnosis and prognosis of HF [10]. Moreover, miR-129-5p mimic effectively suppressed angiotensin II-induced cardiomyocyte hypertrophic responses and oxidative stress [11]. Therefore, we postulated that the alleviating role of MSC-Exos in HF is elicited through the delivery of miR-129-5p. To explore this possibility, in this paper, we applied MSC-Exos to treat a mouse model with HF and mouse cardiomyocytes HL-1 following OGD.
Materials and Methods
Cell Culture and Transfection
Mouse bone marrow MSCs (CP-M131) were cultured using mouse bone marrow MSC complete medium (CM-M131). Mouse cardiomyocytes HL-1 (CL-0605) were cultured using HL-1 cell-specific medium (CM-0605). Both cell lines were grown to be adherent and fibroblast like in a culture environment of 37 °C and 5% CO2. These cells and culture media were purchased from Procell (Wuhan, Hubei, China).
miR-129-5p inhibitor, small interfering RNA (si)-TRAF3, and negative controls (inhibitor NC and si-NC) were designed by Shanghai GenePharma Co., Ltd. (Shanghai, China). The constructed miR-129-5p inhibitor, si-TRAF3, and controls were transfected or co-transfected into well-grown cells using the Lipofectamine 2000 transfection kit (11,668,019, Thermo Fisher Scientific Inc., Waltham, MA, USA) according to the instructions. After 12 h of transfection, the medium was replaced with fresh medium and incubated for another 24 h. The cells were harvested for subsequent experiments.
HL-1 cells were washed with PBS when they reached an 80% confluence, and the cell model of HF was constructed by OGD treatment. The cells were placed in an anaerobic incubator with saturated humidity at 37 °C and cultured in serum-free medium in the presence of 95% N2 and 5% CO2 for 6 h, followed by growth in HL-1 cell-specific medium at 37 °C and 5% CO2 for 12 h, as the OGD group. The cells in the control group were treated normally.
Isolation and Characterization of MSCs
When the MSCs at passage three grew to an 80–90% confluence, they were dissociated with trypsin and filtered through a 100-µm cell filter. Subsequently, MSCs were resuspended in pre-chilled 1 × PBS buffer and incubated with allophycocyanin-coupled monoclonal antibodies to CD44 (103,011, 1:100, BioLegend, San Diego, CA, USA), CD29 (102,215, 1:50, BioLegend), CD34 (128,611, 1:200, BioLegend), and CD45 (103,111, 1:100, BioLegend) or with isotype control IgG (ab232814, 1:200, Abcam, Cambridge, UK). Surface antigens were analyzed using a Guava easyCyte™ flow cytometer (Millipore, Billerica, MA, USA), and graphs were generated by Flow-Jo software (Tree Star Inc., Ashland, Oregon, USA).
Isolation and Characterization of MSC-Exos
Exos were removed from the MSCs medium by ultracentrifugation at 100,000×g, and MSCs were incubated with Exo-free medium for 2 day. The medium supernatant was then centrifuged at 300×g for 10 min, 2000×g for 10 min, and 10,000×g for 0.5 h at 4 °C to remove residual cells and cell debris. The supernatant was then collected and subjected to ultracentrifugation at 100,000×g for 70 min. Transmission electron microscopy (BD Biosciences, San Diego, CA, USA) was applied to observe the morphology of MSC-Exos, and Western blot was applied to identify MSC-Exo surface marker antigens, including CD81 and TSG101. The protein content of MSC-Exos was determined using the bicinchoninic acid assay (BCA) protein assay kit (Solarbio, Beijing, China). The MSC-Exos were stored at − 80 °C for subsequent experiments.
Exosome Uptake Assay
After thawing, the isolated MSC-Exos were labeled using a PKH67 Cell Linker Kit for General Cell Membrane Labeling (MX4023, Shanghai Maokang Biotechnology Co., Ltd., Shanghai, China). The labeled MSC-Exos were subsequently resuspended in Dulbecco’s modified Eagle medium (31,600, Solarbio). Labeled exosomes were then co-cultured with 1 × 105 HL-1 cells (80–90% confluence) seeded in 24-well culture dishes. After 24 h of incubation, internalization was observed by a fluorescence microscope (Eclipse TE2000-U, Nikon Instruments Inc., Melville, NY, USA).
Enzyme-Linked Immunosorbent Assay (ELISA)
Cell supernatants and myocardial tissue homogenates were collected for analysis of lactate dehydrogenase (LDH, A020-1–2, Nanjing JianCheng Bioengineering Institute, Nanjing, Jiangsu, China), superoxide dismutase (SOD, A001-3–2, JianCheng), malondialdehyde (MDA, K454, BioVision, Inc., Exton, PA, USA), and glutathione peroxidase (GSH-Px, K464, BioVision) according to the manufacturer's instructions.
Flow Cytometry
Apoptosis rates of HL-1 cells were measured by flow cytometry using the Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) apoptosis assay kits (A10788, Invitrogen Inc., Carlsbad, CA, USA). Briefly, logarithmic growth phase cells were taken, and the cell concentration was adjusted to 2 × 106 cells/mL by adding PBS containing 10% calf serum. The cell suspension (200 μL) was washed with 1 mL cold PBS and centrifuged at 1000 rpm for 10 min at 4 °C. The cells were resuspended in 200 μL labeling buffer, incubated with 10 µL Annexin V-FITC, and 5 µL PI for 15 min at room temperature in the dark and loaded onto a flow cytometry (Beckman Coulter, Fullerton, CA, USA). The data analysis was conducted using FlowJo software (Tree Star).
Microarray Analysis
Microarray analysis was performed using an Agilent Mouse miRNA array 21.0 (8 × 60 K) (Agilent Technologies, Santa Clara, CA, USA) to detect the effect of MSC-Exos treatment on miRNA expression in HL-1 cells (n = 3). Sample labeling, microarray hybridization, and washing were conducted following the manufacturer’s instructions. Microarray data were analyzed using the miRNA 4.0 platform from Affymetrix (Santa Clara, CA, USA), and genes with ≥ 2-fold changes were considered to be genes with differential expression.
Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was isolated from the cells using TRIzol reagent (15,596,026, Invitrogen). The purity of RNA was subsequently detected by UV–Vis spectrophotometer UV5 (Mettler Toledo, Shanghai, China) (purity requirement: 1.9–2.1 for OD260/280). The extracted RNA was then reverse transcribed to cDNA using the Prime Script™ RT kit (RR036Q, Takara, Dalian, Liaoning, China). In addition, for miRNA, reverse transcription was performed using the miRNA Reverse Transcription Kit (638,315, Takara). RT-qPCR was performed on an ABI 7500 Fast PCR System (Carlsbad, CA, USA) using 25 μL SYBR® Green Real-time PCR Master Mix (Toyobo Life Science, Shanghai, China) containing cDNA 5 μL (50 ng), distilled water 16 μL, forward primers 2 μL, and reverse primers 2 μL. PCR program was set as follows: pre-denaturation: 95 °C for 15 s, denaturation: 95 °C for 15 s, annealing/extension: 72 °C 45 s, and repeated denaturation and annealing for 40 cycles. The 2−ΔΔCt method was used to calculate relative expression. miRNA expression was normalized to U6 snRNA, and mRNA expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primer sequences in the current study are listed in Table 1.
Western Blot
Total proteins were extracted from cells or tissues with radioimmunoprecipitation assay lysis buffer (P0013E, Beyotime, Shanghai, China) on ice. Protein concentrations were determined using a BCA protein assay kit (Solarbio). Proteins were separated by 10% SDS-PAGE and then transferred to polyvinylidene fluoride membranes (Millipore). After being sealed with 5% bovine serum albumin (Gibco, Carlsbad, CA, USA), the membranes were incubated overnight at 4 °C with the primary antibody and treated with horseradish peroxidase-labeled secondary antibody IgG (1:5000, ab6721, Abcam) for 2 h at room temperature. Protein bands were visualized with the aid of an enhanced chemiluminescence fluorescence test kit (Abcam), and relative protein expression was measured by using Quantity One software (Bio-Rad) with GAPDH as an internal reference. The primary antibodies were as follows: CD81 (1:2000, ab109201, Abcam), TSG101 (1:1000, GTX118736, GeneTex, Inc., Alton Pkwy Irvine, CA, USA), cleaved-caspase-3 (1:2000, ab214430, Abcam), Bax (1:2000, GTX109683, GeneTex), Bcl2 (1:1000, #3498 T, Cell Signaling Technologies, Beverly, MA, USA), p-IκBα (1:1000, #2859S, Cell Signaling Technologies), IκBα (1:1000, #9242S, Cell Signaling Technologies), p-p65 (1:1000, ab76302, Abcam), p65 (1:2000, ab16502, Abcam), CTGF (1:1000, GTX124232, GeneTex), and GAPDH (1:10,000, ab181602, Abcam).
Dual-Luciferase Assay
A potential binding site for TRAF3 to miR-129-5p was obtained from StarBase (http://starbase.sysu.edu.cn/). The amplified TRAF3 mutant (MUT) sequence and wild sequence (WT) fragment were cloned into pmirGLO luciferase vector (E1330, Promega, Madison, WI, USA) to construct TRAF3-WT or TRAF3-MUT luciferase vector. TRAF3-WT or TRAF3-MUT was co-transfected with miR-129-5p mimic or mimic NC into HL-1 cells, respectively. After 48 h of incubation, luciferase activity was measured by a dual-luciferase reporter system (Promega). The relative luciferase activity was calculated as the ratio between the firefly and renilla luciferase activity.
RNA Immunoprecipitation (RIP) Assay
RIP assays were performed using the Bersin BIO RIP kit (BersinBio, Guangzhou, Guangdong, China) according to the manufacturer’s protocol. HL-1 cells were collected and lysed with RIP lysis buffer. HL-1 cell lysates (100 μL) were treated with RIP buffer and incubated with Proteinase K and magnetic beads conjugated with anti-Ago2 (1:500, ab186733, Abcam) or IgG (1:1000, ab199376, Abcam). Next, the immunoprecipitated RNA was extracted, and the results were measured by DNA agarose gel electrophoresis and RT-qPCR.
Establishment of a HF Mouse Model
Animal experiments were performed using approved protocols by the Institutional Animal Care and Use Committee of the Second Hospital of Hebei Medical University. All animal procedures were implemented according to the NIH guidelines (NIH Publication No. 86–23). Twenty-four healthy male C57BL/6 J mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). A 12–12-h lights on–off cycle was maintained in an environmentally controlled room. Standard rodent food and water were freely accessible. The mice were randomly divided into four groups of six mice each: the sham, HF, PBS (HF + PBS), and MSC-Exos (HF + MSC-Exos) groups.
We used permanent ligation of the left anterior descending (LAD) coronary artery to construct a mouse model of HF. The mice were anesthetized by intraperitoneal injection with a mixture of xylazine (5 mg/kg, X854786, Shanghai Macklin Biochemical Co., Ltd., Shanghai, China) and ketamine (100 mg/kg, R923159, Macklin). The surgery was performed under aseptic conditions. The chest cavity of mice was sterilized, and the heart was exposed to perform LAD ligation with a 7–0 silk ligature at the fourth rib space. The chest cavity was closed quickly, and the heart was gently massaged to restore the breathing. The incision was finally closed layer by layer. The hearts of rats in the sham group were not ligated. Postoperatively, 400,000 U of penicillin was administered to all mice to prevent infection. Mice in the MSC-Exos group were postoperatively injected with 50 μL MSC-Exos at a concentration of 100 μg/mL through the tail vein once a week for three times (injection of exosomes did not cause an inflammatory response in mice). Mice in the PBS group were injected with an equal volume of PBS through the tail vein. The cardiac function of mice was assessed by echocardiography at an interval of one week postoperatively. After the end of the fourth week, the mice were euthanized (intraperitoneal injection of sodium pentobarbital 200 mg/kg), and heart tissues were obtained for subsequent experiments.
Analysis of Cardiac Function by Echocardiography
After modeling, the mice were anesthetized with a mixture of xylazine (5 mg/kg) and ketamine (100 mg/kg) and immobilized in a supine position on a wooden board. M-mode ultrasound images were acquired using a small animal ultrasound imaging system (VisualSonics, Toronto, Canada). The echocardiography allowed us to measure the LV end-diastolic volume (LVEDV), LV end-systolic volume (LVESV), LV internal dimension-diastole (LVIDd), LV internal dimension in systole (LVIDs) and stroke volume (μL), and to average these indexes over three consecutive cardiac cycles. LV ejection fraction (LVEF), LV fractional shortening (LVFS), LV end-systolic diameter (LVDs, mm), and LV end-diastolic diameter (LVDd, mm) were then calculated, where LVEF = (LVEDV-LVESV)/LVEDV × 100% and LVFS = (LVIDd-LVIDs)/LVIDd × 100%.
Assessment of Infarction Size Using 2,3,5-Triphenyltetrazolium Chloride (TTC) Staining
For TTC staining, three mice in each group were randomly chosen for the injection of Evans Blue staining (2.0% solution in 1 mL) into the coronary circulation through a carotid catheter. Remaining mice were euthanized and used for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). The mice were euthanized, and hearts were rapidly excised, stored at − 80 °C, cut into 1-mm-thick sections, and counter-stained at 37 °C for 15 min with 1% (wt/vol) TTC (Sigma) solution. Representative images were captured. Image-Pro Plus software was used to assess the volumes of non-ischemic (area not at risk), ischemic (area at risk, AAR), and infarcted (INF) regions, and the results were considered as INF/AAR × 100%.
Assessment of Apoptosis Using TUNEL
Heart tissues were fixed overnight in 10% formalin at 4 °C, and 5-μm sections were stained with TUNEL kit (C1089, Beyotime) according to the manufacturer’s protocol to assess cardiomyocyte apoptosis in heart sections. The sections were dewaxed in xylene, soaked in different concentrations of ethanol for 2 min, treated with proteinase K for 20 min, and incubated with TUNEL detection solution for 1 h at room temperature in the dark, counter-stained with DAPI, and finally sealed with anti-fluorescence quenching sealing solution and observed under a fluorescence microscopy.
Immunohistochemistry
The paraffin-embedded sections were soaked twice in xylene for dewaxing and rehydrated in ethanol at 100%, 95%, 90%, 85%, 80%, and 70% concentrations for 5 min, respectively. Subsequently, endogenous peroxidase was removed with 3% peroxide. The sections were heated with citrate buffer in a microwave oven for antigen retrieval as well as exposure of antigen clusters, washed and then sealed with 5% BSA at room temperature for 30 min. After washing, the sections were incubated overnight with diluted CD31 antibody (1:500, GTX130274, GeneTex) or CD206 antibody (1:500, 24595 T, Cell Signaling Technologies) at 4 °C, followed by a 2-h incubation with goat anti-rabbit secondary antibody (1:1000, ab6721, Abcam) at room temperature. The sections were subjected to color development reaction with DAB chromogenic solution for 5 min, followed by immersion in hematoxylin staining solution for nuclear counter-staining for 30 s. The sections were sequentially immersed in different concentrations of ethanol and xylene for dehydration and permeabilization and finally sealed with neutral gum. The number of positive brown cells was observed under the microscope, and the positive rate was calculated.
Data Analysis
For data conforming to the normal distribution, one-way or two-way analysis of variance was used to calculate the statistical significance, and all pairwise multiple comparison procedures were done by Tukey’s post hoc test. For experiments with three replications that did not conform to the normal distribution, the Kruskal–Wallis or Mann–Whitney test was conducted for nonparametric test analysis. At least three independent experiments were performed for each assay, and the values were expressed as mean ± SD. GraphPad Prism 8.0 (GraphPad, San Diego, CA, USA) was used for graphs and data analysis. Statistical significance was defined as p value < 0.05.
Results
Characterization of MSCs and MSC-Exos
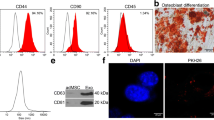
MSCs were observed under an inverted fluorescence microscope in a fibroblast-like morphology (Fig. 1A). Cell surface marker proteins were analyzed by flow cytometry. MSCs were found to be positive for the markers CD44 and CD29, while cell differentiation markers CD34 and CD45 were negative (Fig. 1B).

Culture and characterization of MSCs and MSC-Exos. A Observation of the morphology of MSCs under an optical microscope. B Flow cytometric analysis of surface marker proteins of MSCs. C TEM observation of exosome morphology and structure. D Particle size analysis of exosomes. E Western blot analysis of CD81 and TSG101 expression in MSC-Exos. F Confocal fluorescence microscopy observation of the uptake of MSC-Exos by HL-1 cells. Data are depicted as the mean ± SD from three independent experiments
To characterize MSC-Exos, we extracted exosomes from the medium of MSCs using gradient centrifugation. The morphology of exosomes can be observed by transmission electron microscope (TEM). The isolated exosomes showed as a typical elliptical shape (Fig. 1C). Particle size analysis showed that the diameter of exosomes ranged from 30 to 140 nm (Fig. 1D). The expression of MSC-Exos marker proteins was detected by Western blot, and the expression of CD81 and TSG101 proteins was found to be much higher in MSC-Exos than that in MSCs (Fig. 1E). Furthermore, confocal microscopy results showed that after co-culture of HL-1 cells with PKH-67-labeled MSC-Exos, the isolated MSC-Exos was successfully internalized by HL-1 cells (Fig. 1F). In summary, MSCs and MSC-Exos were successfully isolated, and the isolated MSC-Exos were internalized by HL-1 cells.
MSC-Exos Inhibits Oxidative Stress and Apoptosis in OGD-Treated HL-1 Cells
To investigate the role played by MSC-Exos in oxidative stress and apoptosis, we constructed a cellular model of HF by treating mouse cardiomyocytes HL-1 with OGD, followed by MSC-Exos co-culture. LDH and MDA contents in HL-1 cells were significantly increased, whereas SOD and GSH-PX levels were significantly decreased after OGD treatment by ELISA. After co-culture of OGD-treated HL-1 cells and MSC-Exos, LDH and MDA contents were significantly decreased, while SOD and GSH-PX levels were significantly increased (Fig. 2A). The apoptosis rate was analyzed by flow cytometry, and it was found that OGD treatment significantly promoted apoptosis, and this promotion was significantly reversed by MSC-Exos (Fig. 2B). Western blot presented that the levels of the pro-apoptotic proteins cleaved caspase-3 and Bax were significantly boosted, and the expression of the anti-apoptotic protein Bcl 2 was significantly reduced in OGD-induced HL-1 cells. However, these trends were effectively reversed by treatment with MSC-Exos (Fig. 2C). These results suggest that MSC-Exos can inhibit oxidative stress and apoptosis in OGD-treated HL-1 cells.

MSC-Exos inhibits oxidative stress and apoptosis in OGD-treated HL-1 cells. A The release of LDH, MDA, SOD, and GSH-PX in cells measured using ELISA. B Apoptosis rate (number of apoptotic cells/total number of cells) in cells measured using flow cytometry. C The protein expression of apoptosis-related genes cleaved caspase-3, Bax, and Bcl2 in cells measured using Western blot. Data are depicted as the mean ± SD from three independent experiments. **p < 0.01. Statistical significance was determined using Kruskal–Wallis test
MSC-Exos Enhances the Expression of miR-129-5p in HL-1 Cells
To investigate the specific mechanism of MSC-Exos in OGD-treated HL-1 cells, we analyzed the differentially expressed miRNAs in HL-1 cells treated with MSC-Exos or PBS in the presence of OGD using microarrays. We plotted the heat map with |Log2Fold change |> 2 as the screening condition, showing the top seven differentially expressed miRNAs and found that miR-129-5p was the most significantly elevated one (Fig. 3A). The expression of miR-129-5p in MSC-Exos was examined by RT-qPCR, and the results showed that miR-129-5p expression was significantly enriched in the purified exosomes relative to the supernatant (Fig. 3B). After that, we inhibited the expression of miR-129-5p in MSCs by transfection with miR-129-5p inhibitor, and the miR-129-5p level was significantly reduced in MSCs, as measured by RT-qPCR (Fig. 3C). In addition, exosomes derived from the MSCs after transfection were collected. It was revealed by RT-qPCR that the expression of miR-129-5p was significantly suppressed in exosomes derived from MSCs with miR-129-5p inhibitor (herein termed as MSC-Exos-inhibitor) relative to those from MSCs with NC inhibitor (herein termed as MSC-Exos-NC) (Fig. 3D).

MSC-Exos enhances the expression of miR-129-5p in HL-1 cells. A Microarray analysis of the effect of MSC-Exos treatment on miRNA expression in HL-1 cells. B miR-129-5p enrichment in exosomes by RT-qPCR. C The transfection efficiency of miR-129-5p inhibitor in MSCs by RT-qPCR. D The miR-129-5p expression in MSC-Exos-NC and MSC-Exos-inhibitor by RT-qPCR. Data are depicted as the mean ± SD from three independent experiments. **p < 0.01, ***p < 0.001. Statistical significance was determined using Mann–Whitney test
Inhibition of miR-129-5p Attenuates the Effects of MSC-Exos on OGD-Treated HL-1 Cells
As shown in Fig. 3D, we successfully knocked down the level of miR-129-5p in exosomes and co-cultured these exosomes with OGD-treated HL-1 cells. The RT-qPCR results established that the expression of miR-129-5p was significantly decreased in OGD-treated HL-1 cells after co-culture with MSC-Exos-inhibitor (Fig. 4A). Subsequent ELISA analysis revealed that LDH and MDA levels were considerably increased, whereas SOD and GSH-PX levels were decreased in the MSC-Exos-inhibitor group of cells compared to the MSC-Exos-NC group (Fig. 4B). Flow cytometric assays in Fig. 4C showed that knockdown of miR-129-5p in exosomes promoted apoptosis of HL-1 cells. Western blot assay displayed that the levels of pro-apoptotic protein cleaved caspase-3 and Bax were significantly augmented, and the expression of anti-apoptotic protein Bcl2 was significantly reduced in HL-1 cells after silencing of miR-129-5p (Fig. 4D). The above results indicated that inhibition of miR-129-5p in MSC-Exos significantly attenuated the therapeutic effect of MSC-Exos on OGD-treated HL-1 cells.

Inhibition of miR-129-5p attenuates the protective effect of MSC-Exos on OGD-treated HL-1 cells. A Expression of miR-129-5p in HL-1 cells co-cultured with MSC-Exos-NC and MSC-Exos-inhibitor by RT-qPCR. B The release of LDH, MDA, SOD, and GSH-PX in cells measured using ELISA. C Apoptosis rate (number of apoptotic cells/total number of cells) in cells measured using flow cytometry. D The protein expression of apoptosis-related genes cleaved caspase-3, Bax and Bcl2 in cells measured using Western blot. Data are depicted as the mean ± SD from three independent experiments. *p < 0.05, **p < 0.01. Statistical significance was determined using Mann–Whitney test or Kruskal–Wallis test
miR-129-5p Targets TRAF3 in HL-1 Cells
To investigate the specific mechanisms by which miR-129-5p plays a role in oxidative stress and apoptosis, we need to find downstream targets of miR-129-5p that act in HL-1 cells. We predicted the target genes of mmu-miR-129-5p in miRDB (http://mirdb.org/), TargetScan (http://www.targetscan.org/), miRWalk (http://mirwalk.umm.uni-heidelberg.de/), and StarBase (http://starbase.sysu.edu.cn/). Six intersecting genes, namely BRPF3, KLF7, TRAF3, ERBB4, PTPN4, and ACTR3 came out (Fig. 5A). We transfected miR-129-5p mimic and its control into HL-1 cells, and effective transfection was confirmed using RT-qPCR (Fig. 5B). We examined the expression of six predicted target genes in the cells after transfection using RT-qPCR and found that only TRAF3 was significantly downregulated by miR-129-5p mimic (Fig. 5C). TRAF3 expression was found to be significantly elevated in OGD-treated HL-1 cells and decreased after MSC-Exos treatment by RT-qPCR. By contrast, TRAF3 expression was significantly restored in OGD-treated HL-1 cells after co-culture with MSC-Exos inhibitor (Fig. 5D).

Exosomal miR-129-5p released by MSCs inhibits the expression of TRAF3. A Potential target genes for miR-129-5p predicted using bioinformatics websites. B The transfection efficiency of miR-129-5p mimic in HL-1 cells examined using RT-qPCR. C Effect of miR-129-5p mimic on mRNA expression of potential target genes by RT-qPCR. D RT-qPCR analysis of TRAF3 expression in HL-1 cells under different treatments. E Potential binding sites of miR-129-5p to TRAF3. F The effect of overexpression of miR-129-5p on TRAF3-WT or TRAF3-MUT luciferase activity measured using dual-luciferase assay. G The binding relationship between miR-129-5p and TRAF3 measured using RIP assay. Data are depicted as the mean ± SD from three independent experiments. *p < 0.05, **p < 0.01. Statistical significance was determined using unpaired Mann–Whitney test or Kruskal–Wallis test
We predicted the binding site of TRAF3 to miR-129-5p from StarBase (Fig. 5E). miR-129-5p mimic was found to significantly inhibit the luciferase activity of TRAF3-WT by dual-luciferase assay, while it had no significant effect on the luciferase activity of TRAF3-MUT (Fig. 5F). Consistently, RIP experiments showed that anti-Ago2 significantly enriched miR-129-5p and TRAF3 compared to anti-IgG (Fig. 5G). These results suggest that miR-129-5p targets TRAF3 in HL-1 cells.
Overexpression of TRAF3 Reverses the Therapeutic Effect of MSC-Exos Through Activation of NF-κB Signaling Pathway
We analyzed TRAF3-related genes by KEGG enrichment and found that TRAF3-related genes could be enriched in the NF-κB signaling pathway (Fig. 6A, B). We then transfected si-TRAF3 and control (si-NC) into OGD-treated HL-1 cells co-cultured with MSC-Exos inhibitor, and effective transfection was confirmed using RT-qPCR (Fig. 6C). We then examined the phosphorylation of IκBα and p65, marker proteins of the NF-κB signaling pathway, by Western blot in cells after different treatments. We found that OGD induction led to a significant increase in the levels of p-IκBα and p-p65 in the cells, while MSC-Exos treatment significantly reduced the extent of IκBα and p65 phosphorylation. Poor expression of TRAF3 reversed the effects of miR-129-5p inhibitor to impair the NF-κB signaling pathway again (Fig. 6D).

Overexpression of TRAF3 reverses the therapeutic effect of MSC-Exos by activating the NF-κB signaling pathway. A, B KEGG enrichment analysis of genes associated with TRAF3 and visualization by bubble mapping. C Detection of TRAF3 mRNA expression in HL-1 cells transfected with si-TRAF3 and co-cultured with MSC-Exos-NC and MSC-Exos-inhibitor by RT-qPCR. D Western blot analysis of p-IκBα, IκBα, p-p65, and p65 expression in cells under different treatments. E The release of LDH, MDA, SOD and GSH-PX in cells measured using ELISA. F Apoptosis rate (number of apoptotic cells/total number of cells) in cells measured using flow cytometry. G The protein expression of apoptosis-related genes cleaved caspase-3, Bax and Bcl2 in cells measured using Western blot. Data are depicted as the mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. Statistical significance was determined using Kruskal–Wallis test or Mann–Whitney test
ELISA and flow cytometry assays revealed that the loss of TRAF3 resulted in a significant decrease in LDH and MDA levels and a significant increase in SOD and GSH-PX levels (Fig. 6E), which was occurred concomitant with hampered apoptosis (Fig. 6F). The results of Western blot showed that deficiency of TRAF3 inhibited the expression of cleaved caspase-3 and Bax proteins and promoted the expression of Bcl2 protein (Fig. 6G).
MSC-Exos Alleviates Ventricular Dysfunction and Oxidative Stress via miR-129-5p/TRAF3/NF-κB Axis in Mice with HF
To further investigate whether MSC-Exos could ameliorate HF and oxidative stress of cardiomyocytes in vivo, we constructed a mouse HF model and injected MSC-Exos or PBS into HF mice through the tail vein. We then tested the cardiac function of mice at an interval of one week and found that LVEF and LVFS were significantly lower in the HF mice. The improvement effect of MSC-Exos injection was not significant in the first three weeks, and LVEF and LVFS were significantly higher in the HF mice upon MSC-Exos injection at the fourth week (Fig. 7A). At the fourth week of the experiment, we found that the stroke volume of mice in the HF group decreased significantly, and the level of stroke volume increased significantly after MSC-Exos injection (Fig. 7B), indicating that exosome injection could alleviate the damage to cardiac function caused by LAD ligation. Therefore, we chose to euthanize the mice after the fourth week of the experiment and isolated the myocardial tissues to make sections. TTC staining showed that the infarct area was much higher in the HF mice compared with the sham-operated mice, and MSC-Exos treatment significantly reduced the infarct area (Fig. 7C). The expression of miR-129-5p was significantly decreased and the mRNA level of TRAF3 was increased in cardiomyocytes of HF mice by RT-qPCR; and this was reversed after MSC-Exos injection (Fig. 7D). The ELISA results showed that the levels of LDH and MDA were significantly increased, and the levels of SOD and GSH-PX were inhibited in the myocardial tissue homogenate of HF mice, whereas the oxidative stress was alleviated after MSC-Exos treatment (Fig. 7E).

MSC-Exos alleviates ventricular dysfunction and oxidative stress of cardiomyocytes via miR-129-5p/TRAF3/NF-κB axis in mice with HF. A Echocardiographic assessment of LVEF and LVFS in mice. B Echocardiographic assessment of stroke volume in mice. C Heart damage in mice measured using TTC staining. D Expression of miR-129-5p and TRAF3 mRNA in myocardial tissue of mice by RT-qPCR. E The release of LDH, MDA, SOD, and GSH-PX in myocardial tissue homogenate measured using ELISA. Data are depicted as the mean ± SD (n = 6 for A, B, D, and E; n = 3 for C). **p < 0.01. Statistical significance was determined using Kruskal–Wallis test, one-way or two-way ANOVA, and Tukey’s multiple comparison test
MSC-Exos Alleviates Apoptosis, Inflammation, and Fibrosis in Myocardial Tissues via miR-129-5p/TRAF3/NF-κB Axis in Mice with HF
The results of TUNEL assay in Fig. 8A demonstrated that cardiomyocyte apoptosis induced by HF was significantly reduced by MSC-Exos injection. Immunohistochemical staining of myocardial tissues showed that the levels of angiogenesis-related factor CD31 and anti-inflammatory factor CD206 were significantly reduced. After MSC-Exos treatment, the expression of CD31 and CD206 was restored (Fig. 8B). Meanwhile, we performed western blot assays on the fibrosis-associated protein CTGF in the infarct margin region and the distal infarct in the myocardial tissues. A significant increase was found in CTGF expression in the infarct margin region of HF mice, which was reversed by MSC-Exos injection. By contrast, there was no significant difference regarding CTGF expression in the distal infarct region (Fig. 8C). Western blot assay revealed elevated expression of cleaved caspase-3 and Bax proteins and decreased expression of Bcl2 protein in cardiomyocytes of HF mice (Fig. 8D) and activated phosphorylation levels of IκBα and p65 proteins in myocardial tissues (Fig. 8E). However, MSC-Exos administration suppressed the pro-apoptotic protein expression and elevated anti-apoptotic protein expression by blocking the NF-κB pathway.

MSC-Exos alleviates apoptosis, inflammation, and fibrosis in myocardial tissues via miR-129-5p/TRAF3/NF-κB axis in mice with HF. A Apoptosis of myocardial tissues in mice measured using TUNEL staining. B Immunohistochemical detection of CD31 and CD206 expression in myocardial tissues. C The protein expression of fibrosis-associated protein CTGF in myocardial tissues measured using Western blot. D The protein expression of apoptosis-related genes cleaved caspase-3, Bax, and Bcl2 in myocardial tissues measured using Western blot. E Western blot analysis of p-IκBα, IκBα, p-p65, and p65 expression in myocardial tissues of mice. Data are depicted as the mean ± SD (n = 6 for C, D, and E; n = 3 for A and B). **p < 0.01. Statistical significance was determined using Kruskal–Wallis test, one-way or two-way ANOVA, and Tukey’s multiple comparison test
Discussion
Over the last 10 years, the potential roles of exosomes as effective inter-tissue communicators in cardiovascular physiology and pathology by transferring bioactive RNAs, proteins, and lipids from donor to recipient cells and influencing the biological properties of the latter have been gradually appreciated [12]. In the present work, MSC-Exos were identified as cardiac protectors due to their functions in reducing inflammation, fibrosis, apoptosis, and oxidative stress. Knockdown of miR-129-5p mitigated these protective effects on HL-1 cells exposed to OGD and HF mice induced by LAD ligation, as evidenced by increased cell apoptosis and oxidative stress. Mechanistically, we determined that miR-129-5p targeted TRAF3 to elicit its protective effects via blocking the NF-κB signaling.
Exo released by human umbilical cord MSCs reduced oxidative injury of HK-2 cells induced by oxalate and calcium oxalate monohydrate, as evidenced by lowered MDA and LDH activities [13]. In addition, miR-512-3p enriched in MSC-Exos markedly inhibited oxidized low-density lipoprotein-mediated endothelial cell damage, namely, inhibited Caspase-3 activation and cell apoptosis, inhibited the oxidative factor MDA, and increased the contents of SOD and GSH-PX [14]. In line with these reports, we observed that MSC-Exos significantly enhanced SOD and GSH-PX levels and Bcl2 expression, whereas repressed MDA and LDH activities and cleaved caspase-3 and Bax expression in HL-1 cells. Using microarray analysis on HL-1 cells exposed to OGD and co-cultured with MSC-Exos or PBS, we found that miR-129-5p was at least partially responsible for the protective effects of MSC-Exos. For substantiation, we conducted loss-of-function assays using exosomes derived from MSCs transfected with miR-129-5p inhibitor for co-culture. As expected, the protective effects of MSC-Exos were perturbed. Consistently, miR-129-5p overexpression dramatically alleviated myocardial injury in mice subjected to ischemia/reperfusion, as evidenced by reduced LDH activity, MDA content, and infract size [15]. More specifically, downregulation of miR-129-5p were observed in the serum of chronic HF patients, and administration of miR-129-5p improved heart function and hemodynamic parameters, as well as attenuated oxidative stress in rats with HF [16]. However, the role of miR-129-5p enriched in MSC-Exos in HF, to the best of our knowledge, has been rarely reported before, which underlines the novelty of our study.
Subsequently, we applied bioinformatics websites to predict the downstream targets of miR-129-5p in HF. Even though six genes were screened out, only TRAF3 showed significant downregulation in HL-1 cells following miR-129-5p mimic transfection. The binding relation between miR-129-5p and TRAF3 3’UTR was corroborated using dual-luciferase and RIP assays. TRAF3, an adaptor protein in tumor necrosis factor-related signaling, was identified as a main controller of cardiac hypertrophy in response to pressure overload [17]. Moreover, NF-κB pathway was one of the major downstream pathways mediated by the TRAF family members [18]. This was in line with our KEGG enrichment analysis and western blot assay results. Previously, TRAF3 has been revealed to be bound by miR-155, thus participating in the regulation of oxidative stress and necroptosis achieved by methionine selenium [19]. Das et al. also reported that silencing of TRAF3-interacting protein 2 could inhibited NF-κB activation, inflammatory cytokine expression, collagen expression, and secretion, thus blunting myocardial hypertrophy and contractile dysfunction [20]. Here, we presented that loss of TRAF3 reduced oxidative stress and cell apoptosis in HL-1 cells in the presence of MSC-Exo-inhibitor by blocking the NF-κB pathway. Quite in line with our results, Rac1 inhibition could impair the NF-κB signaling and antagonize the promotion of miR-96 knockdown on doxorubicin-induced myocardial toxicity [21]. Exosomal miR-30e from rat bone marrow MSCs markedly inhibited LOX1 expression, thereby downregulating the activity of the NF-κB p65 signaling and ameliorating HF in rats [22]. miR-129 inhibitor antagonized the anti-tumor effect of magnolol and partly offset the magnolol-induced decline of p-IκBα and p-p65 expression in multiple myeloma cells [23], which also hinted the possible interaction between miR-129-5p and the NF-κB pathway.
Conclusion
Recapitulating these in vitro and in vivo results, exosomal miR-129-5p from MSC-Exos may repress the oxidative stress and cell apoptosis by targeting TRAF3 and blocking the NF-κB signaling. However, there are limitations to the clinical application of MSC-Exos. First, the minimum effective dose of exosomes needs to be optimized. Second, exosome extraction methods need to be further optimized to obtain purer and higher volumes of exosomes. Moreover, cardiac metabolism is highly adaptive to changes in fuel availability and the energy demand, and this metabolic flexibility is of great importance for the heart to maintain its output during the development and in response to stress [24]. The high rates of ATP production are critical in maintaining cardiac contractility. Cardiac ATP is mainly derived from fatty acid oxidation, with glucose metabolism contributing less under normal conditions, whereas fatty acid oxidation may be reduced, which is concomitant with increased glucose utilization under stress conditions [25]. Meanwhile, miR-129-5p has been reported to suppress glucose consumption, lactate production, cellular ATP levels, and glucose uptake of gastric cancer cells [26]. Therefore, the effect of miR-129-5p on glucose metabolism in the HF development would be further explored in the future due to time and funding constraints.
Data Availability
All data generated or analyzed during this study are included in this published article.
References
Islam, M. S. (2018). Heart failure: From research to clinical practice. Advances in Experimental Medicine and Biology, 1067, 1–3. https://doi.org/10.1007/5584_2018_181
Inamdar, A. A., & Inamdar, A. C. (2016). Heart failure: Diagnosis management and utilization. Journal of Clinical Medicine. https://doi.org/10.3390/jcm5070062
Tanai, E., & Frantz, S. (2015). Pathophysiology of heart failure. Comprehensive Physiology, 6(1), 187–214. https://doi.org/10.1002/cphy.c140055
Valaei, K., Taherkhani, S., Arazi, H., & Suzuki, K. (2021). Cardiac oxidative stress and the therapeutic approaches to the intake of antioxidant supplements and physical activity. Nutrients. https://doi.org/10.3390/nu13103483
Suzuki, E., Fujita, D., Takahashi, M., Oba, S., & Nishimatsu, H. (2017). Therapeutic effects of mesenchymal stem cell-derived exosomes in cardiovascular disease. Advances in Experimental Medicine and Biology, 998, 179–185. https://doi.org/10.1007/978-981-10-4397-0_12
Majka, M., Sulkowski, M., Badyra, B., & Musialek, P. (2017). Concise review: Mesenchymal stem cells in cardiovascular regeneration: Emerging research directions and clinical applications. Stem Cells Translational Medicine, 6(10), 1859–1867. https://doi.org/10.1002/sctm.16-0484
Nikdoust, F., Pazoki, M., Mohammadtaghizadeh, M., Aghaali, M. K., & Amrovani, M. (2021). Exosomes: Potential player in endothelial dysfunction in cardiovascular disease. Cardiovascular Toxicology. https://doi.org/10.1007/s12012-021-09700-y
Wang, X., Tang, Y., Liu, Z., Yin, Y., Li, Q., Liu, G., & Yan, B. (2021). The application potential and advance of mesenchymal stem cell-derived exosomes in myocardial infarction. Stem Cells Int, 2021, 5579904. https://doi.org/10.1155/2021/5579904
Ji, Y., Ji, J., Yin, H., Chen, X., Zhao, P., Lu, H., & Wang, T. (2021). Exosomes derived from microRNA-129-5p-modified tumor cells selectively enhanced suppressive effect in malignant behaviors of homologous colon cancer cells. Bioengineered, 12(2), 12148–12156. https://doi.org/10.1080/21655979.2021.2004981
Zhang, H., Zhang, N., Jiang, W., & Lun, X. (2021). Clinical significance of the long non-coding RNA NEAT1/miR-129-5p axis in the diagnosis and prognosis for patients with chronic heart failure. Experimental and Therapeutic Medicine, 21(5), 512. https://doi.org/10.3892/etm.2021.9943
Ye, H., Xu, G., Zhang, D., & Wang, R. (2021). The protective effects of the miR-129-5p/keap-1/Nrf2 axis on Ang II-induced cardiomyocyte hypertrophy. Annals of Translational Medicine, 9(2), 154. https://doi.org/10.21037/atm-20-8079
Han, C., Yang, J., Sun, J., & Qin, G. (2021). Extracellular vesicles in cardiovascular disease: Biological functions and therapeutic implications. Pharmacology & Therapeutics. https://doi.org/10.1016/j.pharmthera.2021.108025
Li, D., Zhang, D., Tang, B., Zhou, Y., Guo, W., Kang, Q., Wang, Z., Shen, L., Wei, G., & He, D. (2019). Exosomes from human umbilical cord mesenchymal stem cells reduce damage from oxidative stress and the epithelial-mesenchymal transition in renal epithelial cells exposed to oxalate and calcium oxalate monohydrate. Stem Cells International, 2019, 6935806. https://doi.org/10.1155/2019/6935806
Chen, S., Zhou, H., Zhang, B., & Hu, Q. (2021). Exosomal miR-512-3p derived from mesenchymal stem cells inhibits oxidized low-density lipoprotein-induced vascular endothelial cells dysfunction via regulating Keap1. Journal of Biochemical and Molecular Toxicology, 35(6), 1–11. https://doi.org/10.1002/jbt.22767
Ma, R., Chen, X., Ma, Y., Bai, G., & Li, D. S. (2020). MiR-129-5p alleviates myocardial injury by targeting suppressor of cytokine signaling 2 after ischemia/reperfusion. Kaohsiung Journal of Medical Sciences, 36(8), 599–606. https://doi.org/10.1002/kjm2.12211
Xiao, N., Zhang, J., Chen, C., Wan, Y., Wang, N., & Yang, J. (2019). miR-129-5p improves cardiac function in rats with chronic heart failure through targeting HMGB1. Mammalian Genome, 30(9–10), 276–288. https://doi.org/10.1007/s00335-019-09817-0
Jiang, X., Deng, K. Q., Luo, Y., Jiang, D. S., Gao, L., Zhang, X. F., Zhang, P., Zhao, G. N., Zhu, X., & Li, H. (2015). Tumor necrosis factor receptor-associated factor 3 is a positive regulator of pathological cardiac hypertrophy. Hypertension, 66(2), 356–367. https://doi.org/10.1161/HYPERTENSIONAHA.115.05469
Shi, J. H., & Sun, S. C. (2018). Tumor necrosis factor receptor-associated factor regulation of nuclear factor kappaB and mitogen-activated protein kinase pathways. Frontiers in Immunology, 9, 1849. https://doi.org/10.3389/fimmu.2018.01849
Zhirong, Z., Qiaojian, Z., Chunjing, X., Shengchen, W., Jiahe, L., Zhaoyi, L., & Shu, L. (2021). Methionine selenium antagonizes LPS-induced necroptosis in the chicken liver via the miR-155/TRAF3/MAPK axis. Journal of Cellular Physiology, 236(5), 4024–4035. https://doi.org/10.1002/jcp.30145
Das, N. A., Carpenter, A. J., Yoshida, T., Kumar, S. A., Gautam, S., Mostany, R., Izadpanah, R., Kumar, A., Mummidi, S., Siebenlist, U., & Chandrasekar, B. (2018). TRAF3IP2 mediates TWEAK/TWEAKR-induced pro-fibrotic responses in cultured cardiac fibroblasts and the heart. Journal of Molecular and Cellular Cardiology, 121, 107–123. https://doi.org/10.1016/j.yjmcc.2018.07.003
Lei, B., Wu, X., Xia, K., Sun, H., & Wang, J. (2021). Exosomal micro-RNA-96 derived from bone marrow mesenchymal stem cells inhibits doxorubicin-induced myocardial toxicity by inhibiting the Rac1/nuclear factor-kappaB signaling pathway. Journal of the American Heart Association, 10(17), e020589. https://doi.org/10.1161/JAHA.120.020589
Pu, L., Kong, X., Li, H., & He, X. (2021). Exosomes released from mesenchymal stem cells overexpressing microRNA-30e ameliorate heart failure in rats with myocardial infarction. American Journal of Translational Research, 13(5), 4007–4025.
Jin, W., Wang, X., Zeng, Y., Lan, Y., & Wang, X. (2021). Magnolol suppressed cell migration and invasion and induced cell apoptosis via inhibition of the NF-kappaB signaling pathway by upregulating microRNA-129 in multiple myeloma. Neoplasma, 68(2), 404–415. https://doi.org/10.4149/neo_2020_200923N1010
Ritterhoff, J., & Tian, R. (2017). Metabolism in cardiomyopathy: Every substrate matters. Cardiovascular Research, 113(4), 411–421. https://doi.org/10.1093/cvr/cvx017
Tran, D. H., & Wang, Z. V. (2019). Glucose metabolism in cardiac hypertrophy and heart failure. Journal of the American Heart Association, 8(12), e012673. https://doi.org/10.1161/JAHA.119.012673
Chen, D., Wang, H., Chen, J., Li, Z., Li, S., Hu, Z., Huang, S., Zhao, Y., & He, X. (2018). MicroRNA-129-5p regulates glycolysis and cell proliferation by targeting the glucose transporter SLC2A3 in gastric cancer cells. Frontiers in Pharmacology, 9, 502. https://doi.org/10.3389/fphar.2018.00502
Acknowledgements
Not applicable.
Funding
None.
Author information
Authors and Affiliations
Contributions
FY: conceptualization, methodology, data curation and writing—original draft; ZYC: software, resources, formal analysis, supervision and validation; WC: project administration, investigation visualization and writing—review and editing; all authors contributed to the manuscript preparation, read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Ethical Approval
Animal experiments were performed using approved protocols by the Institutional Animal Care and Use Committee of the Second Hospital of Hebei Medical University. All animal procedures were implemented according to the NIH guidelines (NIH Publication No. 86-23).
Additional information
Handling Editor: Vincent FM Segers.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yan, F., Cui, W. & Chen, Z. Mesenchymal Stem Cell-Derived Exosome-Loaded microRNA-129-5p Inhibits TRAF3 Expression to Alleviate Apoptosis and Oxidative Stress in Heart Failure. Cardiovasc Toxicol 22, 631–645 (2022). https://doi.org/10.1007/s12012-022-09743-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-022-09743-9