
Abstract
Boron-doped hydroxyapatite/tricalcium phosphates (BHTs) were synthesized to study boron uptake and correlate structural alterations of incremental boron addition (0 to 10 mol%). BHTs with a Ca/P ratio of 1.6 were prepared by a wet precipitation/microwave reflux method, sieved (< 70 μm) and characterized. XRD and FTIR analyses revealed that boron slightly distorted apatite crystal, increased crystallinity (95.78 ± 2.08% for 5BHT) and crystallite size (103.39 ± 23.47 nm for 5BHT) and still, boron addition did not show any further detrimental effects. Total surface area (4.05 ± 0.82 m2/g for 10BHT) and mesoporosity (23.90 ± 7.92 μL/g for 10BHT) were expanded as boron content was increased. Moreover, boron addition made grains become smaller (0.21 ± 0.06 μm for 5BHT) and ordered while hardness (10.51 ± 0.86 GPa for 10BHT) increased. Boron incorporation enhanced bioactivity with significantly highest calcium phosphate deposition and protein adsorption (135.29 ± 29.58 μg on 10BHT). In return, boron favored highest alkaline phosphatase activity (4.80 ± 0.40 MALP/ngDNA.min), intracellular calcium (23.61 ± 0.68 g/gDNA), phosphate (31.84 ± 4.68 g/gDNA), and protein (23.70 ± 3.46 g/gDNA) storage in 5BHT without cytotoxicity (128 ± 18% viability compared to pure HT). Compared to literature, it can be pointed out that we successfully employed an optimal procedure for production of BHTs and incorporated significantly higher boron content in HT (5.23 mol%). Additionally, results tended to conclude that 5BHT samples (5 mol% boron in HT) demonstrated a very high potential to be used in composite bone tissue constructs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bone tissue engineering is an interdisciplinary field aiming to produce biocompatible and multifunctional materials and devices supporting bone regeneration [1]. With the use of bone similar calcium phosphates (CaP), a bioactive ceramic can be fabricated as a stand-alone biomaterial, a bone implant coating, or a reinforcement in a composite scaffold [2]. Due to remarkable osteoconduction ability, CaPs have been extensively employed in orthopedic and dental applications since the middle of twentieth century [3]. In spite of demonstrating good bioactivity, CaPs require use of dopants to their improve structural, topographical, physical, and biological properties [4]. CaP-based implants displayed poor regenerative capacity due to slow rate of osteogenic ions release resulting in limited osteoinductivity [5, 6]. Consequently, CaPs have been designed to acquire tailored properties allowing enhanced bioactivity such as rapid mineral deposition, protein adsorption, and release of ions involving in downstream of signaling cascades leading to osteoblastogenesis [7, 8]. Therefore, insertion of trace elements such as zinc (Zn2+), magnesium (Mg2+), copper (Cu2+), chromium (Cr2+), lithium (Li+), cobalt (Co2+), strontium (Sr2+), and fluoride (F−) [9,10,11,12,13,14] has been predominantly studied to achieve desired osteogenic properties for the last two decades.
Recently, trace elements in natural bone have been the focus of attention in preparation of biomaterials capable of osseointegration. Although there are many studies focusing on boron (B) as main dopant in multidisciplinary fields such as tissue engineering, the importance of B is not fully appreciated in bone tissue engineering (BTE) field.
B is a trivalent ion naturally present in bone as a trace element (< 1%) [15]. B inclusion in diet was shown to improve rabbit tibial mineral volume and reverse ion deficiency [16]. Furthermore, osteoblastic lineage-related bone morphogenic protein-1, collagen type 1, osteocalcin, Runx-2, and osteopontin upregulation was observed when preosteoblastic cell line (MC3T3-E1) was exposed to 100 and 1000 ng/mL in boric acid (H3BO3) form [17]. In a similar study, Gümüşderelioğlu et al. demonstrated that the MC3T3-E1 produced an increased alkaline phosphatase (ALP) concentration after treated with encapsulated H3BO3 in chitosan nanoparticles [18]. In B deficiency-triggered mice after prohibiting B uptake in diet, intracellular lacunae density and femoral rigidity were significantly decreased [19]. Furthermore, a daily B supplementation of 1 mg/L in post-menopausal women was found to improve bone osteocalcin level significantly [20].
In this study, we used H3BO3 as B source in biphasic hydroxyapatite/tricalcium phosphate (HT) structure. Precursor Ca/P ratio was selected as 1.60 to increase B uptake [21] and B-doped HT (BHT) was prepared by wet precipitation method followed by microwave reflux process. Moreover, cell culture studies were conducted with dental pulp stem cells (DPSCs) that were isolated from human third molar tooth. These fairly new abundant source of mesenchymal stem cells [22, 23] were employed to illustrate biological properties of BHTs and ability to prompt production of bone-related factors such as ALP, intracellular calcium (ICa), and phosphate ions (IPa).
We hypothesized that microwave reflux parameters as well as precursor Ca/P ratio (Ca/P = 1.6) could enable us to incorporate B in higher concentrations compared to literature and the effect of B on microstructural, mechanical, chemical, and biological properties of HT could be effectively explored. The change in lattice properties, crystallinity, crystallite size, cell volume, and functional groups were analyzed by X-ray diffraction spectroscopy (XRD) and Fourier transform infrared spectroscopy (FTIR). Elemental analysis and the empirical concentration of B in HT after doping were determined by induced coupled plasma–optical emission spectroscopy (ICP-OES). Microhardness, surface area, and mesoporosity of HT and BHTs were also determined. The effect of B presence on bioactivity was demonstrated by both mineral deposition and protein adsorption studies. Finally, in vitro cytotoxicity of BHTs and their ability to trigger osteogenic differentiation using human DPSCs were tested.
Materials and Methods
Calcium nitrate tetrahydrate (Ca (NO3)2.4H2O; ammonium phosphate (NH4)2HPO4 and boric acid (H3BO3); ethanol; 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT); dimthyl sulfoxide (DMSO); dexamethasone (Dex); L-ascorbic acid (L-AA); and β-glycerophosphate (β-GP), para-nitrophenyl phenol, 4-nitrophenol were acquired from Sigma (USA). Nitric acid (HNO3), acetone, methanol, and magnesium chloride hexahydrate (MgCl2.6H2O) were purchased from Merck (USA) and Picogreen® assay kit from Invitrogen (USA).
Preparation of BHT
Biphasic HT and BHTs were prepared by wet precipitation/microwave reflux method. Briefly, Ca(NO3)2.4H2O was dissolved in deionized water (dH2O) in 25 °C and then added either (NH4)2HPO4 alone or together with (NH4)2HPO4 and H3BO3 to obtain HT and BHTs, respectively (Table 1). After that, pH of the mixture was set to 10 using ammonia (NH4OH) and mixture was vigorously stirred (1000 rpm) for 30 min. The mixture was then placed in a microwave oven, aged for 15 min under 800 W, filtered, and dried under 150 °C in an oven overnight. Finally, samples were sintered at 1100 °C for 2 h with a heating rate of 10 °C/min (Protherm, Turkey) and slowly cooled down to room temperature overnight. Unless specifically indicated, only sintered samples in powder form were employed in the studies.
Prior to further analysis, samples were grounded, ball milled for 1 h at 30 Hz and ultrasonicated continuously for 3 min at 25% amplitude in acetone (250 mg/5 mL) using a probe type ultrasonicator (Branson, USA). Then, they were sieved through No. 200 mesh (~ 70 μm openings) and dried at 75 °C under vacuum (− 10 psi) overnight. Wet particle size analysis (with 1 g of each sample) in double-distilled water (ddH2O) was conducted (Malvern, UK).
Structural and Morphological Properties
Lattice parameters, phases, crystallinity percent (Xc), crystallinity index (CIXRD), and crystallite size of samples (with three different batches) were determined by XRD analysis under 40 kV and 30 mA CuKα radiation and 2θ between 20 and 60° at a step size of 0.2°/min (PANalytical Empyrean, the Netherlands). Phases were ascertained from the intensity ratio of the peaks at (0210) for β-tricalcium phosphate (β-TCP) and (211) for hydroxyapatite (HA). Xc was found by the intensity ratio of the peak at (300) over the hollow (V112/300) between (112) and (300). Finally, CIXRD was determined by constant 0.24 for HA and employing full width at half maximum of peak at (002), and crystallite size was calculated by Scherrer equation using the Scherrer’s constant as 0.94, full width at half maximum of peak at (002), wavelength of λ = 1.54056 Å and 2θ° of (211) reflections in radian. Following equations are used to determine the amount of HA phase, crystallinity, crystallite size, and cell volume:
Functional groups were detected by FTIR in transmission mode between 400 and 4000 cm−1 mid-IR range (Bruker IFS66/S, USA).
Moreover, 1 g of each sample was degassed at 200 °C for 12 h and nitrogen (N2) adsorption-desorption isotherms were obtained at − 196 °C with a surface characterization device (Autosorb II-6B, Austria). Multi-point Brunauer-Emmett-Teller (BET) method (0.05 < P/P0 < 0.25) for total surface area (TSA) and Barrett-Joyner-Halenda (BJH) method for mesoporosity using desorption isotherm were conducted. BET and BJH studies were performed in triplicates. Additionally, samples were dissolved in HNO3 and analyzed with ICP-OES to ascertain calcium to phosphate ratio (Ca/P) and total boron content (B%) (n = 3).
Physical and Mechanical Properties
To determine grain size, microhardness, and density, powders were compacted under 250 MPa into pellets (diameter of 13 cm and thickness of 1 cm), sintered at 1100 °C for 2 h and rinsed thoroughly with dH2O. Then, samples were coated with gold/palladium for scanning electron microscopy (NanoSEM, FEI, USA). Average grain size of each sample was calculated after 50 measurements. Moreover, apparent density of each sample (n = 5) was measured with liquid displacement method applying the following equation:
In equation (5), samples were initially weighed in dry form (wd), then completely wetted in vacuum forcing air to replace with acetone (dacetone = 0.784 g/cm3) and weighed after suspended in acetone (ww). Relative density (ρRel) was determined according to Equation 6 by taking ρtheo of HA as 3.16 g/cm3 and ρtheo of β-TCP as 3.07 g/cm3:
Sample pellets (n = 5, two measurements for each sample) were further tested by microindentation. A 19.63 N force was applied with a 10-s dwelling time and hardness values were calculated from mean diagonal length (d) of indentation:
Mineral Deposition and Protein Adsoprtion Ability
In order to fully characterize bioactivity, mineral deposition and protein adsorption ability of the materials synthesized were tested. After sintering, pellets were rinsed with 70% dH2O: 30% ethanol solution and sterilized at 200 °C for 2 h. ISO 23317 protocol was used for bioactivity test. Pellets (132 mm2) were placed in 50-mL tubes horizontally, introduced with 13.2 mL × 1 simulated body fluid (SBF, pH 7.4) and incubated for 1 and 7 days at 37 °C under 100 rpm constant shaking. SBF was prepared in accordance with Kokubo’s method [24]. After incubation period, samples were removed, rinsed with dH2O and acetone, and dried at 100 °C overnight. To observe deposited minerals, pellets were analyzed with SEM and energy dispersive X-ray (EDX), which was attached to a SEM equipment.
For protein adsorption study, sterilized pellets were placed vertically in a 50-mL tube having 10 mL 0.1% v/v FBS solution in dH2O and incubated for 1, 6, and 24 h at 37 °C under 100 rpm constant shaking (n = 3). At each time point, pellets were removed, rinsed with dH2O, and treated with 0.1 M sodium dodecyl sulfate (SDS) to collect attached proteins. Two hundred and fifty microliters of SDS solution was collected from each sample, introduced to 1715 μL bicinchoninic acid: 35 μL 4% w/v cupric sulfate pentahydrate (BCA solution) and incubated for 1 h at 37 °C. At the end, absorbance of sample BCA solution was measured at 562 nm. The amount of protein adsorbed was determined using the calibration curve constructed with different concentrations of BSA solution (0 to 250 μg/mL)
Cellular Viability Analysis and Colony Formation Assay
Samples were sterilized at 200 °C for 2 h and placed in a growth medium (94.9% v/v Dulbecco’s MEM with 1 g/L glucose and 2.5 mM L glutamine, 5% v/v fetal bovine serum (FBS, heat inactivated), and 0.1% v/v penicillin-streptomycin) at a concentration of 100 mg/mL. Samples were then incubated at 37 °C for 24 h following the ISO-10993:5/12 protocol for sample preparation and cytotoxicity analysis. In the meantime, dental pulp stem cells (DPSC, p. 4) were seeded into cell culture plates at a density of 10,000 cells/well and incubated for 24 h for attachment. After incubation period, samples were centrifuged at 5000 g for 5 min and filtered through a 0.22-μm syringe filter. Extract media obtained were added as media for DPSCs and cells were incubated for 24 h with extracts of HT and BHTs. After a 24-h incubation, media were discarded, and wells were rinsed with phosphate-buffered saline (PBS, 0.1 M, pH 7.4). After PBS removal, cells were incubated for 3 h with MTT (0.5 mg/mL in culture medium without phenol red) at 37 °C. Precipitated formazan salts were then dissolved with DMSO and OD570 was measured for each sample. HT was selected as positive control for cellular viability. In addition, effect of direct contact of particles on colony forming ability of DPSCs (n = 2) was also studied. DPSCs were incubated in the presence of samples (100 μg/mL) for 1 and 7 days. Afterwards, cells were gently rinsed with PBS and incubated with 0.2% w/v crystal violet (CV) in 20% methanol: 80% dH2O for 30 min at the end of each incubation period to observe morphology of the cells. Then, DPSCs were rinsed twice with PBS slowly to remove excess CV, and alive cells stained with blue-purple color were examined under inverted microscope.
Osteogenic Differentiation Analysis
Samples (n = 4, duplicate study) with a 100-mg/mL concentration in osteogenic differentiation medium were placed on transwell membranes having a 1-μm pore size. Osteogenic differentiation medium was prepared by adding 10−7 M Dex, 50 μg/mL L-AA, and 10 mM β-GP. Wells were seeded with DPSCs (10,000 cells/well). DPSCs were allowed to attach for 24 h. After that, transwells with samples were placed on top and incubated for 3 weeks. At the end of each week, transwells were removed from DPSCs, cells were rinsed with PBS. Then, 0.1% TX in carbonate buffer (CB, 0.1 M, pH 10.2) was added and cells underwent freeze-thaw cycle twice (− 80 °C and 20 °C, respectively). Once DPSCs were completely lysed, aliquots were collected and analyzed for alkaline phosphatase activity (ALP), total DNA concentration with Picogreen® assay, total intracellular protein concentration (IPro), intracellular calcium concentration (ICa), and intracellular phosphate concentration (IPa). Briefly, aliquots were incubated with pNpp and 5 mM MgCl2.6H2O solution at 37 °C for 1 h. pNpp was converted to 4-nitrophenol in accordance with ALP concentration and the conversion was measured under OD405. ALP activity was normalized to total DNA. In addition, IPro, ICa, and IPa were determined according to procedure explained in our another study [25].
Statistical Analysis
All data were represented as average ± standard deviation and evaluated for normal distribution (Shapiro-Wilk) prior to one-way analysis of variance (ANOVA). Tukey’s post hoc test was applied following ANOVA. Difference among and between groups were considered significant at p < 0.05.
Results and Discussion
Microstructural Characterization
Scanning electron microscopy micrographs of the produced samples are presented in Fig. 1. Assuming spherical morphology for all particles, volume mean diameter (d [4,3]) was calculated and found to be around 30 μm and was not significantly different among groups (Table 2). Moreover, close-up SEM analysis on the powders revealed grain boundaries and provided a qualitative visual for effective surface area (Fig. 1). It can be noted that particle area of B-doped groups appeared to be greater than that of HT, and 10BHT had a more compact structure similar to that of HT. On the other hand, 2BHT, 3BHT, and 5BHT displayed fine grains, and 5BHT appeared to have a large surface area (Fig. 1). In addition, HT and 10BHT samples showed similar morphologies with larger grains than other B-doped counterparts in the pellet form (Fig. 1). In detail, HT and 1BHT had an average grain size of 1.92 μm and as 1.59 μm, respectively while 2BHT and 3BHT displayed finer grains with a grain size of 0.74 μm and 0.53 μm, respectively. Concurrently, 5BHT exhibited significantly (p < 0.05) smallest grain size (0.21 ± 0.06 μm) and 10BHT appeared much coarser at 0.84 ± 0.26 μm. Consequently, it can be said that B-doping has a limiting condition by which B incorporation results in a fine and ordered grain structure with a highly uniform size distribution, thus granting a control over the final product up to a certain dopant concentration (5%).

SEM electographs of powder and pellet samples (scale bar, 2 μm)
Employing the method of microwave reflux at 800 W for 15 min in a single run and utilizing only one sintering temperature and pH (1100 °C for 2 h, pH 10), we designated B concentration as the sole parameter that allowed us to determine the efficiency of B-doping and overall effect of B on the microstructural characteristics of HT. According to XRD patterns presented in Fig. 2a andbB, all samples showed HA characteristic reflections [26], namely (002), (211), (112), and (300) and unique β-TCP reflection (0210), except for 10BHT. Focusing on the peaks at 2θ of 30.72° for (0210) and 32.20° for (211), HT was found to have the lowest intensities for all reflections and maintained 86.3% HA and 13.7% β-TCP phases (Table 2). It was further observed that B incorporation brought about an increase in I(211) while a steep decrease in I(0210). β-TCP (0210) reflection was diminished in successive B additions and completely lost at 10BHT (Fig. 2a). HT and 1BHT were determined to have similar HA phase concentrations, and it was found to be significantly small when compared with other BHTs. Moreover, a distinct (002) reflection was utilized to determine Xc of HT, and it was found as 89.03 ± 4.65%. In the case of BHTs, the peaks shifted in direct correlation with B content in samples in addition to the altered intensities (Fig. 2b). Especially HA-related reflections were shifted to lower 2θ°, which indicated that B was incorporated in the lattice via B-type substitution [27]. V112/300 value showing the hollow between (112) and (300) reflections was gradually shifted to a smaller distance in BHTs (Fig. 2b). Moreover, (211) and (002) peaks attained greatest intensities for 5BHT. When calculated, 5BHT was found to have the highest Xc (95.77 ± 2.08%). However, no significant difference was detected among samples (Table 2). The increase in Xc towards 5BHT could be a result of slight distortion in the structure [28]. Since the sinterability depends also on diffusion in the solid crystal, expanded lattice up to a threshold level could provide a faster maturation. Thus, an increment in the Xc was observed.

Phases (a), HA- and β-TCP-related reflections (b) found by XRD, FTIR results (c), and change in relative density and hardness values (d, n = 3 for ρRel and n = 10 for hardness analysis)
In parallel with previous data, B-doping in HT caused distortion in the lattice of biphasic structure in a way that a = b-axes shrinked while c-axis expanded (Table 2). A similar finding was presented by Barheine et al. [29]. They concluded that structural disorder was increased as more B replaces PO43− and OH− in HA lattice resulting in a significant change in c axis. In addition, Vsample initially increased as B substitution increased until a threshold of B concentration in HT was reached (Table 2). From HT to 5BHT, a slightly increasing trend for Vsample was observed while the lowest value was obtained for V10BHT (611Å3). In the meantime, we did not observe any newly formed peaks in the XRD study in accordance with the change in a = b and c-axes upon doping with B. In a study by Kolmas et al., strontium and B co-doped HA was prepared. They reported that absence of an extra peak could be interpreted as B units might be incorporated in the HA lattice without an undesirable crystal formation [30]. Therefore, it can be stressed that only HA and β-TCP phases were present and B was succesfully incorporated in HT lattice (Fig. 2a). Crystallite sizes were obtained to be close to each other for all samples (Table 2). Although not significant, 10BHT was found to have the smallest crystallite size (78.30 ± 12.51 nm). As a result, it can be stated that B encouraged formation of HA phase, increase in Xc and improved the rate of crystal maturation. However, over a limiting concentration, presence of B suppressed the growth of crystallite and led to formation of Ca-deficient HA.
In FTIR study (Fig. 2c), peaks at 472 cm−1, 557 cm−1, 601 cm−1, 960 cm−1, 1034 cm−1, and 1086 cm−1 showed the presence of PO43−, and they were in good correlation with literature [31]. Furthermore, peak observed at 646 cm−1 confirmed the presence of OH− [32]. However, the intensity of the OH−-related peak as the B content increases in the structure implied that OH− was replaced with an ion supplied by H3BO3. According to a study conducted by Albayrak, it was reported that B-doped HA was found to contain both BO2− and BO33− as dissociation products of H3BO3 and they substituted PO43− and OH− [33]. In addition, peaks at 746 cm−1, 787 cm−1, 1233 cm−1, and 1374 cm−1 showed the presence of BO33− [34, 35]. According to XRD and FTIR results, it is possible to state that HT structure can be doped with B successfully without formation of additional phases.
Relative densities of HT and BHTs are given in Fig. 2d. All groups showed significantly different ρRel and 10BHT displayed the highest ρRel while HT had the smallest ρRel (p < 0.05). The gradual rise of ρRel from 95.76 g/cm3 for HT to 98.72 g/cm3 for 10BHT could be resulted from formation of pure HA phase as the incorporated B concentration increased in the lattice. Such increase in dense HA phase might allow establishment of a more rigid nanostructure [36]. Since ρRel is inversely correlated with the porosity in the bulk (pellet in our case), it can also be suggested that porosity decreased via B-doping. Poinern et al. investigated the structural and mechanical effects of 50 W ultrasound mixing and various sintering temperatures after compacting the samples under 70 MPa for 1 h [37]. In parallel with our findings, they reported that increase in ρRel was a consequence of improved crystallinity and diminished porosity could lead to enhanced compaction by which high microhardness can be achieved. Moreover, Hasan et al. studied the effect of plasma spray process parameters on physical and mechanical properties of the commercial spherical HA particles having 20–70 μm size [38]. As a result of increased plasma spray power (20 kW, 8 cm distance to 40 kW, 16 cm distance), porosity of HA particles decreased to ca. 5% while hardness surged to a higher value twice as much. In addition to ρRel results, 10BHT displayed 10.51 ± 0.86 GPa hardness, which was significantly high when compared with other BHTs. Moreover, 5BHT also showed a significantly high hardness (8.04 ± 0.63 GPa) while HT was the weakest sample (5.82 ± 0.24 GPa). A biphasic titanium dioxide (TiO2)-doped HT sample was prepared by Nie et al. via isostatic compaction under 285 MPa and sintered at 1250 °C in a microwave oven [39]. They determined that a threshold value of 4% TiO2 dopant in HT structure was optimal for highest ρRel, and thus greatest hardness (4.69 ± 0.34 GPa) was achieved. However, high sintering temperature over 1100 °C led to extensive β-TCP phase. Correspondingly, hardness of the final product was clearly lower than that of B-doped HT produced in our study probably due to lower β-TCP phase presence in our final BHT products. In another study, Reger et al. prepared multi-ion-oped HA containing strontium (Sr), silver (Ag), zinc (Zn), and fluoride (F) ions by a wet precipitation method followed by a range of sintering temperatures between 800 and 1250 °C [40]. They observed that negatively charged dopants, such as F-, prohibited crystal growth and improved Xc over a concentration range (up to 2.5%). Comparably, we observed an overall increase in crystallite size (except for 10BHT), hardness and HA phase stability increased as the B-dopant was incorporated in the HT crystal. However, B=doped HT demonstrated a clearly higher hardness (8.04 ± 0.63 GPa) than that of FHA that was reported by Reger et al. Besides the already mentioned effect of phase stability, B might also have improved the mechanical properties owing to its ability to prevent formation of large grains. A study by Itatani et al. evaluated ball mill blended boron oxide (B2O3) particles (1 to 10 mol%) and commercial HA that was pulse-current sintered up to 1200 °C after isostatically compaction at 100 MPa [41]. They have selected 3% mol B2O3-doped HA for comparison. They determined the grain size after sintering at 1100 °C for 5 h and tested for tensile strength of pellets under 1100 °C, 50 MPa uniaxial deformation. It was observed that grain size (0.56 μm) of ball milled B2O3 and commercial HA was close to that of 3BHT (0.53 ± 0.11 μm) with similar ρRel (96% and 97.12%, respectively). They concluded that B2O3-dopant enabled grain boundaries slide over each other to reach superplastic deformation ,and therefore it acted as a nanoreinforcement augmenting mechanical strength.
In order to verify B presence in the crystal structure and determine Ca/P ratio, samples were analyzed by ICP-OES (Table 3). Although not significantly different, increment in B content resulted in lower Ca/P ratio. Parallel to literature, B-dopants appeared to increase the disorder in lattice leading to Ca deficient CaP formation as previously discussed. Barheine et al. (2009) demonstrated that borate (BO2− or BO33−) substitution in HA lattice results in a formation of two intricate phases in the crystal [42]. They further verified that highly crystalline pure HA phase resists borate incorporation as well as Ca2+ uptake by the core lattice. Therefore, borate-doped HA, borohydroxyapatites, were shown to possess both lower than initially reacted B and Ca deficiency in the final product. In good agreement with literature, B-dopant concentration increase was followed by a sharp decrease of P/B ratio owing to PO43− to BO33−substitution (Table 3). P/B ratio and B content in mineral (%) displayed significant differences among samples (p < 0.05)
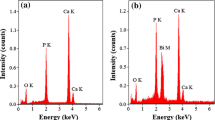
BET and BJH analyses were conducted to characterize TSA and mesoporosity of B-doped HT. It was observed that 5BHT and 10BHT possessed statistically higher TSA than other groups and the highest mesoporosity was acquired for 10BHT (Table 4). Wang et al. conducted BET analysis for differentially sintered HA, and reported that mass transport phenomenon affected growth of the crystallite size and TSA [43]. In this study, B was shown to impact sinterability, thus improving crystallization by temperature treatment and limiting crystallite growth in the meantime. Moreover, Wang et al. discussed that higher TSA and mesoporosity would enable fast apatite mineralization [44]. We also examined the mineral deposition ability of the samples in a × 1 SBF environment. B-doped samples showed evidently better ability to induce CaP deposition of the samples (Fig. 3). To elaborate, we employed EDX and determined Ca/P ratios (Fig. 4a). HT demonstrated significantly lower Ca/P ratio at the end of 1st and 5th days of SBF incubation. On the contrary, B-doped samples, especially 5BHT (1.69 ± 0.02) and 10BHT (1.69 ± 0.02), performed far better and permitted higher Ca/P precipitate formation on the surface than other samples. Hence, it was concluded that B promoted a more competent surface to prompt Ca and P precipitation on the pellets, and favored stoichiometric HA formation in 7 days (Fig. 4a).

Mineral deposition on samples after 1 and 7 days of incubation in SBF (long scale bars, 100 μm; short scale bars, 2 μm)

Ca/P curves after mineral deposition study and protein adsorption results. All groups had significantly higher Ca/P than HT (n = 4, p < 0.05)
Bioactivity is a complex phenomenon that can be evaluated by both HA deposition and protein adsorption [45]. All samples reached to maximum protein adsorption as early as in 1 h (Fig. 4b). Although no significant difference was present, B-doped HT samples had slightly higher protein adsorption capacity (Fig. 4b). Close adsorption capacities were found for all samples. This may be due to the loss of mesoporosity after compaction [46]. Nevertheless, ability to anchor plasma proteins (from FBS) and induce stoichiometric HA deposition confirmed high bioactivity of BHTs.
There is a very limited number of literature covering B-doping in HA or biphasic HT. Furthermore, these studies demonstrated highly detrimental distortion occurred in the structure of the product. Albayrak used H3BO3 as B precursor and employed wet precipitation process which was followed by aging at 40 °C for 48 h [33]. Pure HT obtained had 83.1% HA and 16.9% β-TCP, comparable to that of HT prepared in this study (86.3% HA, 13.7% β-TCP). However, B-doping brought about a significant decrease in HA phase, density, and hardness. Moreover, a substantial intensity of BO2− or BO33− bands in the FTIR spectra was not observed. Therefore, absence of energy application during aging process (only 40 °C) could have prevented B incorporation. In a study by Tunçay et al. involving microwave sintering, H3BO3 was dissolved at a pH of 6.8–7.4 and introduced to × 10 SBF. The mixture was then subjected to microwave working at 600 W or 1200 W for 7.5 min at maximum [47]. They showed a noteworthy amount of B was incorporated into the HT (P/B was 4.75). However, Ca/(P + B) was around 1.16 and XRD patterns showed a highly amorphous CaP instead of HA or HT. Arslan et al. fabricated B-doped CaP employing 600 W microwave sintering process following biomimetic precipitation process [48]. It was again observed that the structure was highly amorphous. However, there was no further investigation in the work of Arslan et al. [48].
Compared to literature, it can be pointed out that the conditions selected for B-doping play a critical role in incorporation of B into the structure and production of a highly bioactive B-doped HT. The BHTs produced in this study showed that B reinforced the structural, mechanical, physical, and chemical properties of the samples. It is further stressed that the wet precipitation process using Ca/(P + B) as 1.60 along with a highly energetic aging procedure (800 W, 15 min) could improve B incorporation rate while allowing crystallization of HA and minimizing the distortion in the crystal structure.
Biological Characterization
Samples were characterized for their biocompatibility (Fig. 6a). Briefly, samples in powder form were sterilized and placed in cell culture medium. Fully immersed particles were incubated for 24 h. For all of the samples, no abrupt pH change was observed (pH was around 7.8). Filtered media were then placed in DPSCs seeded wells. DPSCs treated with extracts of BHTs showed a nonsignificant increase (except for 1BHT) in cellular viability relative to HT (Fig. 6a). In addition, amount of B released into the incubation medium after interacting with cells for 24 h was determined (Table 3). A significantly higher B release was observed in successive dopings, reaching as high as 105 μg/L for 5BHT and 185 μg/L for 10BHT. A study by Burguera et al. showed that the blood B level is in between 90 and 260 μg/L without any supplements [49]. Additionally, 0.5 g B supplementation did not increase blood B levels but raised B level in bone (from 2.0 ± 0.1 to 8.2 ± 0.2 μg/g). Therefore, it can be stated that the changes in structural properties of HT after B-doping did not lead to higher B release than the natural safe limit and could support bone repair. Clonogenic study, in addition to MTT assay, exhibited similar results (Fig. 5). Colony formation was used as a mean to determine particulate effect on cytotoxicity and thus 100 μg/mL particles of each sample was simply placed in the wells seeded with DPSCs [50]. Change in size of the colonies was examined under the phase contrast microscope from 1st day to 7th day (Fig. 5). Additionally, it was also observed that morphology of DPSCs was not altered. DPSCs have spindle morphology without any flat and spread shape. Therefore, it can be said that BHTs were considered cytocompatible.

CV staining of DPSCs after treating with 100 μg/mL HT and BHTs in direct contact for 1 day (D1) and 7 days (D7). Red bar, 100 μm. Smaller images in D7 are closer (× 10) images of DPSCs under phase contrast micrsocope
Considering blood B levels, mechanical strength and ability to induction of apatite deposition, 5BHT and 10BHT samples were selected for further characterization in vitro. In order to mimic natural conditions, we used osteogenic medium for the analysis of osteogenic differentiation [51]. According to the results presented in Fig. 6b, ALP activity showed a significant increase and achieved peak levels of ALP after 2 weeks. This result was in good agreement with the literature where DPSCs were shown to differentiate into osteogenic lineage in as early as 2 weeks [52]. Moreover, it was observed that this increase was further amplified by the presence of B (Fig. 6b) and DPSCs treated with 5BHT demonstrated significantly the highest ALP activity (4.80 ± 0.40 MALP/ngDNA.min). Ojansivu et al. observed that B-doped S53P4 commercial bioglass generated high B ion release and concluded that it might be the reason for downregulated ALP [53]. They suggested that continuous media flow could help avoid the immense ion accumulation. On the other hand, Taşlı et al. employed 20 mg/L sodium borate (NaB) to induce differentiation of DPSCs and determined that B was not cytotoxic and increased ALP production [54]. Najafabi et al. demonstrated that 6 mg/L of H3BO3 was optimal to achieve the highest ALP activity of cells [55]. However, we observed that 5BHT samples in 100 mg/mL had the optimal B presence for highest ALP activity. When calculated, 5BHT contained 23.3 mg/g HT and could release 2.33 g/L B when completely hydrolyzed. Therefore, it can be speculated that 5BHT could provide continuous release of osteogenic ions below cytotoxic limit for longer period of time compared to 10BHT. These results were also validated by the steady increase in IPro (Fig. 6e) and DNA content (Fig. 6f). Increase in DNA amount suggested that cells were healthy and increased in number which also showed that there was no sign of cytotoxicity. The peak value for IPro was observed in the 2nd week which indicated osteogenic differentiation along with increase in ALP activity. Davies et al. observed that exosomes and vesicles filled by microRNAs and osteogenic ions were vigorously produced during differentiation to establish extracellular matrix, during which IPro content of cells increased [56]. Moreover, upregulation of integrin-associated signaling molecules additionally boosts IPro [57].

MTT results (n = 12, (a)), ALP activity (n = 8, (b)), ICa (n = 8, (c)), IPa (n = 8, (d)), IPro (n = 8, (e)), and total DNA collected form lysates during osteogenic differentiation (n = 8, (f)). Significantly highest groups at a particular time point are marked by α. β shows significantly highest groups compared to the 1st week of the given analysis (p < 0.05)
In parallel ALP activity findings, ICa and IPa results demonstrated that commitment to osteogenic phenotype involved Ca2+ and PO43− intracellular ion storage (Fig. 6c). The peak ICa was achieved at the end of 2nd week. Birmingham et al. displayed that ICa and ALP activity increase occur at the same time exhibiting osteoblastic commitment [58]. As the storage increases in the cell, Ca2+ and PO43− forms CaP and it is transferred to extracellular environment to trigger biomineralization. Concurrently, IPa concentration was built up further implying osteogenic differentiation (Fig. 6d). In a study by Shih, PO43− was shown to be a part of signaling cascade maintaining biomineralized matrix thus contributing to osteogenesis in addition to CaP deposition [59]. Corresponding to these results, 5BHT resulted in highest ALP activity, ICa (23.61 ± 0.68 g/gDNA), IPa (31.84 ± 4.68 g/gDNA), and IPro (23.70 ± 3.46 g/gDNA) at 2nd week of osteogenic differentiation study.
Conclusion
B incorporation in biphasic HT structure was studied in the present study. It was observed that the crystal lattice showed dissimilar changes as reported in literature. Doping with B allowed formation and crystallization of HA phase, and increased Xc and Lc when doped up to 5% molar B. Hardness was determined as 10.51 GPa, TSA was risen to 4.05 m2/g and mesoporosity reached its maximum value of 23.90 μL/g when doped by 10% B. Ability to induce apatite deposition and protein adsorption were significantly improved as incorporated B content was increased. Furthermore, B-doping showed no cytotoxicity, encouraged colony formation, and enhanced osteogenic properties. However, 10BHT displayed a decrease in ALP activity, ICa and IPa when compared to 5BHT which demonstrated significantly highest values for each of the osteogenesis markers. Therefore, we deduced two important outcomes: First, the method employed to incorporate B into well-known HT is fairly critical to obtain a B-doped product either having enhanced material properties or completely altered crystal structure. Second, 5BHT may be used as a novel implant coating material or as a reinforcement overhauling mechanical, chemical, and biological functions of composite scaffolds for BTE. As future studies, the effect of B on osteoblastogenic and osteoclastogenic pathways will be investigated. Additionally, comparison of HT and 5BHT in terms bactericidal activity on Staphylococcus aureus will be conducted to elaborate multifunctionality conferred by B-doping. On the other hand, the major limitation of the presented study is that HT and 5BHT were not studied in vivo. Therefore, in vivo studies can be planned as a future study.
References
Nguyen DT, Burg KJ (2015) Bone tissue engineering and regenerative medicine: targeting pathological fractures. J Biomed Mater Res A 103(1):420–429
Tite T, Popa A-C, Balescu LM, Bogdan IM, Pasuk I, Ferreira JM, Stan G (2018) Cationic substitutions in hydroxyapatite: current status of the derived biofunctional effects and their in vitro interrogation methods. Materials. 11(11):2081
Dorozhkin SV (2010) Calcium orthophosphates as bioceramics: state of the art. J Funct Biomater 1(1):22–107
Evis Z, Webster T (2011) Nanosize hydroxyapatite: doping with various ions. Adv Appl Ceram 110(5):311–321
Hoornaert A, Maazouz Y, Pastorino D, Aparicio C, de Pinieux G, Fellah BH, Ginebra MP, Layrolle P (2019) Vertical bone regeneration with synthetic biomimetic calcium phosphate onto the calvaria of rats. Tissue Eng C Methods 25(1):1–11
Zhang BG, Myers DE, Wallace GG, Brandt M, Choong PF (2014) Bioactive coatings for orthopaedic implants—recent trends in development of implant coatings. Int J Mol Sci 15(7):11878–11921
Renaudin G, Gomes S, Nedelec J-M (2017) First-row transition metal doping in calcium phosphate bioceramics: a detailed crystallographic study. Materials. 10(1):92
Bohner M, Galea L, Doebelin N (2012) Calcium phosphate bone graft substitutes: failures and hopes. J Eur Ceram Soc 32(11):2663–2671
Bose S, Fielding G, Tarafder S, Bandyopadhyay A (2013) Understanding of dopant-induced osteogenesis and angiogenesis in calcium phosphate ceramics. Trends in Biotechnology 31(10):594–605
Hidouri M, Dorozhkin SV (2018) Structure and thermal stability of sodium and carbonate-co-substituted strontium hydroxyfluorapatites. New J Chem 42(11):8469–8477
Kulanthaivel S, Roy B, Agarwal T, Giri S, Pramanik K, Pal K, Ray SS, Maiti TK, Banerjee I (2016) Cobalt doped proangiogenic hydroxyapatite for bone tissue engineering application. Mater Sci Eng C 58:648–658
Wang Y, Yang X, Gu Z, Qin H, Li L, Liu J, Yu X (2016) In vitro study on the degradation of lithium-doped hydroxyapatite for bone tissue engineering scaffold. Mater Sci Eng C 66:185–192
Gao J, Wang M, Shi C, Wang L, Wang D, Zhu Y (2016) Synthesis of trace element Si and Sr codoping hydroxyapatite with non-cytotoxicity and enhanced cell proliferation and differentiation. Biol Trace Elem Res 174(1):208–217
Bernhardt A, Schamel M, Gbureck U, Gelinsky M (2017) Osteoclastic differentiation and resorption is modulated by bioactive metal ions Co2+, Cu2+ and Cr3+ incorporated into calcium phosphate bone cements. PLOS ONE 12(8):e0182109
Doğan A, Demirci S, Bayir Y, Halici Z, Karakus E, Aydin A, Cadirci E, Albayrak A, Demirci E, Karaman A, Ayan AK, Gundogdu C, Şahin F (2014) Boron containing poly-(lactide-co-glycolide) (PLGA) scaffolds for bone tissue engineering. Mater Sci Eng C 44:246–253
Hakki SS, Dundar N, Kayis SA, Hakki EE, Hamurcu M, Kerimoglu U, Baspinar N, Basoglu A, Nielsen FH (2013) Boron enhances strength and alters mineral composition of bone in rabbits fed a high energy diet. J Trace Elem Med Biol 27(2):148–153
Hakki SS, Bozkurt BS, Hakki EE (2010) Boron regulates mineralized tissue-associated proteins in osteoblasts (MC3T3-E1). J Trace Elem Med Biol 24(4):243–250
Gümüşderelioğlu M, Tunçay EÖ, Kaynak G, Demirtaş TT, Aydın ST, Hakkı SS (2015) Encapsulated boron as an osteoinductive agent for bone scaffolds. J Trace Elem Med Biol 31:120–128
Gorustovich AA, Nielsen FH (2019) Effects of nutritional deficiency of boron on the bones of the appendicular skeleton of mice. Biol Trace Elem Res 188(1):221–229
Boyacioglu O, Orenay-Boyacioglu S, Yildirim H, Korkmaz M (2018) Boron intake, osteocalcin polymorphism and serum level in postmenopausal osteoporosis. J Trace Elem Med Biol 48:52–56
Basu S, Basu B (2019) Doped biphasic calcium phosphate: synthesis and structure. J Asian Ceram Soc 7(3):265–283
Ajlan SA, Ashri NY, Aldahmash AM, Alnbaheen MS (2015) Osteogenic differentiation of dental pulp stem cells under the influence of three different materials. BMC Oral Health 15(1):132
Mori G, Brunetti G, Oranger A, Carbone C, Ballini A, Muzio LL, Colucci S, Mori C, Grassi FR, Grano M (2011) Dental pulp stem cells: osteogenic differentiation and gene expression. Ann N Y Acad Sci 1237(1):47–52
Kokubo T, Takadama H (2006) How useful is SBF in predicting in vivo bone bioactivity? Biomaterials. 27(15):2907–2915
Pazarçeviren AE, Evis Z, Keskin D, Tezcaner A (2019) Resorbable PCEC/gelatin-bismuth doped bioglass-graphene oxide bilayer membranes for guided bone regeneration. Biomedical Materials 14(3):035018
Li Y, Liu C, Zhai H, Zhu G, Pan H, Xu X, Tang R (2014) Biomimetic graphene oxide–hydroxyapatite composites via in situ mineralization and hierarchical assembly. RSC Adv 4(48):25398–25403
Madupalli H, Pavan B, Tecklenburg MMJ (2017) Carbonate substitution in the mineral component of bone: discriminating the structural changes, simultaneously imposed by carbonate in A and B sites of apatite. J Solid State Chem 255:27–35
Mansour SF, El-Dek SI, Ahmed MK (2017) Physico-mechanical and morphological features of zirconia substituted hydroxyapatite nano crystals. Sci Rep 7:43202
Barheine S, Hayakawa S, Jäger C, Shirosaki Y, Osaka A (2011) Effect of disordered structure of boron-containing calcium phosphates on their in vitro biodegradability. J Am Ceram Soc 94(8):2656–2662
Kolmas J, Velard F, Jaguszewska A, Lemaire F, Kerdjoudj H, Gangloff SC, Kaflak A (2017) Substitution of strontium and boron into hydroxyapatite crystals: effect on physicochemical properties and biocompatibility with human Wharton-Jelly stem cells. Mater Sci Eng C Mater Biol Appl 79:638–646
Reyes-Gasga J, Martínez-Piñeiro EL, Rodríguez-Álvarez G, Tiznado-Orozco GE, García-García R, Brès EF (2013) XRD and FTIR crystallinity indices in sound human tooth enamel and synthetic hydroxyapatite. Mater Sci Eng C 33(8):4568–4574
Bollino F, Armenia E, Tranquillo E (2017) Zirconia/hydroxyapatite composites synthesized via Sol-Gel: Influence of hydroxyapatite content and heating on their biological properties. Materials, 10(7):757
Albayrak O (2016) Structural and mechanical characterization of boron doped biphasic calcium phosphate produced by wet chemical method and subsequent thermal treatment. Mater Charact 113:82–89
Obayes HK, Wagiran H, Hussin R, Saeed MA (2016) Strontium ions concentration dependent modifications on structural and optical features of Li4Sr(BO3)(3) glass. J Mol Struct 1111:132–141
Narwal P, Dahiya MS, Yadav A, Hooda A, Agarwal A, Khasa S (2018) Improved white light emission in Dy3+ doped LiF-CaO-Bi2O3-B2O3 glasses. J Non-Cryst Solids 498:470–479
Uysal I, Severcan F, Tezcaner A, Evis Z (2014) Co-doping of hydroxyapatite with zinc and fluoride improves mechanical and biological properties of hydroxyapatite. Prog Nat Sci Mater Int 24(4):340–349
Poinern GEJ, Brundavanam RK, Le X, Fawcett D (2012) The mechanical properties of a porous ceramic derived from a 30 nm sized particle based powder of hydroxyapatite for potential hard tissue engineering applications. Am J Biomed Eng 2(6):278–286
Hasan MF, Wang J, Berndt CC (2013) Effect of power and stand-off distance on plasma sprayed hydroxyapatite coatings. Mater Manuf Process 28(12):1279–1285
Nie J, Zhou J, Huang X, Wang L, Liu G, Cheng J (2019) Effect of TiO2 doping on densification and mechanical properties of hydroxyapatite by microwave sintering. Ceram Int 45(11):13647–13655
Reger NC, Bhargava AK, Ratha I, Kundu B, Balla VK (2019) Structural and phase analysis of multi-ion doped hydroxyapatite for biomedical applications. Ceram Int 45(1):252–263
Itatani K, Tsuchiya K, Sakka Y, Davies IJ, Koda S (2011) Superplastic deformation of hydroxyapatite ceramics with B2O3 or Na2O addition fabricated by pulse current pressure sintering. J Eur Ceram Soc 31(14):2641–2648
Barheine S, Hayakawa S, Osaka A, Jaeger C (2009) Surface, interface, and bulk structure of borate containing apatitic biomaterials. Chem Mater 21(14):3102–3109
Wang A-J, Lu Y-P, Zhu R-F, Li S-T, Xiao G-Y, Zhao G-F, Xu WH (2008) Effect of sintering on porosity, phase, and surface morphology of spray dried hydroxyapatite microspheres. J Biomed Mater Res A 87A(2):557–562
Wang X, Zhang Y, Lin C, Zhong W (2017) Sol-gel derived terbium-containing mesoporous bioactive glasses nanospheres: In vitro hydroxyapatite formation and drug delivery. Colloids Surf B: Biointerfaces 160:406–415
Nga NK, Thuy Chau NT, Viet PH (2018) Facile synthesis of hydroxyapatite nanoparticles mimicking biological apatite from eggshells for bone-tissue engineering. Colloids Surf B: Biointerfaces 172:769–778
Zhu XD, Fan HS, Xiao YM, Li DX, Zhang HJ, Luxbacher T, Zhang XD (2009) Effect of surface structure on protein adsorption to biphasic calcium-phosphate ceramics in vitro and in vivo. Acta Biomater 5(4):1311–1318
Tunçay EÖ, Demirtaş TT, Gümüşderelioğlu M (2017) Microwave-induced production of boron-doped HAp (B-HAp) and B-HAp coated composite scaffolds. J Trace Elem Med Biol 40:72–81
Arslan A, Çakmak S, Gümüşderelioğlu M (2018) Enhanced osteogenic activity with boron-doped nanohydroxyapatite-loaded poly(butylene adipate-co-terephthalate) fibrous 3D matrix. Artif Cells Nanomed Biotechnol 46(sup2):790–799
Burguera M, Burguera JL, Rondón C, Carrero P (2001) Determination of boron in blood, urine and bone by electrothermal atomic absorption spectrometry using zirconium and citric acid as modifiers. Spectrochim Acta B At Spectrosc 56(10):1845–1857
Tavassoli H, Javadpour J, Taheri M, Mehrjou M, Koushki N, Arianpour F, Majidi M, Izadi-Mobarakeh J, Negahdari B, Chan P, Ebrahimi Warkiani M, Bonakdar S (2018) Incorporation of nanoalumina improves mechanical properties and osteogenesis of hydroxyapatite bioceramics. ACS Biomater Sci Eng 4(4):1324–1336
Langenbach F, Handschel J (2013) Effects of dexamethasone, ascorbic acid and β-glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem Cell Res Ther 4(5):117
Monterubbianesi R, Bencun M, Pagella P, Woloszyk A, Orsini G, Mitsiadis TA (2019) A comparative in vitro study of the osteogenic and adipogenic potential of human dental pulp stem cells, gingival fibroblasts and foreskin fibroblasts. Sci Rep 9(1):1761
Ojansivu M, Mishra A, Vanhatupa S, Juntunen M, Larionova A, Massera J, Miettinen S (2018) The effect of S53P4-based borosilicate glasses and glass dissolution products on the osteogenic commitment of human adipose stem cells. PLoS One 13(8):e0202740
Taşlı PN, Doğan A, Demirci S, Şahin F (2013) Boron enhances odontogenic and osteogenic differentiation of human tooth germ stem cells (hTGSCs) in vitro. Biol Trace Elem Res 153(1-3):419–427
Movahedi Najafabadi B-A-H, Abnosi MH (2016) Boron induces early matrix mineralization via calcium deposition and elevation of alkaline phosphatase activity in differentiated rat bone marrow mesenchymal stem cells. Cell J 18(1):62–73
Davies OG, Cox SC, Azoidis I, McGuinness AJ, Cooke M, Heaney LM, Grover LM (2019) Osteoblast-derived vesicle protein content is temporally regulated during osteogenesis: implications for regenerative therapies. Front Bioeng Biotechnol 7(92)
Becerra-Bayona S, Guiza-Arguello V, Qu X, Munoz-Pinto DJ, Hahn MS (2012) Influence of select extracellular matrix proteins on mesenchymal stem cell osteogenic commitment in three-dimensional contexts. Acta Biomater 8(12):4397–4404
Birmingham E, Niebur GL, McHugh PE, Shaw G, Barry FP, McNamara LM (2012) Osteogenic differentiation of mesenchymal stem cells is regulated by osteocyte and osteoblast cells in a simplified bone niche. Eur Cells Mater 23:13–27
Shih Y-RV, Hwang Y, Phadke A, Kang H, Hwang NS, Caro EJ, Nguyen S, Siu M, Theodorakis EA, Gianneschi NC, Vecchio KS, Chien S, Lee OK, Varghese S (2014) Calcium phosphate-bearing matrices induce osteogenic differentiation of stem cells through adenosine signaling. Proc Natl Acad Sci U S A 111(3):990–995
Acknowledgments
Authors would also like to thank Milad Fathi-Achachelouei for donating DPSCs.
Funding
Authors would like to acknowledge National Institute of Boron (Ankara, Turkey) for providing financial support by Project No. 2018-31-07-25-001.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pazarçeviren, A.E., Tezcaner, A., Keskin, D. et al. Boron-doped Biphasic Hydroxyapatite/β-Tricalcium Phosphate for Bone Tissue Engineering. Biol Trace Elem Res 199, 968–980 (2021). https://doi.org/10.1007/s12011-020-02230-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-020-02230-8