
Abstract
The aim of the present study was to investigate the effects of high concentrations of copper (Cu) on the cecum and rectum of intestinal microbiota in female mice. Twenty-four Kunming mice were weighed and randomly divided into two groups (n = 12 per group) including the control group and Cu group. Cu group was given drinking water with 5 mg/kg-bw copper chloride (CuCl2), while the control group was treated with drinking water without CuCl2. At the 90th day, results showed that compared with the control group, mice in the treatment group had a lower body weight, and the feces turned yellow and had a lower pH value. Histopathological lesions showed that the intestinal tissue from the treatment group had increased thickness of outer muscularis and smoothed muscle fiber, widened submucosa, decreased goblet cells, and showed blunting of intestinal villi and severe atrophy of central lacteal. In addition, at the genus level, 16S rRNA gene sequencing from the Cu group showed that Corynebacterium were significantly increased whereas Staphylococcaceae, Odoribacter, Rikenella, and Jeotgalicoccus were significantly decreased in the cecum. Dehalobacterium, Coprococcus, and Spirochaetales increased significantly whereas Salinicoccus, Bacillales, Staphylococcus, and Lactobacillales decreased sharply in the rectum. This study demonstrated that high concentrations of Cu could induce tissue injury and interrupt the homeostasis of microbiota.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
During the past several decades, the ecological environment has been severely polluted or degraded due to heavy metal contamination from vehicle emissions, industrial discharges and other activities [1]. It has been shown that heavy metals at certain concentrations can have long-term toxic effects within ecosystems [2]. Despite the fact that it is indispensable, excessive amount of Cu can impose a long-term burden on the biogeochemical cycle in the ecosystem such as the contamination of soil and water, especially the enrichment of the food chain [3]. Over-exposure to Cu may lead to adverse effects on animal organisms such as respiratory and nervous disorders. Moreover, it can cause the development of a number of diseases including gastrointestinal bleeding and hepatorenal failure [4,5,6]. Although there is a close relationship between the overdose of Cu and many organogenic diseases in animals, little is known about these diseases in the perspective of changing the intestinal flora.
Gastrointestinal microbiota is essential for maintaining homeostasis both inside and outside the intestinal tract, which plays a key role in intestinal physiology, energy metabolism and immunity [7,8,9]. As signaling molecules, the metabolite of microorganism can impact the host’s metabolism such as short-chain fatty acid (SCFA) production and vitamin synthesis [10,11,12]. Such health-relevant functions may be considered as ecosystem services provided by the gut microbiota for the host. As key ecosystem service providers (ESPs) which are indispensable to the host’s well-being, healthy animals can reduce the risk of developing diseases such as allergic disorders and chronic immune-mediated inflammatory diseases, whereas diseased animals may have aberrant patterns leading to the state of “dysbiosis.” Some examples are that the deregulation of gut homeostasis by certain bacteria could lead to diseases or even death because of the poisoning, which was found in hydrobiology [13, 14]. However, there is little research on high doses of Cu-causing diseases by changing the gastrointestinal microbiota on mammals. In other words, whether the gut microbiota can be impacted by high doses of Cu and thereby causes further adverse effects on host health remains unknown.
The present study was designed to investigate the influence of high doses of Cu on the intestinal metabolism and intestinal microbiological community in the caecum and rectum of mice. To achieve this, the intestinal bacterial composition was investigated to evaluate the differences of intestinal microbiota using 16S rRNA sequencing method. It may provide a theoretical basis for more accurate assessment of the risk in intestinal diseases induced by a high dose of Cu.
Materials and Methods
Ethics Statement and Samples Collection
All animal experiments were done according to the Guidelines of Animal Experiments by the Institutional Animal Care and Use Committee of Jiangxi Agricultural University. The feeding experiment and sample collection were prepared in the Institute of Animal Population Health, College of Animal Science and Technology, Jiangxi Agricultural University.
The treatment procedure in this experiment was similar to Mitra et al. [15]. Twenty-four 8-week-old Kunming female mice were fed with the basal diet (Table 1) during the 7-day adaptation period, and then they were weighed and randomly divided into two groups (n = 12 per group): the control check group (CCK group, average weight = 27.66 g, 0 mg/kg-bw Cu) and Cu check group (CCu group, average weight = 27.71 g, 5 mg/kg-bw Cu). After 90 days, twelve mice from each group were reweighted and killed sacrificed by cervical dislocation, and then divided into four groups with 6 mice each group: cecal control check group (CCK), rectal control check group (RCK), cecal Cu check group (CCu) and rectal Cu check group (RCu). Cecum and rectum contents were collected aseptically into 1.5 ml EP tubes and then stored at − 20 °C until further analysis. Meanwhile, the cecum and rectum were collected and put in paraformaldehyde for pathological section preparation.
Histopathological Examination
After sacrifice of mice at 90th day, the cecal and rectal tissues were rapidly removed and fixed in 4% paraformaldehyde. The fixed tissues were dehydrated in a graded series of ethanol, cleared in xylene, and then embedded in paraffin. Histological slices were prepared using a microtome and stained with hematoxylin and eosin (H&E). The stained slices were evaluated using the bio-microscope.
Isolation of Intestinal Bacterial DNA
The bacterial DNA in the cecum and rectum were extracted using the Agilent High Sensitivity DNA Kit (Agilen, China) according to the manufacturer’s protocol. The quantities of extracted DNA were measured using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and qualities were examined by electrophoresis on 1% agarose and visualized by the GelDoc XR System (Bio-Rad, USA) respectively.
Gene Sequencing of 16S rRNA
The V4 region (the length of DNA is 280 bp) flanked by the evolutionary conserved regions were used to design polymerase chain reaction (PCR) primer and it was then amplified using PCR with the primers. The bacterial 16S rRNA universal prime sequences synthesized at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China) are: 520F (5′-AYTGGGYDTAAAGNG-3′) and 802R (5′-TACNVGGGTATCTAATCC-3′). The PCR components contained 5 μl of Q5 reaction buffer (5 times), 5 μl of Q5 high-fidelity GC buffer (5 times), 0.25 μl of Q5 high-fidelity DNA polymerase (5 U/μl), 2 μl (2.5 mM) of dNTPs, 1 μl (10 μM) of each forward and reverse primer, 2 μl of DNA template, and 8.75 μl of ddH2O. PCR assay was performed with the following amplification configurations: denaturation (30 s, 98 °C), 25 cycles (15 s, 98 °C; 30 s, 50 °C; 30 s, 72 °C), and final elongation (5 min, 72 °C). PCR products were purified with Agencourt AMPure Beads (Beckman Coulter, Indianapolis, IN) and quantified using the PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA). After the individual quantification step, amplicons were pooled in equal amounts, and paired-end 2 × 280 bp sequencing was performed using the Illlumina MiSeq platform with MiSeq Reagent Kit v3 at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China).
High-Throughput Sequencing
The quality of the DNA library was examined using the Agilent High Sensitivity DNA Kit and Agilent Bioanalyzer. The DNA library was then used on the Illumina MiSeq platform (CA, USA). The collected raw sequencing data were processed using the following steps: (1) the sequences of barcode and primers were first removed from the raw sequencing data; (2) the sequencing data were then analyzed using the software FLASH (version 1.2.7, http://ccb.jhu.edu/software/FLASH); (3) the sequencing fragments of each sample were aligned and connected to form the raw sequencing tags; (4) the raw sequencing tags with low quality were removed and the chimeric sequences were removed from the final sequencing tags. The resulting sequences were effective sequencing tags used for bacterial taxonomic annotation.
Sequence Analysis
The identified 16S rRNA sequences (the effective sequencing tags) were analyzed using Uparse software. Sequences sharing ≥ 97% identity were considered at the same taxonomic position, which were assigned to one operational taxonomic unit (OTU). An OTU table was generated to record the abundance of each OTU in each sample and the taxonomy of these OTUs. OTUs containing less than 0.001% of total sequences across all samples were discarded. To minimize the difference of sequencing depth across samples, an averaged, rounded rarefied OTU table was generated by averaging 100 evenly resampled OTU subsets under the 90% of the minimum sequencing depth for further analysis. When OTUs were constructed, the Uparse software automatically selected representative sequences. The representative OTUs were annotated with taxonomic information using the 16S rRNA gene sequence database Green-Gene database (Release 13.8, http://greengenes.secondgenome.com/). Finally, the sequences were annotated as follows: Kingdom, Phylum, Class, Order, Family, Genus, and Species. Additionally, Venn diagrams were plotted to show the shared and unique OTUs in each specified group using R software (Version 2.11.1) [16].
Measures of Microbial Diversity
The evaluation of microbial diversity in a natural environment involves two aspects: species richness and evenness information. To assess whether the 16S rRNA sequences could include all the bacteria in the samples, we conducted the alpha diversity analysis, represented by rarefaction curves. Alpha diversity was analyzed using the Quantitative Insights into Microbial Ecology (QIIME) software package (Version 1.7.0). To evaluate alpha diversity, we conducted the analysis of community richness with the Chao 1 estimator, the abundance-based coverage estimator (ACE), the analysis of community diversity with the Shannon index and Simpson index [17]. Rarefaction curves indicate that the number of unique bacterial species does not increase as the number of identified sequences increases [18]. Different from alpha diversity, beta diversity is used to investigate the community structure of the similarity between different samples. The total diversity (gamma diversity) is determined by the diversity in the two aspects: alpha diversity and beta diversity. Principal component analysis (PCA), multidimensional scaling analysis (MDS), and clustering analysis methods were used to measure the difference between samples. The complete dataset was submitted to the NCBI short read archive database with the accession number PRJNA415103.
Results
Body Weight
The body weights of mice after 90 days were analyzed using the t test. The values were expressed in the format of the mean ± standard error. The weight of mice in Cu check group (29.25 ± 0.25 g) was significantly lower than that in the control check group (31.49 ± 0.37 g) (p value < 0.01).
Histopathology
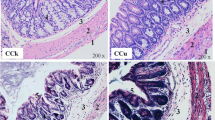
We compared Cu group with control group, and the results are shown in Fig. 1. The color of intestinal contents of the feces was brassy, and the fecal pH level (6.2–7.0) was significantly lower in Cu group compared with the control group (7.5–8.0). Cu group showed that mice had lower body weights, and feces turned yellow and showed lower pH values (Fig. 2). Moreover, histopathological lesions in Cu group showed increased thickness of outer muscularis and smooth muscle fiber, widened submucosa, decreased goblet cells, blunting of intestinal villi and severe atrophy of central lacteal.

Feces from different sources. a CCK group; b CCu group; c RCK group; d RCu group

Pathological analysis of cecal and rectal tissues in different groups (HE. 400×). a CCK group; b CCu group; c RCK group; d RCu group. Arrows indicate pathological changes from a to b, and c to d. 1) the outer muscularis thickness; 2) the smooth muscle fiber; 3) submucosa; 4) the goblet cells; 5, 6) the intestinal villus and central lacteal
Results of 16s RNA Sequencing Data Analysis
In total, 769,766 sequences were generated from 22 samples (two samples failing to qualify requirements were removed). Among them, 634,430 (82.4%) sequences were considered to be high-quality sequence. The 634,430 reads were used for abundance analysis, diversity analysis, and taxonomic comparison. Summary information of the sequencing data was presented in Table 2. OTUs were annotated at the levels of phylum, class, order, family, genus, species, and unclassified by the RDP Classifier and the 16S rRNA sequence database, and the GreenGene database (Fig. 3). Each oval represented a group (Fig. 2), and samples in overlapping regions were symbolized as the total of OTUs. There were 1115 (42.36%) OTUs shared among the four groups. The percentages of OTUs uniquely in CCu group, CCK group, RCK group, and RCu group were respectively 0.61%, 2.01%, 1.60%, 0.84%. The order of magnitude was CCK > RCK > RCu > CCu (Fig. 4).

Statistical results of OTU’s identification and classification. The numbers of OTUs of phylum, class, order, family, genus, and species were reported. The number of OTUs that could not be classified into any known group was designated as “unclassified”

Venn diagrams representation of the OTUs shared among the samples from different sources. Venn diagram showing the unique and shared OTUs in the samples from different sources: CCu group, CCK group, RCK group, and RCu group
Alpha Diversity Analysis
The rank abundance curves of the 22 samples directly reflected the abundance and rarity of OTUs in the community. As shown in Fig. 4, the curves became gradually stable, indicating a uniform species distribution (Fig. 5).

OTU rank abundance curves. The x-coordinate is the OTU of the bearing size, and the y-coordinate is the sample abundance. Each curve represents a sample OTU distribution, and the longer the broken curve, the bigger the number of OTUs in the sample; the smoother the broken curve, the higher the evenness of the community
Beta Diversity Analysis
In Fig. 6, RCk group, CCK group, RCu group and CCu group were studied using Partial Least Squares Discriminant analysis (PLS-DA) method. Samples of each group were well classified together using the PLS-DA method, indicating good performance of our classification method.

Principal component analysis (PCA) of the samples. Top two PCs were used as the x and y coordinates. Each sample was represented as a point. Samples from different sources were colored differently: (1) CCK group (blue); (2) CCu group (orange); (3) RCK group (gray); (4) RCu group (green)
Results of the microbial composition analysis at the order level were represented in Fig. 7. In RCK group, Bacteroidales, Lactobacillales, Bacillales, Pseudomonadales were the dominant intestinal community and Bifidobacteriales, Desulfovibrionales, Coriobacteriales, Burkholderiales, and CW040 were in low abundance. In RCu group, the three top most abundant genera were Bacteroidales, Clostridiales, Erysipelotrichales, and the genera with decreased abundance were Bifidobacteriales, Desulfovibrionales, Coriobacteriales, Burkholderiales, and CW040. In CCK group, we found that Bacteroidales, Clostridiales, and Pseudomonadales were the dominant intestinal and Bifidobacteriales, Desulfovibrionales, Coriobacteriales, Burkholderiales, and CW040 were in low abundance. In CCu group, the community was mainly composed of Bacteroidales, Clostridiales, and Erysipelotrichales, whereas Bifidobacteriales, Desulfovibrionales, Coriobacteriales, Burkholderiales, and CW040 were significantly lower in abundance. There was a significant difference between the RCK group and RCu group. However, there is no significant difference between the CCK and the CCu group.

Relative abundances at the bacterial order level for the four groups
To further investigate the difference in intestinal microbiota composition among the different groups, we used the LEFSE method to test samples for relative abundance. In Fig. 8A, at the phylum level, we observed no significant difference in the CCu group and CCK group. But at the genus level, there was a significant increase in Corynebacterium and Dorea and a significant decrease in Staphylococcaceae, Jeotgalicoccus, Odoribacter, and Rikenella in CCu group. In Fig. 8B, in comparison of control group and Cu group, at the phylum level, there was a significant decrease in Spirochaetales. At the genus level, there was a significant increase in Dehalobacterium, and Coprococcus, whereas there was a significance decrease in Bacillus, Jeotgalicoccus, Salinicoccus, Staphylococcus, and Ignatzschineria in the RCu group.

Significant difference between groups of OTUs: CCK group versus CCu group (Panels A1, A2); RCK group versus RCu group (Panels B1, B2); Panels A1, B1 are the cladograms for the comparisons. Panels A2, B2 show LDA scores for differently abundant taxa preceding and following Cu treatment are in panel (A2, B2,). (Only taxa meeting LDA ≥ 2.5 are shown). In each cladogram, changes in relative abundance of bacterial taxa identified by LefSe are shown in phylogenetic trees from the Phylum to the Genus (from the inner to the outer ring). In each cladogram, the node circle size is proportional to the taxa relative abundance. The node size corresponds to the average relative abundance of the OTUs. Yellow color was used to indicate that there were no significant difference between groups, and other colors (such as green and red) were used to show that OTUs had a significant difference between groups
Discussion
Cu is a crucial trace element component in humans and animals [19, 20]. Small amounts of Cu can be regarded as nutritional additives, whereas excessive amounts of Cu can lead to liver diseases and gastrointestinal tract diseases. Damaged gastrointestinal tract affects the digestion and absorption of nutrient substances, leading to weight loss in animals [21]. In our experiment, we found that the mice in Cu group had copper-colored feces, lower pH values, and lower body weights. When exposed to excessive amounts of Cu, it was found that microbial active substances could be impacted in that the microorganisms produced a simple single organic compound, macromolecular humic acid, rich acid, or microbial exudates etc. [22]. The decrease in fecal pH could be due to the production of a large amount of acidic substances. Meanwhile, Cu affects bioactive substances and can also cause intestinal damage, and metabolic disorders, and reduce nutrient decomposition and absorption, which affects mouse weight loss. In addition, the thickness of the adventitia wall and muscularis was elevated, the submucosa became thickened, and the intestinal villus atrophied significantly in CCu group and RCu group were observed. These results were supported by another study in the literature, in which the dysfunction of bile excretion of Cu could lead to the retention of Cu in tissues, individual enterocyte necrosis with enterocytes in the lumen and blunt of the villi when exposed to excessive Cu [23]. These evidences suggested that the mice were affected by Cu. To further study the composition of the microbial community and diversity of the samples from female mice at the phylum and genus level, high-throughput 16S rRNA gene sequencing were used to investigate the bacterial diversity effects of high amounts of Cu on mice.
Effect of Copper on Cecum Intestinal Microbial Community
In our study, CCu group was compared with CCK group in the cecum. Corynebacterium and Dorea showed a significant increase at the genus level. Corynebacterium glutamicum is the Corynebacterium-Mycobacterium-Nocardia group of actinomycetes, which has a significant regulatory system named CopRS for the extracytoplasmic sensing of elevated Cu-ion concentrations and for the induction of a set of genes capable of Cu efflux [24]. Interestingly, CopRS is activated only when Cu concentrations rise. If the anti-Cu regulatory system is activated in a copper-contaminated environment, it can protect the Corynebacterium from Cu damage [25]. Therefore, Corynebacterium can tolerate relatively high levels of Cu and be grown in the environment with Cu pollution. In addition, a few studies have also reported that Corynebacterium have the highest tolerance to high concentrations of Cu, and may have good adaptability and can still be grown in the situation of Cu pollution [2, 26]. Additionally, Dorea belongs to Akkermansia, and the increase in Akkermansia is related to the proliferation of intestinal epithelial cells induced by high heme iron diet, and can destroy the intestinal mucous layer, because Cu stimulates erythropoiesis and heme formation. Therefore, high abundance of Akkermansia can aggravate intestinal damage [23, 27]. In our study of CCu group, similar lesions that intestinal villus atrophied significantly in pathological sections. In summary, the increase in Dorea has a great effect on the destruction of the intestines.
On the contrary, Bacteroidales, Pseudomonadales, Rikenella, Odoribacter, Straphylococcaceae, and Jeotgalicoccus decreased significantly, and they are described as strictly anaerobic, mesophilic, and rod-shaped bacteria that usually ferment carbohydrates or proteins. Excessive amounts of Cu ions stimulate reactive oxygen species (ROS) production via the Fenton reaction, in which hydrogen peroxide interacts with iron to generate hydroxyl radicals [21, 27]. In order to overcome cellular damage with increased levels of ROS, organisms activate an antioxidant defense system, composed of antioxidant enzymes such as superoxide dismutase (SOD), the enzyme catalyzed the dismutation of superoxide radicals (O2−) to molecular oxygen (O2) and hydrogen peroxide (H2O2) [28]. Furthermore, Cu exposure promotes the activities of antioxidant enzymes [29]. This reaction can produce oxygen which can inhibit the growth of the strictly anaerobic bacteria. Hence, the abundance of this species of bacteria could decrease in the environment with Cu contamination.
Effect of Copper on Rectum Intestinal Microbial Community
Compared with the RCK group in the rectum, Spirochaetales, Clostridiales, Erysipelotrichales, Dehalobacterium, and Coprococcus increased significantly. Jeotgalicoccus, Salinicoccus, Lactobacillales, Bacillales, and Staphylococcus were significantly decreased at the genus level. Interestingly, Spirochaetales, Clostridiales, and Erysipelotrichales belong to Firmicutes, which are inversely correlated with obesity. A few researchers have shown that, compared with lean mice, there is a 50% reduction in the abundance of Bacteroidetes and a proportional increase in Firmicutes in the obese mice [30, 31]. Besides, a few studies have found that many species of Clostridiales and Spirochaetales can be pathogenic, such as leading to infectious diseases and poisoning in mice models [32, 33]. The above argument is consistent with the pathological changes and emaciation in mice caused by Cu poisoning in RCu group. It may be related to the increase in these bacteria.
The changes in Jeotgalicoccus, Salinicoccus, Lactobacillales, Bacillus, and Staphylococcus should not be neglected. A previous study has shown a few similar features between Jeotgalicoccus and Salinicoccus in phylogenetic tree analyses [34]. In addition, the two species of bacteria have a crucial role in the body health and were seen as a potential beneficial body service provider, which could be one of the core functional groups of intestinal microbiota [14]. Additionally, a research has shown that chronic Cu exposure can disturb the composition of intestinal microbiota related to lipid metabolism and immunity in freshwater fish [35]. Therefore, in our results, the decrease of these bacteria could also affect core functions of normal microbiota in the rectum, impact on the rectum metabolism and lead to digestive function disorder. In addition, we also found that Lactobacillus is an anaerobic bacterium, and its reduction may be similar to the reduction of anaerobic bacteria in the cecum under high Cu conditions. Furthermore, the microorganisms of the genus Bacillus are parts of the probiotic preparations such as Bacillus subtilis and Bacillus licheniformis. A report shows that β-gal-acto-sidase can catalyze the hydrolysis ONPG (o-nitrophenyl-β-D-galactopyranoside) to produce galactose ando-nitrophenol (ONP), and can be generated from Bacillus subtilis without the assistance of any inducers. When high amounts of Cu ions exist, the activity of β-galactosidase is inhibited and the concentration of bacillus subtilis is decreased [36]. This may be an important reason for the reduction of Bacillus, which directly provides an opportunity for the development of pathogenic bacteria. In addition, changes of other genera, including Coprococcus were observed. However, these genera have been rarely reported to be associated with Cu poisoning in humans or in animal models.
Conclusion
High doses of Cu decreased the mice’s body weight and reduced feces pH. Furthermore, it caused obvious histopathological lesions and changed the diversity of microbiota in mice in this study. It could provide evidences and a theoretical basis in explaining the mechanisms of high-concentration Cu poisoning.
References
Sun R, Chen L (2016) Assessment of heavy metal pollution in topsoil around Beijing metropolis. PLoS One 11:e155350
Lee IS, Kim OK, Chang YY, Bae B, Kim HH, Baek KH (2002) Heavy metal concentrations and enzyme activities in soil from a contaminated Korean shooting range. J Biosci Bioeng 94:406–411
Papa S, Bartoli G, Pellegrino A, Fioretto A (2010) Microbial activities and trace element contents in an urban soil. Environ Monit Assess 165:193–203
Sakhaee E, Emadi L, Azari O, Kheirandish R, Esmaili Nejad MR, Shafiei Bafti H (2016) Effects of Cuminum cyminum L. essential oil on some epididymal sperm parameters and histopathology of testes following experimentally induced copper poisoning in mice, vol 48, pp 542–547
Sakhaee E, Abshenas J, Emadi L, Azari O, Kheirandish R, Samaneh A (2014) Effects of vitamin C on epididymal sperm quality following experimentally induced copper poisoning in mice. Comp Clin Pathol 23:181–186
O'Hara AM, Shanahan F (2006) The gut flora as a forgotten organ. EMBO Rep 7:688–693
Kanno T, Matsuki T, Oka M, Utsunomiya H, Inada K, Magari H, Inoue I, Maekita T, Ueda K, Enomoto S, Iguchi M, Yanaoka K, Tamai H, Akimoto S, Nomoto K, Tanaka R, Ichinose M (2009) Gastric acid reduction leads to an alteration in lower intestinal microflora. Biochem Biophys Res Commun 381:666–670
Arredondo M, Nunez MT (2005) Iron and copper metabolism. Mol Asp Med 26:313–327
Ognik K, Stepniowska A, Cholewinska E, Kozlowski K (2016) The effect of administration of copper nanoparticles to chickens in drinking water on estimated intestinal absorption of iron, zinc, and calcium. Poult Sci 95:2045–2051
Backhed F, Manchester JK, Semenkovich CF, Gordon JI (2007) Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A 104:979–984
Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, DiDonato AJ, Fu X, Hazen JE, Krajcik D, DiDonato JA, Lusis AJ, Hazen SL (2015) Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163:1585–1595
Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439:484–489
Breton J, Massart S, Vandamme P, De Brandt E, Pot B, Foligne B (2013) Ecotoxicology inside the gut: impact of heavy metals on the mouse microbiome. BMC Pharmacol Toxicol 14:62
Zhang J, Guo Z, Xue Z, Sun Z, Zhang M, Wang L, Wang G, Wang F, Xu J, Cao H, Xu H, Lv Q, Zhong Z, Chen Y, Qimuge S, Menghe B, Zheng Y, Zhao L, Chen W, Zhang H (2015) A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. ISME J 9:1979–1990
Mitra S, Keswani T, Ghosh N, Goswami S, Datta A, Das S, Maity S, Bhattacharyya A (2013) Copper induced immunotoxicity promote differential apoptotic pathways in spleen and thymus. Toxicology 306:74–84
Zaura E, Keijser BJ, Huse SM, Crielaard W (2009) Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol 9:259
Fan LM, Barry K, Hu GD, Meng SL, Song C, Wu W, Chen JZ, Xu P (2016) Bacterioplankton community analysis in tilapia ponds by Illumina high-throughput sequencing. World J Microbiol Biotechnol 32:10
Zhao M, Du S, Li Q, Chen T, Qiu H, Wu Q, Chen S, Zhou Y, Zhang Y, Hu Y, Su Y, Shen L, Zhang F, Weng D, Li H (2017) High throughput 16SrRNA gene sequencing reveals the correlation between Propionibacterium acnes and sarcoidosis. Resp Res 18:28
Bost M, Houdart S, Oberli M, Kalonji E, Huneau JO, Margaritis IN (2016) Dietary copper and human health: current evidence and unresolved issues. J Trace Elem Med Bio 35:107–115
Su R, Wang R, Cao H, Pan J, Chen L, Li C, Shi D, Tang Z (2011) High copper levels promotes broiler hepatocyte mitochondrial permeability transition in vivo and in vitro. Biol Trace Elem Res 144:636–646
Gaetke LM, Chow CK (2003) Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 189:147–163
Kviatkovskaia IIa (1981) Effect of microbial preparations used in agriculture on the intestinal microflora. Gig Sanit (6):82–83
Malinak CM, Hofacre CC, Collett SR, Shivaprasad HL, Williams SM, Sellers HS, Myers E, Wang YT, Franca M (2014) Tribasic copper chloride toxicosis in commercial broiler chicks. Avian Dis 58:642–649
Schelder S, Zaade D, Litsanov B, Bott M, Brocker M (2011) The two-component signal transduction system CopRS of Corynebacterium glutamicum is required for adaptation to copper-excess stress. PLoS One 6:e22143
Chang FJ, Coyne HJ, Cubillas C, Vinuesa P, Fang X, Ma Z, Ma D, Helmann JD, García-de Los Santos A, Wang Y, Dann CE, Giedroc DP (2014) Cu(I)-mediated allosteric switching in a copper-sensing operon repressor (CsoR). J Biol Chem 289:19204–19217
Hussein EI, Jacob JH, Jahmani ASF, Yousef ND (2013) Dissemination of heavy metals and tolerant bacteria along Zarqa river (Jordan). J Biol Sci 13:100–111
Leonard SS, Harris GK, Shi X (2004) Metal-induced oxidative stress and signal transduction. Free Radic Biol Med 37:1921–1942
McCord JM, Fridovich I (1969) Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244:6049–6055
Martins I, Goulart J, Martins E, Morales-Roman R, Marin S, Riou V, Colaco A, Bettencourt R (2017) Physiological impacts of acute cu exposure on deep-sea vent mussel Bathymodiolus azoricus under a deep-sea mining activity scenario. Aquat Toxicol 193:40–49
Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102:11070–11075
Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, Flint HJ (2000) Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol 66:1654–1661
Fitzpatrick LR, Small JS, Greene WH, Karpa KD, Farmer S, Keller D (2012) Bacillus coagulans GBI-30, 6086 limits the recurrence of Clostridium difficile-induced colitis following vancomycin withdrawal in mice. Gut Pathog 4:13
Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, Wang GP (2013) Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol 51:2884–2892
Schwaiger K, Lzel CH, Mayer M, Bauer J: Notes on the almost unknown genus Jeotgalicoccus. Lett Appl Microbiol 50(4):441-4
Meng X, Li S, Qin C, Zhu Z, Hu W, Yang L, Lu R, Li W, Nie G (2018) Intestinal microbiota and lipid metabolism responses in the common carp (Cyprinus carpio L.) following copper exposure. Ecotox Environ Safe 160:257–264
Fang D, Yu Y, Wu L, Wang Y, Zhang J, Zhi J (2015) Bacillus subtilis-based colorimetric bioassay for acute biotoxicity assessment of heavy metal ions. RSC Adv 5:59472–59479
Acknowledgements
This project was supported by the National Natural Science Foundation of China grant #31492266 awarded to PL, the Natural Science Foundation of Jiangxi Province grant #20171ACB21026 awarded to PL, the Technology R&D Program of Jiangxi Province grant #2014BBF60035 awarded to PL, and the Department of Education Science and Technology Research of Jiangxi Province grant # GJJ170243.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
All authors have read the manuscript and agreed to submit it in its current form for consideration for publication in the journal
Rights and permissions
About this article

Cite this article
Cheng, S., Mao, H., Ruan, Y. et al. Copper Changes Intestinal Microbiota of the Cecum and Rectum in Female Mice by 16S rRNA Gene Sequencing. Biol Trace Elem Res 193, 445–455 (2020). https://doi.org/10.1007/s12011-019-01718-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-019-01718-2