
Abstract
MiRNAs are related to neuronal proliferation and apoptosis following cerebral ischemia-reperfusion injury (CIRI). This study focused on miR-30c-5p in the disease. An oxygen-glucose deprivation/re-oxygenation (OGD/R) model was prepared in HT22 cells and transfected to overexpress miR-30c-5p and G Protein Subunit Alpha I2 (GNAI2) respectively or co-transfected to silence miR-30c-5p and GNAI2. Meanwhile, a middle cerebral artery occlusion (MCAO) model was constructed in mice, and miR-30c-5p and GNAI2 were silenced in vivo simultaneously. The mice were evaluated for neurological damage, apoptosis, and inflammation. HT22 cells were tested for cytotoxicity, proliferation, apoptosis, and inflammatory factors. The interaction between miR-30c-5p and GNAI2 was predicted, analyzed, and confirmed. MiR-30c-5p was found to be downregulated in both experimental models. miR-30c-5p reduced lactate dehydrogenase production, inflammatory response, inhibit apoptosis, and enhanced neuronal proliferation, while GNAI2 overexpression showed the opposite results. Downregulated miR-30c-5p worsened neurological function, apoptosis, and inflammation of MCAO mice while silencing GNAI2 attenuated the influence of downregulated miR-30c-5p. MiR-30c-5p can improve neuronal apoptosis and inflammatory response caused by CIRI and is neuroprotective by targeting GNAI2, providing a new target for treating CIRI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stroke is recognized as the third most prevalent cause of mortality and the primary contributor to long-term impairment [1]. Cerebral ischemia typically arises from cerebral aortic embolism or thromboembolic occlusion, resulting in inadequate oxygen and nutrient delivery to brain tissue. This condition often leads to long-term disability in approximately 80% of stroke patients [2]. When blood flow and reoxygenation are reestablished, it leads to the occurrence of severe ischemia/reperfusion (I/R) injury, which results in the generation of significant quantities of cellular free oxygen radicals and inflammatory cytokines [3, 4]. I/R injury plays a significant role in the neuroinflammatory response, initiating various cellular death mechanisms such as apoptosis, acute necrosis, and autophagy [5]. Although clinical measures have been carried out to reduce the risk of brain injury, sequelae such as inflammation and neurological function injury caused by reperfusion are the key reasons for the unsatisfactory treatment of patients. Therefore, exploration of novel effective therapeutic targets for cerebral ischemia-reperfusion injury (CIRI) is imperative and time-sensitive.
MicroRNAs (miRNAs) are a class of small, non-coding RNA molecules that play a crucial role in gene regulation. Through base pairing, miRNAs can bind to target mRNAs and negatively regulate their expression. This regulation can occur through various mechanisms, including cleavage of the mRNA, repression of translation, or destabilization of mRNA structure [6, 7]. Following cerebral ischemia-reperfusion injury, the biological events initiated dysregulate different miRNAs, responsible for long-terming progression of neuronal damage [8,9,10]. Due to their capacity to modulate the production of specific proteins, microRNAs (miRNAs) present themselves as a potential therapeutic approach for mitigating neuronal injury subsequent to a cerebral ischemic episode [11, 12]. For instance, miR-19a/b-3p promotes inflammatory responses during I/R via targeting SIRT1/FoxO3/SPHK1 axis [13]. Upregulation of miR-29a provides protection against I/R damage [14]. MiR-7-5p is upregulated in the I/R injury model, degrading Sirtuin 1 and thereby increasing neuronal apoptosis [15]. Recent studies have confirmed that miR-30c is protective against apoptosis and inflammation induced by myocardial ischemia/reperfusion injury (MIRI) [16] and can improve the pathological injury of brain tissue caused by CIRI [17]. However, detailed mechanisms of miR-30c-5p in the inflammatory response following CIRI deserve in-depth investigations.
GNAI2 is a member of the α subunit family of heterotrimer G protein [18], also known as the “inhibitory Gα subunit,” which hampers adenylate cyclase activity and leads to the reduction of intracellular cyclic AMP level [19], and is a key regulator of signal transduction pathways [20], such as cell proliferation, differentiation, and migration [21,22,23]. Emerging documents have clarified that GNAI2 is associated with many pathological processes, including I/R injury, neurodevelopmental disorders, immune deficiency, and atherosclerosis [24,25,26]. In addition, Devanathan et al. confirmed that GNAI2 knockdown could improve brain tissue injury and neural function deficit in MIRI models [27]. However, few reports have focused on the mechanism of GNAI2 in CIRI, and the correlation between miR-30c-5p and GNAI2 in CIRI remains unclear.
In the present study, we sought to investigate the biologic functions and underlying mechanism role of miR-30c-5p in CIRI. Our findings demonstrate that miR-222-3p is a major regulator of GNAI2 expression and improves neural function injury and inflammation in CIRI.
Materials and Methods
Cell Culture
Mouse brain neuron cell line HT-22 (ATCC, USA) was characterized using STR typing. For cell culture, a Dulbecco Modified Eagle Medium (Gibco, USA) was employed. The medium was composed of 10% fetal bovine Serum (Invitrogen, USA), 100 U/mL penicillin (Sigma-Aldrich, USA), and 100 U/mL streptomycin (Invitrogen). The cell culture was conducted in a humid incubator with a controlled environment of 5% CO2 and 37 °C.
Cell Transfection
Si-GNAI2, pcDNA 3.1-GnAI2, si-NC, pcDNA 3.1, miR-30c-5p mimic/inhibitor, and mimic/inhibitor NC were all purchased from GenePharma (Shanghai, China). Lipofectamine 3000 (Invitrogen) was employed for transfection in HT-22 cells in accordance with the manufacturer’s instructions. After a 48-h incubation period, the cells were harvested for subsequent analysis using RT-qPCR or Western blot.
Oxygen Glucose Deprivation/Reperfusion (OGD/R) Cell Model
Cells are subjected to hypoxia and glucose deficiency, followed by reperfusion (OGD/R), in order to simulate neuronal damage that occurs during cerebral ischemia. Cells were cultured in a medium composed of DMEM (Gibco, CA, USA), 10% fetal bovine serum (Gibco, CA, USA), and 1% penicillin-streptomycin. Cells are grown in a cell culture incubator at 37 °C containing 5% CO2. To implement the OGD/R model, the medium is replaced with glucose-free DMEM and the cells in the previous conditions for a duration of 6 h. Following this, the complete medium is reintroduced and the cells are maintained under normal culture conditions [28].
Lactate Dehydrogenase (LDH) Release
Cytotoxicity was determined by a lactate dehydrogenase (LDH) Assay Kit (Beyotime, China). The cell supernatant was collected and LDH release was detected by measuring absorbance at 490 nm using SpectraMax® M5 (Molecular Devices, USA).
CCK-8 Assay
CCK-8 method (DoJindo, Japan) was employed to measure neuronal damage at 6 h after OGD/R. HT-22 cells were placed in a 6-well plate containing 2 mL of complete medium (3.5 × 105 cells/well). CCK-8 reagent was added for 2 h to determine OD450 nm.
Flow Cytometry
Apoptosis was assessed by employing the annexin V-FITC apoptosis detection kit (eBioscience, USA). After OGD/R treatment for a duration of 6 h, HT-22 cells were subjected to centrifugation at a speed of 400 × g for a duration of 5 min. Subsequently, the cells were washed with PBS 3 times, and then inoculated into a 12-well plate for an incubation period of 15 min. During this incubation, the cells were treated with 5 µL of annexin V-FITC and 10 µL of propidium iodide (Sigma-Aldrich). Apoptotic cells were analyzed using BD FACSCalibur flow cytometer (Becton Dickinson, USA) and the BD CellQuest software (Becton Dickinson).
Enzyme-Linked Immunosorbent Assay (ELISA)
The levels of IL-1β, IL-6, and TNF-α in cells and mouse brain tissues were assessed using an enzyme-linked immunosorbent assay (ELISA) kit (MLbio, Shanghai, China), and the OD at 450 nm was read on a microplate reader (Bio-Rad, USA) to generate drawing the standard curve.
RT-qPCR
TRIzol (Invitrogen) was employed to isolate total RNA from tissues and cells. For mature miRNAs, reverse transcription was performed using HiScript Q Select RT SuperMix for qPCR Kit (Vazyme, R133–01) and stem-loop RT primers (RiboBio) and subjected to a quantitative analysis using AceQ qPCR SYBR Green Master Mix (Vazyme, R141) with U6 as an internal control. For mRNA, HiScript Q RT SuperMix for qPCR Kit (Vazyme, R123-01) was utilized for reverse transcription, and SYBR Green Real-time PCR Master Mix (Vazyme, R141-02) was for quantitative analysis, with β-actin as an internal control. Primers (Table 1) for amplification were synthesized by Invitrogen.
Western Blot
Proteins were extracted from tissue and cells with RIPA lysis buffer (P0013B, Beyotime) and analyzed by BCA reagent (Thermo Fisher Scientific, USA). Proteins separated on 10–12% SDS polyacrylamide gel were transferred to a polyvinylidene fluoride membrane (Bio-Rad), left overnight with primary antibody (cleaved caspase-3 [9664, CST], p-p65 [3033, CST], β-actin [A5441, Millipore]) at 4 °C, and incubated with horseradish peroxidase-conjugated secondary antibody (7076P2, CST, USA) before being visualized in the enhanced chemiluminescence detection system (Thermo Fisher Scientific).
Animal Model of MCAO/R
MCAO/R was conducted according to the previous report [29]. Twenty-five adult male C57BL/6J mice, aged 6–8 weeks and weighing 16–22 g, were obtained from Hunan SJA Laboratory Animal Co., Ltd., Changsha, Hunan. The mice were housed in a controlled environment with a temperature of 22 ± 3 °C, humidity of 60 ± 5%, and a 12-h light/dark cycle for adaptation. All experimental designs involving animals were granted approval by the Animal Ethics Committee of Yichun People’s Hospital of Jiangxi Province. Anesthesia induction was achieved using a combination of 3% isoflurane, 30% oxygen, and 70% nitrous oxide. Anesthesia was maintained using 1.5% isoflurane administered via a mask. A 6−0 nylon thread (Doccol, Sharon, MA, USA) was coated with silicone was introduced through the external carotid artery and advanced 9–10 mm along the internal carotid artery to the carotid bifurcation, up to the point of the internal carotid artery. After 1 h, the thread was extended. MCAO was not performed on the normal control group.
miRNA Angomir and Lentivirus Injection
The GNAI2-targeting shRNA lentiviral vector (shRNA-GANI2), miR-30c-5p antagomir, and negative controls (shRNA-NC and antagomir NC) were derived from GenePharma. Lentiviral vectors (2 µl, 1 × 109 TU/ml) or miR-30c-5p antagomir (80 mg/kg) were injected into the ventricles of mice 14 days before MCAO/R. The injection coordinates are as follows: AP, −0.3 mm; L, 1.0 mm; V, 2.2 mm.
Neurological Score
Neurological deficits were assessed 24 h after MCAO/R using a modified neurological severity scoring test [30]. A combination of motor (muscle status, aberrant movement), sensory (visual, tactile, and proprioceptive), and reflex tests make up the neurologic severity score. The severity of the injury was determined by the authors using a score graded from 0 to 14, with a normal score of 0 and a maximal deficit score of 14. For failing to complete the activities or for not having a tested reflex, one point is given. Scores between 5 and 9 denote a mild injury, while those between 10 and 14 denote a severe one. A score between 1 and 4 denotes a minor injury. The highest point total is 14.
TUNEL Staining
Apoptotic cells in brain tissue were observed using the One-Step TUNEL Apoptosis Detection Kit (Roche). Brain tissue blocks were cut into 4 μm after paraffin embedment. After dewaxing and hydration, brain tissues were incubated with protease K, fluorescence-labeled dUTP solution, and TdT enzymes. DNase I was a positive control, while dUTP as a negative control. Next, brain tissues were developed with diaminobenzidine and stained with DAPI (Sigma-Aldrich), followed by dehydration with gradient of ethanol, clearance with xylene, and sealed with neutral balm.
Dual Luciferase Reporter Experiment
GANI2 3′-UTR primers containing predicted miR-30c-5p site were amplified by PCR from human genomic DNA, and the DNA fragments were cloned into the pmiR-RB-REPORT vector (RiboBio) and named GANI2-WT. For the pmiR-RB-GANI2-3′-UTR-MUT vector, miR-30c-5p binding site in GANI2 3′-UTR was mutagenized by PCR-based method. The above luciferase reporter and miR-30c-5p mimic and mimic NC were co-transfected into HT-22 cells using Lipofectamine 3000 (Invitrogen) and luciferase activity was measured 24 h later according to the manufacturer’s protocol (Promega, E2920).
RNA-Pull down
HT-22 cells (2 × 106) were transfected with biotinylated miR-30c-5p or control (GenePharma) at 50 nm for 36 h, lysed in RIPA lysis buffers (Abcam), and incubated with 500 pM antisense oligonucleotides containing RNase inhibitors at 4 °C overnight and homogenized with 10 µL streptavidin agarose beads (Thermo Fisher Scientific) at 4 °C for 2 h. The compound was then centrifuged for 10 min, and RNA was extracted using Trizol and analyzed by RT-qPCR to measure GNAI2 levels.
Data Analysis
All experiments had at least three biological replicates. GraphPad Prism 9.0 was employed for statistical analysis. Data reported as mean ± standard deviation (SD) were compared using the student t-test (two groups) or one-way ANOVA (multiple groups). P < 0.05 emphasizes statistical difference.
Results
MiR-30c-5p Improves OGD/ R-Induced Neuronal Injury
To further investigate the function of MiR-30c-5p in CIRI, we created OGD/R model in HT-22 cells. qRT-PCR showed that OGD/R treatment inhibited miR-30c-5p expression in HT-22 cells (Fig. 1A). Subsequently we successfully transfected NC and miR-30c-5p mimic in OGD/R-treated HT-22 cells (Fig. 1B). LDH detection showed an increase in LDH release in OGD/R-treated HT-22 cells, but miR-30c-5p overexpression reversed this condition (Fig. 1C). CCK-8 assay displayed that OGD/R inhibited cell proliferation, while restoring miR-30c-5p enhanced cell viability (Fig. 1D). Flow cytometry showed that the increased apoptosis rate caused by OGD/R treatment could be attenuated after overexpressing miR-30c-5p (Fig. 1E). Subsequently, cellular inflammatory cytokines were assessed by ELISA. As measured, OGD/R treatment enhanced IL-1β, IL-6, and TNF-α in the cell supernatant, but this effect was improved after miR-30c-5p was overexpressed (Fig. 1F). Meanwhile, apoptotic and inflammatory proteins were evaluated by Western blot, showing that OGD/R increased cleaved caspase-3 and p-p65, but elevation of miR-30c-5p prevented changes in these proteins (Fig. 1G). Altogether, these results suggest that miR-30c-5p overexpression decreases OGD/R-induced cell injury and death, and downregulates the associated inflammatory factors.

Overexpression of miR-30c-5p improves OGD/ R-induced neuronal injury. miR-30c-5p mimic was transfected into OGD/R treated HT-22 cells. A, B RT-qPCR detection of miR-30c-5p in OGD/R-treated HT-22 cells. C LDH release from HT-22 cells. D CCK-8 to detect cell viability of HT-22 cells. E Flow cytometry to detect the apoptosis rate of HT-22 cells. F ELISA to detect the levels of IL-1β, IL-6, and TNF-α in the supernatant of HT-22 cell culture medium. G Western blot analysis of cleaved caspase-3 and p-p65 in HT-22 cells. Data were expressed as mean ± SD (N = 3). *P < 0.05
GNAI2 is Mediated by miR-30c-5p
On the bioinformatics website ENCORI (http://starbase.sysu.edu.cn/index.php), a potential binding site was found between GNAI2 and miR-30c-5p (Fig. 2A). Subsequently, GNAI2-WT/MUT luciferase reporters were produced, and only GNAI2-WT could be affected by miR-30c-5p in regard to luciferase activity (Fig. 2B). RNA-pull down experiments further confirmed that miR-30c-5 targeted GNAI2 (Fig. 2C). OGD/R treatment had a promoting effect on GNAI2 expression, but this influence was rescued by upregulating miR-30c-5p (Fig. 2D). These results speculated that miR-30c-5 negatively regulates GNAI2 expression, implying that GNAI2 might be a downstream target of miR-30c-5.

GNAI2 is the target gene of miR-30c-5p. A Potential binding sites of GNAI2 and miR-30c-5p predicted by bioinformatics website ENCORI (https://starbase.sysu.edu.cn/index.php). B Dual luciferase reporter assay to analyze the targeting relationship between GNAI2 and miR-30c-5p. C RNA-pull down assay to analyze the binding relationship between GNAI2 and miR-30c-5p. D Western blot to detect the effect of overexpression of miR-30c-5p on GNAI2 protein expression in HT-22 cells. Data were expressed as mean ± SD (N = 3). *P < 0.05
GNAI2 Worsens OGD/R-Induced Neuronal Injury
Then, we investigated the functional role of GNAI2 in ischemia. As anticipated, the Western blot analysis revealed a significant increase in the expression level of GNAI2 in cells treated with OGD/R and transfected with pcDNA 3.1-GNAI2. This experimental approach was employed to investigate the functional role of GNAI2 in OGD/R-treated cells. Effectively, pcDNA 3.1-GNAI2 induced GNAI2 expression in neurons (Fig. 3A). LDH detection revealed a significant elevation in LDH release in HT-22 cells treated with OGD/R and transfected with pcDNA 3.1-GNAI2 (Fig. 3B). CCK-8 assay displayed that GNAI2 overexpression inhibited cell proliferation (Fig. 3C). Flow cytometry analysis revealed a significant increase in the rate of apoptosis in cells treated with OGD/R and transfected with pcDNA 3.1-GNAI2 (Fig. 3D). Apoptotic protein cleaved caspase-3 expression was also significantly increased by Western blot (Fig. 3E). Subsequently, cellular inflammatory cytokines were assessed by ELISA, showing an increased level of IL-1β, IL-6, and TNF-α in in OGD/R-treated HT-22 cells transfected with pcDNA 3.1-GNAI2. The expression of p-p65 was also increased after transfecting with pcDNA 3.1-GNAI2. Cell function experiments were conducted to investigate the effects of upregulating GNAI2 on various cellular processes. The results showed that upregulating GNAI2 led to an increase in LDH release (Fig. 3B), a decrease in neuronal proliferation (Fig. 3C), an increase in apoptosis rate (Fig. 3D), and elevated levels of inflammatory cytokines (Fig. 3E) as well as p-p65 (Fig. 3F). These findings suggest that GNAI2 plays a role in promoting cell death induced by OGD/R.

Overexpression of GNAI2 aggravates OGD/R-induced neuronal injury. GNAI2-targeting pcDNA 3.1 overexpression vector was transfected into OGD/R-treated cells. A Western blot to detect GNAI2 in HT-22 cells. B LDH release from HT-22 cells. C CCK-8 to detect cell viability of HT-22 cells. D Flow cytometry to detect the apoptosis rate of HT-22 cells. E ELISA to detect the levels of IL-1β, IL-6, and TNF-α in the supernatant of HT-22 cell culture medium. F Western blot analysis of cleaved caspase-3 and p-p65 in HT-22 cells. Data were expressed as mean ± SD (N = 3). *P < 0.05
MiR-30c-5p Affects OGD/ R-Induced Neuronal Injury Through GNAI2 Targeting
To verify whether MiR-30c-5p affects OGD/R-induced cell damage through GNAI2 targeting, miR-30c-5p inhibitor, and si-GNAI2 were co-transfected into OGD/R-treated neuron cells. qRT-PCR results indicated that miR-30c-5p inhibitor reduced the expression of miR-30c-5p (Fig. 4A). At the same time, we found that GNAI2 expression in miR-30c-5p inhibitor + si-NC group increased significantly due to miR-30c-5p inhibition, while GNAI2 expression in miR-30c-5p inhibitor + si-GNAI2 group did not change significantly due to GNAI2 knockdown (Fig. 4B). LDH detection and CCK-8 assay revealed a significant reversal in LDH release and cell viability in HT-22 cells treated with OGD/R and transfected with miR-30c-5p inhibitor (Fig. 4C-D). Flow cytometry analysis revealed a significant decrease in the rate of apoptosis in in miR-30c-5p inhibitor + si-GNAI2 group compared to the miR-30c-5p inhibitor + si-NC group (Fig. 4E). Correspondingly, the results of Western blot showed that the expression of Apoptotic protein cleaved caspase-3 and inflammatory proteins p-p65 was significantly downregulated after knocking down GNAI2 (Fig. 4F-G). Together, these data indicate that miR-30c-5p inhibitor ameliorates OGD/R-induced cell injury via GNAI2.

miR-30c-5p affects OGD/ R-induced neuron INJURY by regulating GNAI2. miR-30c-5p inhibitor and si-GNAI2 were co-transfected into OGD/R-treated neurons. A RT-qPCR detection of miR-30c-5p in HT-22 cells. B Western blot to detect GNAI2 in HT-22 cells. C LDH release from HT-22 cells. D CCK-8 to detect cell viability of HT-22 cells. E Flow cytometry to detect the apoptosis rate of HT-22 cells. F ELISA to detect the levels of IL-1β, IL-6, and TNF-α in the supernatant of HT-22 cell culture medium. G Western blot analysis of cleaved caspase-3 and p-p65 in HT-22 cells. Data were expressed as mean ± SD (N = 3). *P < 0.05
MiR-30c-5p/GNAI2 Axis Affects Neuronal Injury in MCAO/R Mice
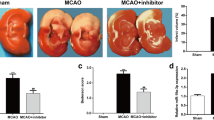
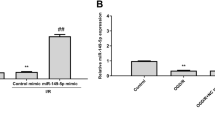
To further support the results of in vitro experiments, a mouse model of MCAO/R was constructed successfully. qRT-PCR and Western blot results showed that the expression of miR-30c-5p was decreased and GNAI2 was increased significantly in MCAO/R mice (Fig. 5A, B). Then, we injected miR-30c-5p inhibitors antagomir (miR-30c-5p anta) and sh-GNAI2 into MCAO/R mice. qRT-PCR results indicated that miR-30c-5p antagomir reduced the expression of miR-30c-5p. Western blot results showed that the GNAI2 expression increased significantly after inhibition of miR-30c-5p, and then this elevation was reversed by GNAI2 knockdown (Fig. 5A, B). In addition, the knockdown of miR-30c-5p increased neural damage scores in MCAO/R mice, but this effect was counteracted by silencing GNAI2 (Fig. 5C). TUNEL staining was used to evaluate neuronal apoptosis in mice. An increased proportion of TUNEL-positive neurons was observed after miR-30c-5p antagomir injection, while neuronal apoptosis was attenuated after sh-GNAI2 injection (Fig. 5D). ELISA and Western blot determined that silencing miR-30c-5p induced inflammation and apoptosis in MCAO/R mice, while depleting GNAI2 abolished these changes (Fig. 5E, F). Taken together, these data show that miR-30c-5p targets regulatory GNAI2 to mediate neuronal injury in MCAO/R mice.

MiR-30c-5p/GNAI2 axis attenuates neuronal damage in MCAO/R mice. miR-30c-5p and GNAI2 were knocked down simultaneously in MCAO/R mice. A RT-qPCR detection of miR-30c-5p in mouse brain tissue. B Western blot to detect GNAI2 in mouse brain tissue. C Neurological injury score. D TUNEL staining to detect neuronal apoptosis in mouse brain tissue. E ELISA to detect IL-1β, IL-6, and TNF-α in mouse brain tissue. F Western blot analysis of cleaved caspase-3 and p-p65 in mouse brain tissue. Data were expressed as mean ± SD (n = 5). Scale bar = 100 μm. *P < 0.05
Discussion
Brain is the most sensitive organ to hypoxia, and hypoxia caused by ischemia will lead to local brain tissue damage, while inflammation caused by reperfusion is the key pathophysiological mechanism of tissue damage and cell necrosis in ischemic stroke [31], which seriously threatens human health and life [32, 33]. Therefore, it is imperative to find effective biological targets to inhibit apoptosis and inflammation induced by ischemia-reperfusion and protect nervous system function and brain tissue. It has been reported that miRNAs can negatively regulate at least 60% of the coding genes [34], including proteins related to cerebral ischemia. Based on this, this study surveyed miR-30c-5p in CIRI, with a view to improving the therapeutic effect and quality of life of patients. Further experiments confirmed that miR-30c-5p-targeted regulation of GNAI2 inhibits neuronal apoptosis, cytotoxicity, and inflammation, and improves brain injury and neurological dysfunction.
Recent miRNA microarray studies have determined that miRNAs expression levels undergo complex and highly variable changes after ischemia-reperfusion, which can be potential diagnostic markers and clinical therapeutic targets for ischemic stroke [35, 36]. For example, in patients with ischemic stroke, serum miR-124 in patients presents differential expression patterns after ischemic stroke onset [37]. At the same time, miR-124 expression elevates in ischemic penumbra, which increases anti-apoptotic protein expression [38, 39]. It is noted that miR-30c is closely related to cerebrovascular diseases with diagnostic and prognostic values [40, 41]. The current work tested downregulated miR-30c-5p in OGD/R cell models. We found that overexpression of miR-30c-5p inhibits LDH release, apoptosis, and related inflammatory factor activity, and promotes cell proliferation. The above results demonstrate miR-30c-5p improves the cellular damage and inflammatory response caused by hypoxia and glucose deficiency in OGD/R cells. In addition, we used MCAO/R mice model to verify the improvement effect of miR-30c-5p on CIRI in vivo. Therefore, in this study used in vivo and in vitro models to identify the important role of miR-30c-5p in cerebral ischemia-reperfusion, and may be used as a novel target for the treatment of ischemia-reperfusion.
MiRNAs exert their effects by suppressing expression of their target genes [34]. In the present study, luciferase reporter assays revealed GNAI2 to be a direct target gene of miR-30c-5p in CI/R. Indeed, GNAI2 can regulate a variety of signaling pathways and maintain homeostasis [42, 43]. Specifically, GNAI2 is increased during ischemia-reperfusion, and hepatocellular specific GNAI2 knockdown can improve ischemia-reperfusion liver injury in part by attenuating inflammatory response and hepatocyte apoptosis [24]. Also, GNAI2 knockdown protects brain tissue from pathological changes and neurological deficits caused by MIRI [27]. In OGD/R cells and MCAO/R mouse models, we found that GNAI2 expression levels were enhanced, GNAI2 overexpression promoted LDH production, inhibited cell activity, and aggravated neuronal apoptosis and inflammation. In addition, knocking down GNAI2 reversed downregulation of miR-30c-5p on ischemia-reperfusion injury. The modified neurological severity scoring test also demonstrated that the inhibition of GNAI2 improved nerve damage in mice subjected to MCAO/R. Our findings indicate that GNAI2 is downregulated by miR-30c-5p. Down-regulation of GNAI2 was shown to exert inhibitory effects on apoptosis and inflammatory responses, ultimately leading to a reduction in CIRI. Therefore, the discovery and verification of MiR-30c-5p/GNAI2 axis in CIRI can provide new strategies and directions for the diagnosis and treatment of CIRI. However, comprehensive investigations regarding the downstream factors associated with GNAI2 have not yet been undertaken. Our study specifically examined the impact of miR-30c-5p on cellular proliferation and associated inflammatory markers, while not exploring additional factors such as autoimmunity and stress resilience. We intend to undertake further comprehensive research in the future. Furthermore, miR-30c-5p functions as a miRNA that plays a crucial role in the regulation of gene expression. Future investigations should delve into the potential involvement of other targets of miR-30c-5p in the process of ischemia-reperfusion, along with elucidating the underlying signal transduction pathways.
Conclusion
MiR-30c-5p can target and regulate GNAI2, thereby inhibiting neuronal apoptosis, inflammatory response and cytotoxicity caused by CIRI, and protecting brain tissue and neurological function, providing a new target and strategy for the treatment of CIRI.
Data Availability
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
References
Lallukka, T. (2018). Trends in diagnosis-specific work disability before and after stroke: A longitudinal population-based study in Sweden. Journal of the American Heart Association, 7(1), e006991.
Moskowitz, M. A., Lo, E. H., & Iadecola, C. (2010). The science of stroke: Mechanisms in search of treatments. Neuron, 67(2), 181–198.
Eltzschig, H. K., & Eckle, T. (2011). Ischemia and reperfusion–From mechanism to translation. Nature Medicine, 17(11), 1391–1401.
Radak, D., Resanovic, I., & Isenovic, E. R. (2014). Link between oxidative stress and acute brain ischemia. Angiology, 65(8), 667–676.
Khoshnam, S. E., Winlow, W., & Farzaneh, M. (2017). The interplay of MicroRNAs in the inflammatory mechanisms following ischemic stroke. Journal of Neuropathology and Experimental Neurology, 76(7), 548–561.
Lu, T. X., & Rothenberg, M. E. (2018). MicroRNA J Allergy Clin Immunol, 141(4): 1202–1207.
Macfarlane, L. A., & Murphy, P. R. (2010). MicroRNA: Biogenesis, function and role in cancer. Curr Genomics, 11(7), 537–561.
Hamzei Taj, S., et al. (2016). Dynamic modulation of microglia/macrophage polarization by miR-124 after focal cerebral ischemia. Journal of Neuroimmune Pharmacology: The Official Journal of the Society on Neuroimmune Pharmacology, 11(4), 733–748.
Zhao, F., et al. (2017). miR-30d-5p plays an important role in autophagy and apoptosis in developing rat brains after hypoxic-ischemic injury. Journal of Neuropathology and Experimental Neurology, 76(8), 709–719.
Kanagaraj, N., et al. (2014). Downregulation of miR-124 in MPTP-treated mouse model of Parkinson’s disease and MPP iodide-treated MN9D cells modulates the expression of the calpain/cdk5 pathway proteins. Neuroscience, 272, 167–179.
Rink, C., & Khanna, S. (2011). MicroRNA in ischemic stroke etiology and pathology. Physiol Genomics, 43(10), 521–528.
Saugstad, J. A. (2010). MicroRNAs as effectors of brain function with roles in ischemia and injury, neuroprotection, and neurodegeneration. Journal of Cerebral Blood Flow and Metabolism, 30(9), 1564–1576.
Zhou, F., et al. (2021). miR-19a/b-3p promotes inflammation during cerebral ischemia/reperfusion injury via SIRT1/FoxO3/SPHK1 pathway. J Neuroinflammation, 18(1), 122.
Ouyang, Y. B., et al. (2013). Astrocyte-enriched miR-29a targets PUMA and reduces neuronal vulnerability to forebrain ischemia. Glia, 61(11), 1784–1794.
Diwan, D., et al. (2021). Sirtuin 1 mediates protection against delayed cerebral ischemia in subarachnoid hemorrhage in response to hypoxic postconditioning. J Am Heart Assoc, 10(20), e021113.
Sun, M., et al. (2021). MicroRNA-30c-5p protects against myocardial ischemia/reperfusion injury via regulation of Bach1/Nrf2. Toxicol Appl Pharmacol, 426, 115637.
Zhang, M., et al. (2020). Neuroprotective effects of miR-30c on rats with cerebral ischemia/reperfusion injury by targeting SOX9. Pathology, Research and Practice, 216(12), 153271.
Downes, G. B., & Gautam, N. (1999). The G protein subunit gene families. Genomics, 62(3), 544–552.
Wong, Y. H., et al. (1991). Mutant alpha subunits of Gi2 inhibit cyclic AMP accumulation. Nature, 351(6321), 63–65.
Hilger, D., Masureel, M., & Kobilka, B. K. (2018). Structure and dynamics of GPCR signaling complexes. Nature Structural & Molecular Biology, 25(1), 4–12.
Dhanasekaran, N., et al. (1998). Regulation of cell proliferation by G proteins. Oncogene, 17(11 Reviews), 1383–1394.
Feigin, M. E., & Muthuswamy, S. K. (2009). Polarity proteins regulate mammalian cell-cell junctions and cancer pathogenesis. Current Opinion in Cell Biology, 21(5), 694–700.
Luttrell, L. M. (2008). Reviews in molecular biology and biotechnology: Transmembrane signaling by G protein-coupled receptors. Molecular Biotechnology, 39(3), 239–264.
Sun, Q., et al. (2019). Guanine nucleotide-binding protein G(i)α2 aggravates hepatic ischemia-reperfusion injury in mice by regulating MLK3 signaling. The FASEB Journal, 33(6), 7049–7060.
Hamada, N., et al. (2017). Role of a heterotrimeric G-protein, Gi2, in the corticogenesis: Possible involvement in periventricular nodular heterotopia and intellectual disability. Journal of Neurochemistry, 140(1), 82–95.
Boularan, C., et al. (2015). Lymphocyte-specific loss of Ric-8A results in a Gα protein deficit and severe humoral immunodeficiency. The Journal of Immunology, 195(5), 2090–2102.
Devanathan, V., et al. (2015). Platelet Gi protein Gαi2 is an essential mediator of thrombo-inflammatory organ damage in mice. Proceedings of the National Academy of Sciences of the United States of America, 112(20), 6491–6496.
Han, B., et al. (2018). Novel insight into circular RNA HECTD1 in astrocyte activation via autophagy by targeting MIR142-TIPARP: Implications for cerebral ischemic stroke. Autophagy, 14(7), 1164–1184.
Wang, M., et al. (2019). Homocysteine enhances neural stem cell autophagy in in vivo and in vitro model of ischemic stroke. Cell Death and Disease, 10(8), 561.
Li, Y., et al. (2000). Intrastriatal transplantation of bone marrow nonhematopoietic cells improves functional recovery after stroke in adult mice. Journal of Cerebral Blood Flow and Metabolism, 20(9), 1311–1319.
Liu, X. Q., Sheng, R., & Qin, Z. H. (2009). The neuroprotective mechanism of brain ischemic preconditioning. Acta Pharmacologica Sinica, 30(8), 1071–1080.
Sommer, C. J. (2017). Ischemic stroke: Experimental models and reality. Acta Neuropathologica, 133(2), 245–261.
Liu, X. (2019). The potential role of microRNA-124 in cerebral ischemia injury. International Journal Of Molecular Sciences, 21(1), 120.
Lewis, B. P., Burge, C. B., & Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell, 120(1), 15–20.
Di, Y., et al. (2014). MicroRNAs expression and function in cerebral ischemia reperfusion injury. Journal of Molecular Neuroscience, 53(2), 242–250.
Khoshnam, S. E., et al. (2017). Emerging roles of microRNAs in ischemic stroke: As possible therapeutic agents. Journal of Stroke, 19(2), 166–187.
Sun, M., et al. (2019). Dynamic changes in miR-124 levels in patients with acute cerebral infarction. International Journal of Neuroscience, 129(7), 649–653.
Chen, S. H., et al. (2016). Effects of acupuncture at Baihui (GV 20) and Zusanli (ST 36) on peripheral serum expression of microRNA 124, laminin and integrin β1 in rats with cerebral ischemia reperfusion injury. Chinese Journal of Integrative Medicine, 22(1), 49–55.
Sun, Y., et al. (2013). MicroRNA-124 protects neurons against apoptosis in cerebral ischemic stroke. Cns Neuroscience & Therapeutics, 19(10), 813–819.
Sun, T., Li, W., & Ling, S. (2016). miR-30c and semaphorin 3A determine adult neurogenesis by regulating proliferation and differentiation of stem cells in the subventricular zones of mouse. Cell Proliferation, 49(3), 270–280.
Yi, S., et al. (2017). miR-30c promotes Schwann cell remyelination following peripheral nerve injury. Neural Regeneration Research, 12(10), 1708–1715.
Simon, M. I., Strathmann, M. P., & Gautam, N. (1991). Diversity of G proteins in signal transduction. Science, 252(5007), 802–808.
Syrovatkina, V., et al. (2016). Regulation, signaling, and physiological functions of G-proteins. Journal of Molecular Biology, 428(19), 3850–3868.
Funding
None.
Author information
Authors and Affiliations
Contributions
XD designed the study, performed animal experiments, and wrote the manuscript. YZ performed in vitro assays, analyzed, and interpretated data. DD revised the manuscript, provided technical support, and supervised the project. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
The present study was approved by the Animal experiments were approved by Yichun People’Hospital of Jiangxi Province Animal Experimental Ethics Committee. and all procedures complied with the National Institutes of Health Guide for the Use of Laboratory Animals.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Deng, X., Zeng, Y. & Ding, D. MiR-30c-5p-Targeted Regulation of GNAI2 Improves Neural Function Injury and Inflammation in Cerebral Ischemia-Reperfusion Injury. Appl Biochem Biotechnol 196, 5235–5248 (2024). https://doi.org/10.1007/s12010-023-04802-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-023-04802-5