
Abstract
Prolonged exposure to high doses of fluoride causes chronic poisoning called fluorosis, which affects many tissues and causes serious health problems. This study was planned to investigate the apoptotic, autophagic, and necrotic molecular pathways of fluoride. Sodium fluoride (NaF) was administered to normal rat kidney epithelial (NRK-52E) cells. The NaF IC50 value was determined using the MTT assay. The expression of the genes in the autophagic, apoptotic, and necrotic pathways was determined by real-time PCR. It was determined that there were significant changes in NaF-induced molecular pathways depending on the time. There were no increases in apoptotic and necrotic pathway markers except for Atg3, an autophagy gene, at the 3rd and the 12th hours. However, there was an induction in all cell death signaling pathways at 24 h. The molecular mechanisms demonstrated NaF-induced cellular death in the NRK-52E cell line. It was concluded that these molecular mechanisms were activated with NaF, and different mechanisms accelerated the cellular death at the 24th hour.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fluoride (F) is an element with high electronegativity that can be naturally found in soil, water, and various nutrients and many products used in daily life (toothpaste, insecticides, some chemical compounds). Fluoride toxicity, known as fluorosis, occurs in humans and animals due to long-term exposure to high concentrations of fluoride. In fluorosis, several tissues, especially the teeth and the bones, are damaged. Fluoride is mainly excreted through the kidneys. Since the kidney is the primary organ responsible for fluoride excretion, effects on the kidneys are expected. During the filtration of fluoride, the kidneys are exposed to large amounts of fluoride and participate in fluoride homeostasis with calcified tissues. Fluoride can lead to necrosis through various cellular mechanisms; however, its role has not been fully clarified [1,2,3,4].
Several studies have been carried out to clarify fluoride-induced toxicity mechanisms. Oxidative stress–induced DNA damage and activation of apoptotic pathways are important mechanisms [3,4,5,6]. Apoptosis is necessary for the normal functions and survival of multicellular organisms. Each cell lives for a certain period of time and dies. There is a controlled balance between cellular death and proliferation. Oxidative stress plays an important role in the initiation of apoptosis [7, 8]. Autophagy is a physiological mechanism that eliminates damaged cellular proteins and organelles. Thus, the cellular content is broken down to a recyclable state. The autophagy mechanism recycles intracellular molecules in the absence of nutrients. In living organisms, local and sudden death of tissues and organelles is called necrosis. Necrosis is characterized by cell damage and apoptosis plays a major role in necrosis. Necrosis began to be used to describe pathological cell death which causes inflammation [9,10,11,12].
Despite the widespread knowledge about the useful effect of fluoride on tooth decay, it is thought that long-time fluoride exposure may cause chronic toxicity [13, 14]. In recent years, there have been many studies investigating the metabolic pathways of fluoride toxication. In the previous studies of ours, the M30, caspase 3, 8, and 9 levels in the renal epithelial cell line (NRK-52E) were investigated as apoptosis indicators, and it was determined that although the utilized NaF concentration increased the necrosis, it did not properly induce apoptosis. However, it was concluded that other mechanisms were more effective on the observed necrosis other than apoptotic pathways [3, 4]. This study was planned to determine the possible roles of high concentration (IC50) of NaF administration on different molecular mechanisms (apoptotic, autophagic, and necrotic) in the normal rat kidney epithelial cell line NRK-52E, as time-dependent (at 3, 12, 24 h) manner.
Material and Method
Cell Culture
In this study, the normal rat kidney epithelial (NRK-52E) cell line was used to investigate the apoptotic, autophagic, and necrotic effects of NaF. The NRK-52E cells were grown in the RPMI 1640 medium supplemented with 10% FBS, 1% L-glutamine, and 1% penicillin-streptomycin incubated at 37 °C with 5% CO2 and saturated humidity [3].
Preparation of Solutions
The stock solutions of NaF in the study were prepared by referring to the concentrations in our previous study [3]. It was dissolved NaF in the complete medium.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide Cytotoxicity Test
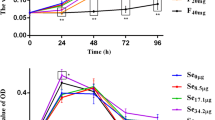
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay to measure the cytotoxic effect of NaF at different times. MTT cell viability tests were carried out and the IC50 concentrations of NaF were determined at the 3rd, 12th, and 24th hours and compared with the control group. The cells were plated in 96-well plates at 7 × 103 cells/well density in 100 μl medium and incubated overnight for attachment to the surface of the plate. The NRK-52E cells were treated with various concentrations of NaF (Table 1, Fig. 1) for 3, 12, and 24 h. The control and the study groups were designated.

MTT results dependent on time
Total RNA Isolation and Quantitative Real-Time PCR
The cells were plated in 25-cm2 flasks with 106 density in 3500 μl medium and incubated overnight for attachment to the surface of the plate. The cells were incubated for 3, 12, and 24 h. The total RNA was extracted using the TRIzol reagent [15] for each incubation time (for 3, 12, and 24 h) and the amount of obtained RNA was measured with the BioDrop spectrophotometer. cDNA synthesis was performed using the Qiagen kit (RT2 PCR master mix). The experiments were repeated in triplicate for each gene, Bax, Bad, Bak1, Bcl2, and caspases 3, 8, and 9 for apoptosis; Map11c3a, Map11c3b, Atg 3, Atg 5, Sqstm 1, and Becn1 for autophagy; and Ripk1 and Ripk3 genes for necrosis.
The expressions of genes were carried out with qPCR. Reference sequence numbers from the GenBank were obtained from Qiagen and used for all primers (genes produced for rats) (Qiagen SABiosciences USA).
NaF was added to cells separately in the determined IC50 values and the cells were incubated for 3, 12, and 24 h to determine the gene expression.
The quantitative relative expression of the target genes was normalized to the GAPDH glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene and calculated using the comparative ΔΔCt method. The fold change was analyzed with the 2−ΔΔCt method. The quantitative real-time PCR (qRT-PCR) data were analyzed using the Gene Globe Data Analysis Center (Qiagen). The raw data based on the CT method were normalized with the GAPDH control gene. The fold regulation represented the fold change results in a biologically meaningful way. The fold change values greater than one indicated a positive or an upregulation, and the fold regulation was equal to the fold change.
Statistical Analysis
The fold change values were accepted as ≤ 2 upregulation and downregulation. The p values were calculated based on the Student t test of the replicate 2−ΔΔCT values for each gene in the control group and the treatment groups, and p values lower than 0.05 were accepted as statistically significant (SPSS 22.0).
Results
The IC50 concentrations of NaF were determined as 9600, 5500, and 3200 μM for the 3rd, 12th, and 24th hours, respectively, and compared to the control group (Table 1, Fig. 1). The PCR results of the analysis conducted on the study findings are presented in Figs. 2, 3, 4, and 5.

qRT-PCR results

Autophagic gene expressions compared to the control gene

Apoptotic gene expressions when compared to the control gene

Necrotic gene expressions when compared to the control gene
According to the MTT test results using Microsoft Excel slope graph, the IC50 concentrations of NaF were determined as 9600 mM, 5500 mM, and 3200 mM concentrations for the 3, 12, and 24 h, respectively.
It was observed that there was no difference in Map11c3a expression between the 3rd and the 24th hours, while there was a decrease at the 12th hour when compared to the other hours. There were no differences in Map11c3b expressions based on time when compared to the control gene. Atg 3 expression increased at the 3rd hour, decreased at the 12th hour despite a 7-fold previous increase, and the expression increased 38-fold at the 24th hour. A 6-fold increase was observed in the Atg 5 expression based on the time, especially at the 24th hour. Sqstm 1 expression decreased by almost half at the 3rd and the 12th hours, while there was a 5-fold increase at the 24th hour. There was no increase in the Becn1 expression at the 3rd hour, while there was a decrease at the 12th hour and a 2.5-fold increase at the 24th hour (Fig. 3). It was determined that autophagic genes were upregulated based on time. The highest increase was observed in the Atg3 gene (p ≤ 0.05).
The highest increase was observed in Bak1 expression at the 3rd hour and in the caspase 3 gene at the 24th hour. Caspase 9 expression increased by 1.4- and 1.8-fold at the 3rd and the 24th hours. There was a decrease in caspase 8 expression at the 3rd and the 12th hours, while there was a 3-fold increase at the 24th hour. Caspase 3 expression decreased by half at the 12th hour and the Bax and Bad gene expressions increased by more than 4-fold at the 24th hour, similar to the caspases, and exhibited a decrease at the 12th hour when compared to the control gene. It was noteworthy that apoptotic genes reacted differently based on time. The level of Bcl-2, an antiapoptotic gene, decreased at the 12th hour and increased twice at the 24th hour (Fig. 4) (p ≤ 0.05).
Ripk 1 expression decreased by almost half at the 3rd and the 12th hours, while there was a 3-fold increase at the 24th hour. Ripk 3 expression decreased at the 3rd hour, while there was an insignificant increase at the 12th hour and a 3-fold increase at the 24th hour. It was observed that necrotic genes were more effective at the 24th hour (Fig. 5) (p ≤ 0.05).
Discussion
High levels of fluoride exposure lead to tissue and organ damage starting at cellular level. Several studies have been conducted on molecular damage and further cellular and experimental studies have been planned. The parameters in apoptotic, autophagic, and necrotic pathways have been reported to be noteworthy, particularly in fluoride-induced necrosis [3, 16, 17]. In the present study, apoptotic, autophagic, and necrotic gene expressions were determined by qRT-PCR at the optimum IC50 concentrations and different durations, and the results were analyzed. In a study by Luo et al. [18], it was reported that the administration of NaF, which exceeded 12 mg/kg for 42 days, induced renal oxidative damage and cell necrosis due to increased ROS level and decreased antioxidant enzyme amount.
Panneerselvam et al. [19] reported that the Bax/Bcl2 ratio changed due to fluoride accumulation caused an increase in cytochrome C and caspase 3 and thus triggered myocardial apoptosis. In this study, it was found that necrosis increased at the 24th hour in the 3200 μM NaF–treated group. Our results are consistent with the literature results of studies with different concentrations and animal models showing that NaF triggers the necrotic pathway [3, 4]. Necrosis was detected at high fluoride concentration, indicating a high magnitude of damage to cellular organelles [20]. However, there are no studies in the literature investigating the expression levels of genes involved in necrosis by fluorine treatment in kidney cells. This is the first study to demonstrate that NaF triggers the necrosis concentration and time dependence using an in vitro model for kidney cells. Our study was found to be compatible with the literature.
It was shown that apoptosis occurs through both extrinsic and intrinsic pathways due to oxidative stress as a result of high levels of ROS formation activated by NaF [21]. It was reported that fluoride causes renal oxidative damage, degeneration, and necrosis in tubular cells, consistent with the changes in renal function parameters, renal tubular hyaline pulses, and distention of the glomeruli. Fluoride interferes with renal anatomy and physiology, which can lead to renal pathogenesis. Several degrees of fluoride-related damage to tubular epithelial architecture such as cell distention and lysis, cytoplasmic vacuum, nuclear condensation, apoptosis, and necrosis were observed [22,23,24].
It has been reported that NaF induces inflammatory responses through activation of renal histopathological lesions and the nuclear factor-kappa B (NF-κB) signaling pathway, and by reducing the expression of anti-inflammatory cytokines [18].
In studies investigating the molecular basis of the damage induced by high-concentration fluoride administration, different tissues and cells were used. NaF was used as a fluoride ion source. It was reported that NaF exposure led to reactive oxygen species (ROS) formation [25], induced endoplasmic reticulum (ER) stress, oxidative stress and mitochondrial damage and apoptosis [22, 26,27,28], and induced apoptosis through involvement of both external and intrinsic apoptotic pathways [29, 30].
Certain studies argued that apoptosis was dependent on the PI3K/AKT signaling pathway in the fluoride-administered skeletal muscle cell (C2C12 cell) model [31], active caspases initiated the apoptosis process through a mitochondria-mediated pathway [6], induced apoptosis by increasing the ROS-dependent NF-κB signal in mouse splenocytes [32], and that NaF-induced apoptosis could be caused by mitochondrial and death receptor pathways [33].
Fluoride induces cellular stress, including endoplasmic reticulum stress and oxidative stress, leading to disruption of ameloblasts responsible for the production of tooth enamel [34]. It was reported that there was a possible correlation between fluoride-induced oxidative stress, DNA damage, and apoptosis in rat hepatocytes [35].
In a study investigating the role of fluorine-induced apoptosis in SH-SY5Y cell–treated NaF at cytotoxic concentration for 24 h, Tu et al. [17] demonstrated that the caspase 3 level was upregulated, and the Bcl-2 protein level decreased.
In previous studies in our laboratory [2,3,4] and in this study, it was found that NaF stopped the cell proliferation depending on time and concentration. Our MTT results are consistent with the literature.
In this study, we used the NRK-52E rat kidney cell line, and the levels of major genes of necrosis, apoptosis, and autophagic cell death pathways were determined by qRT-PCR method. In our literature search for studies using cell line, no other study was found in which IC50 or a cytotoxic concentration of NaF was administered, except for this study in which it was administered for 3 h. No change was detected except for an increase in Bak-1 at the 3rd hour. In studies using different cells from the cell line used in the above literature, it can be considered that the increase in caspase 3 and Bax protein amounts and the decrease in Bcl-2 being incompatible with our study for the 12th hour may be due to the different cell types used and/or the concentrations given in IC50.
The increased caspase 3 and Bax protein levels and the decreased Bcl-2 were not compatible with our study in the literature given above [28, 36]. This may be due to the different cell types used and the concentrations being treated. In this study, the increases in caspase 3 and caspase 8 were consistent with the literature in the NaF group for the 24th hour. However, the fact that caspase 9 and other mitochondrial proapoptotic (Bak-1, Bax, and Bad) and antiapoptotic (Bcl-2) genes do not change according to the control gene is not consistent with the literature. In the literature, although the amount of proapoptotic protein increased, the Bcl-2/Bax ratio decreased and caspase 9 increased, indicating that fluorine may have used the mitochondrial pathway. In our study, it can be said that the receptor death pathway becomes activated due to the increase in caspase 3 and caspase 8.
Despite the determination of the autophagic pathway being completely effective on necrosis at the 12th hour, the death signals were present at the 24th hour in cells without autophagy and the necrotic signal was strong in the cells. Despite the autophagy attempt by cells in order to rest, it was determined that the cells had entered apoptosis and that the cell death had completed [28].
Although fluoride may induce cellular apoptosis, the role of autophagy in fluoride-induced apoptosis remains unclear. Autophagy is a highly preserved intracellular digestion process that disrupts damaged organelles and protein aggregates [16]. Autophagy markers in NaF administration were examined at mRNA and protein expression levels, and data demonstrating autophagy as a potential molecular mechanism in NaF-induced damage were obtained [16, 37, 38].
It was reported that fluoride acts on the nervous system and osteoblast cells by inducing the ER stress and autophagy [17, 20, 36, 39]. Furthermore, the significance of autophagy in dental fluorosis has been demonstrated [40].
Li et al. [41] reported that the levels of autophagic markers (Beclin-1 and LC3II) increased as the NaF concentration increased, whereas the apoptotic markers (caspases 3, 8, 9) increased after 0.5 mM in a study treated with NaF on M3T3-E1 cells line in 24 h.
In the present study, significant genes were selected to represent different necrosis mechanisms in order to reveal the molecular basis of cellular death observed in the renal epithelial cell line NRK-52E at different times. The expressions of these genes were determined by RT-PCR and the results were analyzed. Thus, the cause of the cytotoxicity observed at the 3rd hour was determined as cellular apoptosis through the intrinsic apoptotic pathway. However, it was determined that a certain amount of autophagy and even advanced mitophagy, albeit incomplete, were present before apoptosis. Following the failure of that pathway, it was observed that the intrinsic apoptotic pathway was initiated, and the necrosis began through caspase 9 without the use of the external receptor pathway.
Conclusion
In the present study, molecular mechanisms in NaF-induced necrosis were investigated at optimum IC50 concentrations and different durations. It was determined that apoptosis was induced by the intrinsic apoptotic pathway at the 3rd hour, the autophagic pathway was completely activated at the 12th hour, and necrotic signal became prominent at the 24th hour in cells that could not remain in autophagy. In conclusion, it was concluded that the molecular mechanisms in NaF-dependent necrosis in this cell line were activated in a time-dependent manner; it was concluded that cytotoxic mechanisms were activated at the 3rd hour after NaF administration and necrosis, which is a sign of high damage to cellular organelles, increased at the 12th and the 24th hours.
Change history
11 May 2021
A Correction to this paper has been published: https://doi.org/10.1007/s12011-020-02554-5
References
Yur F, Mert N, Dede S, Değer Y, Ertekin A, Mert H, Yaşar S, Doğan I, Işık A (2013) Evaluation of serum lipid fractions and tissue antioxidant levels in sheep with fluorosis. Fluoride 46(2):90–96
Cetin S, Yur F (2016) Levels of trace elements in muscle and kidney tissues of sheep with fluorosis. Biol Trace Elem Res 174:82–84
Yuksek V, Dede S, Taşpınar M, Çetin S (2017) The effects of certain vitamins on apoptosis and DNA damage in sodium fluoride (Naf) administered renal and osteoblast cell lines. Fluoride 50(3):300–313
Yuksek V, Çetin S, Usta A, Kömüroğlu AU, Dede S (2017) Effect of some vitamins on antioxidant/prooxidant parameters in sodium fluoride (NaF)-treated cell line (hFOB 1.19). Turk J Vet Res 1(1):1–6
Cao J, Chen J, Xie L, Wang J, Feng C, Song J (2015) Protective properties of sesamin against fluoride-induced oxidative stress and apoptosis in kidney of carp (Cyprinus carpio) via JNK signaling pathway. Aquat Toxicol 167:180–190
Tan PP, Zhou BH, Zhao WP, Jia LS, Liu J, Wang HW (2018) Mitochondria-mediated pathway regulates C2C12 cell apoptosis induced by fluoride. Biol Trace Elem Res 185:440–447. https://doi.org/10.1007/s12011-018-1265-6
Kannan K, Jain SK (2000) Oxidative stress and apoptosis. Pathophysiology 7(3):153–163
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
Coskun G, Ozgur H (2011) Molecular mechanism of apoptosis and necrosis. Arch Med Rev J 20:145–158
Öz Arslan D, Korkmaz G, Gözüaçık D (2011) Otofaji: Bir hücresel stres yanıtı ve ölüm mekanizması. Acıbadem Üniv Sağ Bil Derg 2(4):184–194 (in Turkish)
Lee Y, Overholtzer M (2019) After-death functions of cell death. Yale J Biol Med 92(4):687–694
Nirmala JG, Lopus M (2020) Cell death mechanisms in eukaryotes. Cell Biol Toxicol 36(2):145–164
Ismail AI, Hasson H (2008) Fluoride supplements, dental caries and fluorosis: a systematic review. J Am Dent Assoc 139(11):1457–1468
Sharma D, Singh A, Verma K, Paliwal S, Sharma S, Dwivedi J (2017) Fluoride: a review of pre-clinical and clinical studies. Environ Toxicol Pharmacol 56:297–313
Chomczynski P, Mackey K (1995) Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19(6):942–945
Kuang P, Deng H, Liu H, Cui H, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L (2018) Sodium fluoride induces splenocyte autophagy via the mammalian targets of rapamycin (mTOR) signaling pathway in growing mice. Aging (Albany NY). https://doi.org/10.18632/aging.101499
Tu W, Zhang Q, Liu Y, Han L, Wang Q, Chen P, Zhang S, Wang A, Zhou X (2018) Fluoride induces apoptosis via inhibiting SIRT1 activity to activate mitochondrial p53 pathway in human neuroblastoma SH-SY5Y cells. Toxicol Appl Pharmacol 347:60–69
Luo Q, Cui H, Deng H, Kuang P, Liu H, Lu Y, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L (2017) Sodium fluoride induces renal inflammatory responses by activating NF-κB signaling pathway and reducing anti-inflammatory cytokine expression in mice. Oncotarget 8(46):80192–80207
Panneerselvam L, Govindarajan V, Ameeramja J, Nair HR, Perumal E (2015) Single oral acute fluoride exposure causes changes in cardiac expression of oxidant and antioxidant enzymes, apoptotic and necrotic markers in male rats. Biochimie 119:27–35
Wurtz T, Houari S, Mauro N, MacDougall M, Peters H, Berdal A (2008) Fluoride at non-toxic dose affects odontoblast gene expression in vitro. Toxicology 249(1):26–34
Niu Q, Chen J, Xia T, Li P, Zhou G, Xu C, Zhao Q, Dong L, Zhang S, Wang A (2018) Excessive ER stress and the resulting autophagic flux dysfunction contribute to fluoride-induced neurotoxicity. Environ Pollut 233:889–899
Luo Q, Cui H, Deng H, Kuang P, Liu H, Lu Y, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L (2017) Histopathological findings of renal tissue induced by oxidative stress due to different concentrations of fluoride. Oncotarget 8(31):50430–50446
Alhusaini AM, Faddah LM, El Orabi NF, Hasan IH (2018) Role of some natural antioxidants in the modulation of some proteins’ expressions against sodium fluoride-induced renal injury. Biomed Res Int 2018. https://doi.org/10.1155/2018/5614803
Quadri JA, Sarwar S, Sinha A, Kalaivani M, Dinda AK, Bagga A, Roy TS, Das TK, Shariff A (2018) Fluoride-associated ultrastructural changes and apoptosis in human renal tubule: a pilot study. Hum Exp Toxicol 37(11):1199–1206
Sana S, Ghosh S, Das N, Sarkar S, Mandal AK (2017) Vesicular melatonin efficiently downregulates sodium fluoride-induced rat hepato- and broncho-TNF-α, TGF-β expressions, and associated oxidative injury: a comparative study of liposomal and nanoencapsulated forms. Int J Nanomedicine 12:4059–4071
Suzuki M, Everett ET, Whitford GM, Bartlett JD (2017) 4-phenylbutyrate mitigates fluoride-induced cytotoxicity in ALC cells. Front Physiol 8:302
Ying Z, Lin M, Jian L, Ming Z, Kaiqiang Z, Hefeng G (2014) Excessive fluoride inducing calcium overload and apoptosis of ameloblasts. Hua Xi Kou Qiang Yi Xue Za Zhi 32(6):542–546
Wei M, Duan D, Liu Y, Wang Z, Li Z (2014) Autophagy may protect MC3T3-E1 cells from fluoride-induced apoptosis. Mol Med Rep 9(6):2309–2315
Zhang S, Niu Q, Gao H, Ma R, Lei R, Zhang C, Xia T, Li P, Xu C, Wang C, Chen J, Dong L, Zhao Q, Wang A (2016) Excessive apoptosis and defective autophagy contribute to developmental testicular toxicity induced by fluoride. Environ Pollut 212:97–104
Ribeiro DA, Cardoso CM, Yujra VQ, DE Barros VM, Aguiar O Jr, Pisani LP, Oshima CTF (2017) Fluoride induces apoptosis in mammalian cells: in vitro and in vivo studies. Anticancer Res 37(9):4767–4777
Zhang YL, Luo Q, Deng Q, Li T, Li Y, Zhang ZL, Zhong JJ (2015) Genes associated with sodium fluoride-induced human osteoblast apoptosis. Int J Clin Exp Med 8(8):13171–13178
Deng H, Kuang P, Cui H, Luo Q, Liu H, Lu Y, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L (2017) Sodium fluoride induces apoptosis in mouse splenocytes by activating ROS-dependent NF-κB signaling. Oncotarget 8(70):114428–114441
Deng H, Kuang P, Cui H, Chen L, Luo Q, Fang J, Zuo Z, Deng J, Wang X, Zhao L (2016) Sodium fluoride (NaF) induces the splenic apoptosis via endoplasmic reticulum (ER) stress pathway in vivo and in vitro. Aging (Albany NY) 8(12):3552–3567
Suzuki M, Bandoski C, Bartlett JD (2015) Fluoride induces oxidative damage and SIRT1/autophagy through ROS-mediated JNK signaling. Free Radic Biol Med 89:369–378
Campos-Pereira FD, Lopes-Aguiar L, Renosto FL, Nogueira GA, Costa EF, Barbieri-Pulz R, Silva-Zacarin EC, Oliveira CA, Pigoso AA, Severi-Aguiar GD (2017) Genotoxic effect and rat hepatocyte death occurred after oxidative stress induction and antioxidant gene downregulation caused by long term fluoride exposure. Chem Biol Interact 264:25–33
Gu X, Han D, Chen W, Zhang L, Lin Q, Gao J, Fanning S, Han B (2016) SIRT1-mediated FoxOs pathways protect against apoptosis by promoting autophagy in osteoblast-like MC3T3-E1 cells exposed to sodium fluoride. Oncotarget 7(40):65218–65230
Zhang J, Zhu Y, Shi Y, Han Y, Liang C, Feng Z, Zheng H, Eng M, Wang J (2017) Fluoride-ınduced autophagy via the regulation of phosphorylation of mammalian targets of rapamycin in mice leydig cells. J Agric Food Chem 65(40):8966–8976
Feng Z, Liang C, Manthari RK, Wang C, Zhang J (2018) Effects of fluoride on autophagy in mouse sertoli cells. Biol Trace Elem Res 187:499–505. https://doi.org/10.1007/s12011-018-1405-z
Zhao Y, Li Y, Gao Y, Yuan M, Manthari RK, Wang J, Wang J (2018) TGF-β1 acts as mediator in fluoride-induced autophagy in the mouse osteoblast cells. Food Chem Toxicol 115:26–33
Lei S, Zhang Y, Zhang K, Li J, Liu L (2015) Effects of fluoride on the expression of Beclin1 and mTOR in ameloblasts. Cells Tissues Organs 200(6):405–412
Li X, Meng L, Wang F, Hu X, Yu Y (2019) Sodium fluoride induces apoptosis and autophagy via the endoplasmic reticulum stress pathway in MC3T3-E1 osteoblastic cells. Mol Cell Biochem 454:77–85
Funding
This study was supported by the Van Yuzuncu Yil University Directorate of Scientific Research Projects and registered with the project no: TYL-2017-6041.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article unfortunately contained mistakes. The complete list of corrections is given below. * The author’s affiliations in now corrected in the author group. * The correct order of the keywords should be: Apoptosis, Autophagy, In vitro, Naf, Necrosis, NRK-52E cell line * p.2, fourth sentence of the last paragraph under the header “Total RNA Isolation and Quantitative Real-Time PCR”, BCt should be CT * p.3, second sentence of the first paragraph under the Results section, results should be PCR results * The correct bibliographic information for reference [1] should be Yur F, Mert N, Dede S, Değer Y, Ertekin A, Mert H, Yaşar S, Doğan I, Işık A (2013) Evaluation of serum lipid fractions and tissue antioxidant levels in sheep with fluorosis. Fluoride 46(2):90–96
Rights and permissions
About this article

Cite this article
Urut, F., Dede, S., Yuksek, V. et al. In Vitro Evaluation of the Apoptotic, Autophagic, and Necrotic Molecular Pathways of Fluoride. Biol Trace Elem Res 199, 3700–3706 (2021). https://doi.org/10.1007/s12011-020-02491-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-020-02491-3