
Abstract
Polyferric sulfate (PFS), an economical coagulant widely used for removing heavy metal contaminants from water, is susceptible to reduction and transformation by iron-reducing bacteria prevalent in sediments. However, the effect of heavy metal ions adsorbed in PFS flocs on this biological process remains unclear. According to our results, compared with other heavy metal cations (e.g., Cu2+, Cd2+, Zn2+, Ni2+, Pb2+, and Co2+), Cu2+ had a stronger inhibitory effect on PFS floc reduction by Shewanella putrefaciens CN32, a typical dissimilatory iron-reducing bacterium. The presence of Cu2+ remarkably influenced the global transcription of CN32, resulting in 782 upregulated genes and 713 downregulated genes that are mainly annotated in energy production, amino acid metabolism, protein biosynthesis, and oxidation‒reduction processes. The anaerobic TCA cycle for energy (electron) production was significantly activated in the presence of Cu2+, while the transcription of many genes related to the extracellular electron transfer pathway was downregulated, which is responsible for the decreased Fe3+ reduction. Moreover, the pathways of assimilatory sulfate reduction and subsequent cysteine biosynthesis were significantly enriched, which is hypothesized to result in the consumption of abundant energy produced from the enhanced anaerobic TCA cycle, revealing a strategy to address the oxidative stress caused by Cu2+. This work elucidates the unusual suppressive effects of Cu2+ on the microbial reduction of PFS flocs, which reveals the high resistance of PFS flocs to microbial destruction when used to treat Cu2+ pollution in water, thus demonstrating their tremendous practical prospects.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Coagulation is one of the most common processes used in water and wastewater treatment to destabilize and/or adsorb colloids and dissolved contaminants. Chemical coagulation treatment of inorganic and organic pollution has been widely studied by researchers worldwide because of its advantages of rapid treatment processes, high removal efficiency, and simple operation [1, 2]. Polyferric sulfate (PFS), a well-known, economical and readily available coagulant, has been investigated for treating wastewater, particularly wastewater contaminated with heavy metal cations such as Cd2+, Cu2+, and Pb2+ [3,4,5,6]. In China, nearly 300 tons of synthetic PFS flocs are reportedly dumped into rivers every year for emergency treatment of such pollution incidents [7]. However, the degradation of PFS flocs settling into river sediments and the consequent rerelease of pollutants remain a great concern, given the reductive metabolic activities of microorganisms prevalent in such anoxic environments [4, 7].
In anoxic environments such as sediments, microbial iron metabolism, especially dissimilatory iron reduction, is crucial to the biogeochemical cycle of iron and the fate of organic or metallic contaminants [8,9,10]. Dissimilatory iron-reducing microorganisms, most notably those of the genus Shewanella, can reduce extracellular insoluble iron-containing (hydro)oxides [11] and clay minerals [12] through versatile extracellular electron transfer (EET) pathways across the cell envelope [13], coupled with intracellular oxidation of organic matter for energy generation. Moreover, Shewanella are aquatic microorganisms prevalent in a range of natural sedimentary habitats with notable physiological diversity [14, 15]. According to recent reports, Shewanella strains are capable of extracellular reduction and/or biotransformation of PFS flocs [4, 7, 16]. In particular, the reductive dissolution of Cd-loaded PFS flocs resulted in the rerelease of Cd2+ into the solution [4]. However, the effect of the coagulated heavy metal cations in PFS flocs on the extracellular reduction of the flocs by the genus Shewanella has not received sufficient attention, although this is a very important aspect for evaluating the effectiveness and safety of PFS flocs in treating heavy metal contamination.
Herein, to fill this knowledge gap, the effects of several common heavy metal cations (including Cu2+, Cd2+, Zn2+, Ni2+, Pb2+, and Co2+) on PFS floc reduction by Shewanella putrefaciens CN32, a commonly used model strain of the genus Shewanella with available whole-genome sequences, were investigated using the reduction ratio of Fe3+ as an indicator. Then, the mechanism of Cu2+ effects on the metabolism of S. putrefaciens CN32 was explored through transcriptome and metabolic pathway analysis because Cu2+ showed the most obvious and unique inhibition of PFS floc reduction.
Materials and Methods
Bacterial Strain and Culture Conditions
S. putrefaciens CN32 (ATCC, BAA-1097; hereafter referred to as CN32) was routinely cultured in Luria–Bertani (LB) broth (per liter, composed of 10 g of tryptone, 5 g of yeast extract, and 5 g of NaCl, at pH 7.0) until logarithmic growth at 30 °C with shaking at 200 rpm. The bacterial cells were collected by centrifugation at 6000 rpm for 5 min, washed three times with sterilized 20 mM PIPES buffer (per liter, composed of 0.2922 g of NaCl, 0.0755 g of CaCl2, 0.1904 g of MgCl2, 0.2995 g of NH4Cl, and 6.0474 g of PIPES, at pH 7.0), and then resuspended in PIPES buffer supplemented with 20 mM sodium lactate as electron donors to prepare a bacterial suspension with a final cell density (OD600nm) of 0.2 for follow-up use.
Preparation of PFS Flocs Loaded with Heavy Metal Cations
The synthesis of PFS flocs loaded with different heavy metal cations (Cu2+, Cd2+, Zn2+, Ni2+, Pb2+, and Co2+) through a coagulation approach was performed according to previous literature [4]. Typically, taking the preparation of Cu-loaded PFS (referred to as Cu_PFS) floc as an example, 2 g of PFS powder was dissolved in 20 mL of deionized water, and the prepared solution was added to 500 mL of 5 mg L−1 Cu(NO3)2 solution with quick stirring. Then, the pH of the solution was adjusted to 6.8 with 5 M NaOH for flocculation. The size of flocs increased with time, and Cu_PFS flocs were obtained after 1 ~ 2 h of gravity sedimentation. For the control, PFS flocs were prepared similarly, except that no heavy metal cations were added. The contents of all flocs were normalized by the Fe3+ concentration, which was equal to the concentration of total iron.
Reduction of Flocs by CN32
Batch-type experiments for the anaerobic reduction of flocs by CN32 were performed in 100-mL serum bottles. For each type of floc, appropriate aliquots of flocs were added to 50 mL of bacterial cell suspension to achieve a final Fe3+ concentration of 3.0 mM and then transferred into a sterilized serum bottle, followed by nitrogen blowing for 30 min to remove dissolved oxygen. Then, the serum bottle was capped with a butyl rubber closure to maintain an anaerobic environment and placed in a rotator (200 rpm) at 30 °C. The reduction of each floc was performed in triplicate to test the reproducibility. At specific time intervals, 1 mL aliquots were withdrawn from the suspensions using a sterile syringe in an anaerobic glove box (Shanghai Longyue Instrument Equipment Co., Ltd, China) for chemical analyses. The concentrations of Fe2+ and total iron were determined by the o-phenanthroline photometry method [17, 18].
Sampling and RNA-Seq
After anaerobic reduction was conducted for 36 h, the cells were collected from the Cu_PFS and PFS groups using centrifugation for RNA extraction. Each group consisted of three biological replicates. Total RNA was extracted using QIAzol Lysis Reagent (Qiagen, Germany) according to the manufacturer’s protocols. The RNA quality was determined by agarose gel electrophoresis and quantified using a NanoDrop 2000 (Thermo Fisher Scientific, USA). As shown in Fig. S1, the quality of RNA extracted from each sample met the standard of cDNA library construction. rRNA was removed by using a RiboZero rRNA removal kit (Epicentre, Stockholm, Sweden) and fragmented into approximately 200 bp fragments using fragmentation buffer. cDNA synthesis, end repair, A-base addition, and ligation of the Illumina-indexed adaptors were performed according to the Illumina protocol. Libraries were constructed by PCR amplification for 15 cycles and quantified by TBS380, followed by 150 bp*2 paired-end sequencing on an Illumina NovaSeq 6000 by Shanghai Majorbio Biopharm Biotechnology Co., Ltd (Shanghai, China). The transcriptome sequencing data were submitted to the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) with the accession number PRJNA994195.
Data Processing and Differential Gene Expression Analysis
The raw reads after sequencing were quality controlled through the “fastp” bioinformatic program, and clean reads were obtained by removing adapter sequences, low-quality reads, ambiguous “N” nucleotides, and fragments < 25 bp. The clean reads of six samples with a Phred score Q20 > 98% totalled 3.42 Gb (Table S1). Then, the clean reads were mapped to the reference genome (S. putrefaciens 4 H, GenBank: GCA_025402875.1) with Burrows‒Wheeler mode using Bowtie software (http://bowtie-bio.sourceforge.net/index.shtml), and the read mapping percentages of all samples were greater than 94% (Table S2). The expression levels of genes were calculated using RSEM software (http://deweylab.github.io/RSEM/) with default parameters and represented using transcripts per million (TPM). The differentially expressed genes (DEGs) were screened using the DESeq2 package in R software [19]. The DEGs were selected using the following criteria: Log2FoldChange (Log2FC) ≥ 1 or ≤ -1 and p-adjust ≤ 0.01. A DEG with a positive or negative Log2FC value was considered upregulated or downregulated, respectively, in the Cu_PFs vs. PFS comparison. To understand the functions of the DEGs, comprehensive gene annotations, including annotations based on the Clusters of Orthologous Groups of proteins (COG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases, were performed. The GO functional enrichment and KEGG pathway analyses were carried out using Goatools (https://github.com/tanghaibao/GOatools) and KOBAS (http://kobas.cbi.pku.edu.cn/), respectively.
Results and Discussion
Effect of Heavy Metal Cations on PFS Reduction by CN32
The effects of different heavy metal cations coexisting in PFS flocs on the dissimilatory Fe3+ reduction process by strain CN32 in an anaerobic environment were investigated and compared, as shown in Fig. 1a. Strain CN32 showed a strong ability to reduce PFS flocs in the absence of any heavy metal cations, and the produced Fe2+ accumulated to 2.39 mM (i.e., reduction ratio of 80%) after 180 h of reaction. The robust iron reduction in PFS flocs by the genus Shewanella was also observed in previous reports [4, 7]. The presence of individual Zn2+, Co2+, Ni2+, Pb2+, and Cd2+ in PFS flocs only slightly influenced Fe3+ reduction. Surprisingly, however, the presence of Cu2+ robustly inhibited the PFS reduction by CN32 in terms of the concentration of Fe2+ produced. In particular, the concentration of Fe2+ changed only very slightly after 36 h of reaction, indicating that the dissimilated iron reduction ability of CN32 was obviously suppressed. Notably, at the experimental concentration (5 mg/L) tested, these heavy metal cations had little influence on the growth of CN32 (Fig. S2), indicating their negligible biotoxicity. Moreover, the impact of the presence of the above six heavy metal cations was studied (Fig. 1b). It was found that the inhibitory effect on PFS reduction was positively correlated with the Cu2+ concentration ratio, which further suggested the suppression of CN32-mediated reduction of flocs in the presence of Cu2+. The attenuated reduction of PFS flocs in the presence of Cu2+ suggested that Cu_PFS flocs had high stability against dissimilatory iron-reducing bacteria. To explore the mechanism of the effect of Cu2+ on CN32 reduction of PFS flocs, CN32 cells collected from the Cu_PFS and PFS groups after anaerobic incubation for 36 h were subsequently compared in terms of global transcriptome and metabolic pathway analysis.

Effects of coexisting common heavy metal cations on PFS reduction by CN32: a individual and b multiple cations
Global Transcriptional Responses of CN32 to Cu2+
To assess the validity of each group of biological replicates, correlation and PCA analyses were first performed on the gene expression levels of all six samples. As shown in Fig. S3, three biological replicates in the Cu_PFS and PFS groups were clustered together separately, indicating that the sequencing data of triplicates for each group could be used for differential gene expression analysis. Comparison between the samples of the Cu_PFS and PFS groups using DESeq2 resulted in 1495 DEGs (Fig. 2), among which the expression of 782 DEGs was upregulated, and that of 713 DEGs was significantly (p-adjust ≤ 0.01) downregulated.

Volcano plot of DEGs in the Cu_PFS group compared with the PFS group
To elucidate their functions and the metabolic pathways that they were involved in, the identified DEGs were analyzed by COG, GO, and KEGG database annotation. Accordingly, 1478 and 1149 of the 1495 DEGs were annotated in the COG and GO databases, respectively, while only 568 were annotated in the KEGG database. In the COG database annotation, the DEGs were divided into 23 classifications (Fig. S4), which were mainly classified as energy production and conversion (C), amino acid transport and metabolism (E), translation, ribosomal structure, and biogenesis (J), transcription (K), and signal transduction mechanisms (T). The first three classifications contained more upregulated DEGs, and the last two classifications, conversely, contained more downregulated DEGs. Based on the results of GO annotations (Fig. S5), the DEGs were classified into three major functional categories: biological process (BP), cellular component (CC), and molecular function (MF). Moreover, the results of enrichment analyses showed that 1007 DEGs were enriched in 124 GO terms, of which 44 pathways were significantly (p-adjust ≤ 0.01) enriched (Fig. 3). The pathways with enrichment ratios higher than 0.7 included nucleoside diphosphate metabolic process, purine ribonucleoside triphosphate metabolic process, nucleotide phosphorylation, nucleoside diphosphate phosphorylation, ATP metabolic process, tricarboxylic acid cycle, and translation in the BP category; oxidoreductase complex, cytosolic large ribosomal subunit, large ribosomal subunit, ribosomal subunit, ribonucleoprotein complex, and extracellular region in the CC category; rRNA binding, structural constituent of ribosome, structural molecule activity, and 2 iron, 2 sulfur cluster binding in the MF category. These results indicated that the presence of Cu2+ significantly impacted energy production, amino acid metabolism, protein biosynthesis, and oxidation‒reduction processes, which was also confirmed by the KEGG enrichment results (Fig. S6).

GO enrichment analyses of DEGs (BP biological process, CC cellular component, MF molecular function; ** and *** denote p-adjust ≤ 0.01 and ≤ 0.001, respectively)
DEGs for Anaerobic Central Carbon Metabolism
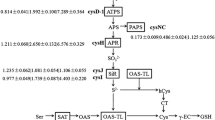
Carbon metabolism is the fundamental pathway for microorganisms to utilize organic substrates, generate energy, and maintain growth, in which the TCA cycle plays a central role [20]. Although for most microorganisms, the TCA cycle functions only in the presence of oxygen, some reports have suggested that the genus Shewanella could use this cycle to couple with alternative electron acceptors such as fumarate, TMAO, and solid electrodes for anaerobic respiration [20,21,22,23]. In these studies, in which Shewanella strains were grown in minimal medium, lactate was of particular interest because it was used as a preferred source of carbon and energy. As shown in Fig. 4, compared with those in the PFS group, the vast majority of genes encoding key enzymes in the TCA cycle in the Cu_PFS group were significantly upregulated at the transcriptional level. This result is consistent with previous reports that changes in electron acceptor types, and their environment under anaerobic conditions could obviously affect the expression of TCA cycle-related genes in Shewanella strains [20, 23, 24]. In particular, the amount of increase in the transcriptional level of the gene cluster lldEFG (ID: N5094_RS13900, N5094_RS13905, N5094_RS13910), encoding flavin adenine dinucleotide (FAD)-dependent lactate dehydrogenases specific for the L-isomer of lactate (used in this study), was much higher than that of the gene dld (ID: N5094_RS13895), encoding lactate dehydrogenases specific for the D-isomer. The transcription of the gene cluster aceEF (ID: N5094_RS02340, N5094_RS02345) encoding pyruvate dehydrogenase was significantly upregulated, while that of the gene plfAB (ID: N5094_RS07990, N5094_RS07995) encoding pyruvate-formate lyase was significantly downregulated, which indicated that the pyruvate derived from lactate dehydrogenation was oxidized to acetyl coenzyme A (acetyl-CoA), followed by entrance into the TCA cycle rather than direct conversion to acetate and formate [21]. Moreover, the gene prpC (ID: N5094_RS01795), encoding 2-methylcitrate synthase, which was reported to control the entrance of acetyl-CoA into the anaerobic TCA cycle in Shewanella strains, also showed a significantly higher transcriptional level (Log2FC = 3.17). In addition, the Log2FC values of the genes acnB (ID: N5094_RS02380) encoding aconitate hydratase, icd (ID: N5094_RS08810) encoding isocitrate dehydrogenase, sucAB (ID: N5094_RS08610, N5094_RS08615) encoding α-ketoglutarate dehydrogenase complex, sucCD (ID: N5094_RS08620, N5094_RS08625) encoding succinyl-CoA synthase and sdhABCD (ID: N5094_RS08590, N5094_RS08595, N5094_RS08600, N5094_RS08605) encoding succinate dehydrogenase all exceeded 2, suggesting the upregulation of their transcription. These results clearly demonstrated that the TCA pathway was significantly enriched in the Cu_PFS group compared with the PFS group and indicated that the presence of Cu2+ was conducive to energy (electron) production in strain CN32 under anaerobic conditions.

Transcriptional levels of genes related to anaerobic central carbon metabolism of strain CN32 (Cu_PFS vs. PFS; enzymes encoded by upregulated and downregulated genes with p-adjust ≤ 0.01 are color-coded; however, they are directly labelled black when p-adjust > 0.01)
DEGs Involved in Extracellular Electron Transfer for Fe3+ Reduction
As a typical class of dissimilated iron-reducing bacteria, Shewanella strains are known to transfer electrons to extracellular insoluble Fe3+ via reduction through the Mtr pathway consisting of by c-type cytochromes (c-Cyts), and endogenously secreted flavins act as either electron shuttles or cofactors of outer-membrane c-Cyts [13, 25,26,27], thus leading to extracellular Fe3+ reduction. To explore the impact of Cu2+ in PFS flocs on the EET of strain CN32, the transcriptional levels of potential c-Cyt-encoding genes associated with the Mtr pathway were analyzed, as shown in Table 1. In particular, the gene cymA was significantly downregulated, suggesting that when Cu2+ was present, EET via the Mtr pathway for PFS reduction was weakened, as the cytoplasmic membrane-bound c-Cyt CymA is the gatekeeper that facilitates the flow of electrons into this pathway [28, 29]. In addition, the transcription of genes that were annotated as encoding OmcA/MtrC family decaheme c-type cytochromes was downregulated to varying degrees, and notably, these cytochromes have been identified as outer-membrane c-Cyts that deliver electrons to extracellular Fe3+ by direct physical contact [13]. Moreover, as mentioned above, c-Cyts are the building blocks of the Mtr pathway, but the genes involved in c-Cyt biosynthesis, such as the ccm cluster, were generally downregulated to different degrees (Table S3). This finding further demonstrated the suppressive effect of Cu2+ on electron transfer via the Mtr pathway. Since flavins also play important roles in EET, the transcription of genes related to flavin biosynthesis was analyzed (Table S4). The results showed that DEGs were not found among almost all potential genes, suggesting that flavin-dependent EET did not differ significantly between the Cu_PFS and PFS groups. In summary, the EET activity of CN32 in the Cu_PFS group was inferior to that in the PFS group, which explained why the degree of Fe3+ reduction in the former group was lower than that in the latter group (Fig. 1).
DEGs for Assimilatory Sulfate Reduction
Considering that sulfate ions in the PFS flocs are potential electron acceptors for the genus Shewanella, transcriptional levels of genes involved in sulfur metabolism were compared between the Cu_PFS and PFS groups. As shown in Table 2 and Fig. S7, eleven genes involved in the assimilatory sulfate reduction pathway were upregulated, which indicated the activation of sulfur assimilation metabolism in the Cu_PFS group, including extracellular sulfate uptake, intracellular sulfate and sulfite reduction, and cysteine biosynthesis [20]. In particular, the transcriptional levels of the genes cysH, encoding sulfate reductase, and cysIJ, encoding sulfite reductase, in the Cu_PFS group were at least one order of magnitude higher than those in the PFS group. The enrichment of assimilatory sulfur metabolism implied that more energy (electrons) was needed for the reduction of large quantities of sulfate, which might explain why the anaerobic TCA cycle was significantly enhanced in the Cu_PFS group despite fewer electron requirements for Fe3+ reduction, compared with that in the PFS group. In addition, the genes encoding cysteine synthase were also obviously upregulated, which could be considered a strategy to address oxidative stress possibly caused by heavy metal cations since cysteine is an essential precursor to the synthesis of biomolecules (e.g., glutathione) with the ability to remove reactive oxygen species. The upregulated transcription of cysteine synthase-encoding genes was also observed when the genus Shewanella was exposed to alkaline and high electrode potential stress [20, 30].
Conclusions
The effects of several common heavy metal cations present in PFS flocs on the ability of S. putrefaciens CN32 to extracellularly reduce Fe3+ in the PFS flocs were studied, and Cu2+ showed a more significant inhibitory effect than the others, suggesting that the PFS flocs chelated with Cu2+ had high stability against dissimilatory iron-reducing bacteria. The coexistence of Cu2+ had a profound influence on the global transcription of CN32, resulting in 782 upregulated genes and 713 downregulated genes, which were mainly enriched in energy production, amino acid metabolism, protein biosynthesis, and oxidation‒reduction processes. Metabolic pathway analysis showed that the anaerobic TCA cycle of CN32 was significantly activated in the Cu_PFS group compared to that in the PFS group, suggesting more energy (electron) production in the former group. The transcription of genes encoding c-Cyts and those involved in c-Cyt biosynthesis were generally downregulated, while the differences in flavin biosynthesis-related genes were slight, indicating that the weakened EET of CN32 was responsible for the decreased Fe3+ reduction in the Cu_PFS flocs. Moreover, the overall assimilatory sulfate reduction pathway of CN32, followed by cysteine biosynthesis, was intensified in the Cu_PFS group, which needed abundant energy (electrons) produced from the anaerobic TCA cycle and could be a strategy to address the oxidative stress caused by Cu2+. Therefore, the mechanism of Cu2+-mediated inhibition of PFS floc reduction by strain CN32 was elucidated in this work, which indicated that PFS flocs would have high stability and safety when used to treat Cu2+ pollution in water.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Tang, X. M., Zheng, H. L., Teng, H. K., Sun, Y. J., Guo, J. S., Xie, W. Y., Yang, Q. Q., & Chen, W. (2016). Chemical coagulation process for the removal of heavy metals from water: A review. Desalination and Water Treatment, 57(4), 1733–1748.
Chai, L. Y., Li, Q. Z., Wang, Q. W., & Yan, X. (2018). Solid-liquid separation: An emerging issue in heavy metal wastewater treatment. Environmental Science and Pollution Research, 25(18), 17250–17267.
Zouboulis, A. I., Moussas, P. A., & Vasilakou, F. (2008). Polyferric sulphate: Preparation, characterisation and application in coagulation experiments. Journal of Hazardous Materials, 155(3), 459–468.
Li, C., Yi, X., Dang, Z., Yu, H., Zeng, T., Wei, C., & Feng, C. (2016). Fate of Fe and Cd upon microbial reduction of Cd-loaded polyferric flocs by Shewanella oneidensis MR-1. Chemosphere, 144, 2065–2072.
Li, Y., Zeng, X., Liu, Y., Yan, S., Hu, Z., & Ni, Y. (2003). Study on the treatment of copper-electroplating wastewater by chemical trapping and flocculation. Separation and Purification Technology, 31(1), 91–95.
Suo, C. Y., Xu, D. D., Yuan, R. F., & Zhou, B. H. (2020). Synchronous removal of cd(II), pb(II), and Cu(II) by coagulation in the presence of polymeric ferric sulfate. Desalination and Water Treatment, 195, 421–434.
Wang, Q., Wei, Z., Yi, X., Tang, J., Feng, C., & Dang, Z. (2019). Biogenic iron mineralization of polyferric sulfate by dissimilatory iron reducing bacteria: Effects of medium composition and electric field stimulation. Science of the Total Environment, 684, 466–475.
Kappler, A., Bryce, C., Mansor, M., Lueder, U., Byrne, J. M., & Swanner, E. D. (2021). An evolving view on biogeochemical cycling of iron. Nature Reviews Microbiology, 19(6), 360–374.
Lovley, D. R. (1991). Dissimilatory Fe(III) and mn(IV) reduction. Microbiological Reviews, 55(2), 259–287.
Jiang, Y., Xi, B. D., Li, R., Li, M. X., Xu, Z., Yang, Y. N., & Gao, S. B. (2019). Advances in Fe(III) bioreduction and its application prospect for groundwater remediation: A review. Frontiers of Environmental Science & Engineering, 13(6), 89.
Neal, A. L., Rosso, K. M., Geesey, G. G., Gorby, Y. A., & Little, B. J. (2003). Surface structure effects on direct reduction of iron oxides by Shewanella oneidensis Geochimica et Cosmochimica Acta, 67(23), 4489–4503.
Yong, S. N., Lim, S., Ho, C. L., Chieng, S., & Kuan, S. H. (2022). Mechanisms of microbial-based iron reduction of clay minerals: Current understanding and latest developments. Applied Clay Science, 228, 106653.
Shi, L., Dong, H., Reguera, G., Beyenal, H., Lu, A., Liu, J., Yu, H. Q., & Fredrickson, J. K. (2016). Extracellular electron transfer mechanisms between microorganisms and minerals. Nature Reviews Microbiology, 14(10), 651–662.
Lemaire, O. N., Méjean, V., & Iobbi-Nivol, C. (2020). The Shewanella genus: Ubiquitous organisms sustaining and preserving aquatic ecosystems. FEMS Microbiology Reviews, 44(2), 155–170.
Hau, H. H., & Gralnick, J. A. (2007). Ecology and biotechnology of the genus Shewanella Annual Review of Microbiology, 61, 237–258.
Hao, X. R., Tang, J., Yi, X. Y., Gao, K., Yao, Q., Feng, C. H., Huang, W. L., & Dang, Z. (2022). Extracellular polymeric substance induces biogenesis of vivianite under inorganic phosphate-free conditions. Journal of Environmental Sciences, 120, 115–124.
Harvey, A. E., Jr, Smart, J. A., & Amis, E. S. (1955). Simultaneous spectrophotometric determination of iron(II) and total iron with 1,10-phenanthroline. Analytical Chemistry, 27(1), 26–29.
Zhu, F., Huang, Y., Ni, H., Tang, J., Zhu, Q., Long, Z. E., & Zou, L. (2022). Biogenic iron sulfide functioning as electron-mediating interface to accelerate dissimilatory ferrihydrite reduction by Shewanella oneidensis MR-1. Chemosphere, 288, 132661.
Love, M. I., Huber, W., & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15(12), 550.
Lian, Y., Yang, Y., Guo, J., Wang, Y., Li, X., Fang, Y., Gan, L., & Xu, M. (2016). Electron acceptor redox potential globally regulates transcriptomic profiling in Shewanella decolorationis S12. Scientific Reports, 6, 31143.
Brutinel, E. D., & Gralnick, J. A. (2012). Anomalies of the anaerobic tricarboxylic acid cycle in Shewanella oneidensis revealed by Tn-seq. Molecular Microbiology, 86(2), 273–283.
Tang, Y. J., Meadows, A. L., Kirby, J., & Keasling, J. D. (2007). Anaerobic central metabolic pathways in Shewanella oneidensis MR-1 reinterpreted in the light of isotopic metabolite labeling. Journal of Bacteriology, 189(3), 894–901.
Grobbler, C., Virdis, B., Nouwens, A., Harnisch, F., Rabaey, K., & Bond, P. L. (2015). Use of SWATH mass spectrometry for quantitative proteomic investigation of Shewanella oneidensis MR-1 biofilms grown on graphite cloth electrodes. Systematic and Applied Microbiology, 38(2), 135–139.
Cao, X. H., Qi, Y. L., Xu, C., Yang, Y. Y., & Wang, J. (2017). Transcriptome and metabolome responses of Shewanella oneidensis MR-1 to methyl orange under microaerophilic and aerobic conditions. Applied Microbiology and Biotechnology, 101(8), 3463–3472.
Zou, L., Zhu, F., Long, Z. E., & Huang, Y. (2021). Bacterial extracellular electron transfer: A powerful route to the green biosynthesis of inorganic nanomaterials for multifunctional applications. Journal of Nanobiotechnology, 19(1), 120.
Zou, L., Huang, Y., Long, Z. E., & Qiao, Y. (2019). On-going applications of Shewanella species in microbial electrochemical system for bioenergy, bioremediation and biosensing. World Journal of Microbiology & Biotechnology, 35(1), 9.
Okamoto, A., Hashimoto, K., Nealson, K. H., & Nakamura, R. (2013). Rate enhancement of bacterial extracellular electron transport involves bound flavin semiquinones. Proceedings of the National Academy of Sciences of the United States of America, 110(19), 7856–7861.
Wu, X., Zou, L., Huang, Y., Qiao, Y., Long, Z. E., Liu, H., & Li, C. M. (2018). Shewanella putrefaciens CN32 outer membrane cytochromes MtrC and UndA reduce electron shuttles to produce electricity in microbial fuel cells. Enzyme and Microbial Technology, 115, 23–28.
Louro, R. O., & Paquete, C. M. (2012). The quest to achieve the detailed structural and functional characterization of CymA. Biochemical Society Transactions, 40, 1291–1294.
Leaphart, A. B., Thompson, D. K., Huang, K., Alm, E., Wan, X. F., Arkin, A., Brown, S. D., Wu, L. Y., Yan, T. F., Liu, X. D., Wickham, G. S., & Zhou, J. Z. (2006). Transcriptome profiling of Shewanella oneidensis gene expression following exposure to acidic and alkaline pH. Journal of Bacteriology, 188(4), 1633–1642.
Funding
This work was supported financially by the National Natural Science Foundation of China (grant no. 32260025), the Open Funding Project of the State Key Laboratory of Biocatalysis and Enzyme Engineering, China (grant no. SKLBEE20220010), the Innovation and Entrepreneurship Training Program for College Students of Jiangxi Province, China (grant no. S202310414090), and the Natural Science Foundation of Jiangxi Province, China (grant no. 20202ACB215001).
Author information
Authors and Affiliations
Contributions
JP: experimentation, data collection, writing—original draft. FF and GZ: data collection and interpretation, manuscript revision. LZ: conceptualization, supervision, data analysis, manuscript revision, and funding acquisition.
Corresponding author
Ethics declarations
Ethical Approval
Not applicable.
Consent to Participate
All authors consented to participate.
Consent for Publication
All authors have provided their consent to publish their work.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 1.77 MB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Peng, J., Feng, F., Zhang, G. et al. Transcriptome Analysis Reveals the Inhibitory Effect of Cu2+ on Polyferric Sulfate Floc Reduction by Shewanella putrefaciens CN32. Appl Biochem Biotechnol 196, 4862–4873 (2024). https://doi.org/10.1007/s12010-023-04787-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-023-04787-1