
Abstract
Linalool is a pleasant-smelling monoterpenoid widely found in the essential oils of most flowers. Due to its biologically active properties, linalool has considerable commercial potential, especially in the food and perfume industries. In this study, the oleaginous yeast Yarrowia lipolytica was successfully engineered to produce linalool de novo. The (S)-linalool synthase (LIS) gene from Actinidia argute was overexpressed to convert geranyl diphosphate (GPP) into linalool. Flux was diverted from farnesyl diphosphate (FPP) synthesis to GPP by introducing a mutated copy of the native ERG20F88W−N119W gene, and CrGPPS gene from Catharanthus roseus on its own and as part of a fusion with LIS. Disruption of native diacylglycerol kinase enzyme, DGK1, by oligo-mediated CRISPR-Cas9 inactivation further increased linalool production. The resulting strain accumulated 109.6 mg/L of linalool during cultivation in shake flasks with sucrose as a carbon source. CrGPPS expression in Yarrowia lipolytica increased linalool accumulation more efficiently than the ERG20F88W−N119W expression, suggesting that the increase in linalool production was predominantly influenced by the level of GPP precursor supply.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Linalool is a terpene alcohol with a pleasant scent, found in the majority of popular essential oils [1]. Linalool has been demonstrated to have antibacterial, antifungal, anticancer, anti-inflammatory, and antioxidant properties, making it interesting for both pharmaceutical and cosmetic applications [2]. This volatile component is also a precursor in the biosynthesis of vitamin E [3]. The most popular use of linalool is as a fragrance, where the proportion of linalool among terpenoids in floral fragrances is up to 70% [4]. The two enantiomers of linalool possess differing odor characteristics. For one, the odor threshold value of the (R)-enantiomer is about nine times less than that of (S)-linalool [5, 6]. Furthermore, (R)-linalool has a smell reminiscent of lavender, while (S)-linalool has a citrus aroma [7]. Global linalool consumption exceeded 11,000 metric tons in 2018, and its global market is projected to reach $12.3 billion in 2024 [8].
At present, several microorganisms have been used for heterologous biosynthesis of linalool, such as Pantoea ananatis, Escherichia coli, Saccharomyces cerevisiae, and Yarrowia lipolytica [9,10,11,12]. Nitta et al. achieved the highest titer of linalool to date using a P. anantis strain constructed a genetically modified strain by damaged endogenous crtEXYIB-crtZ operon responsive to carotenoid synthesis, knocked out glucose dehydrogenase GCD knocked out, and several heterologous genes overexpressed. Specifically, the authors introduced acetyl-CoA acetyltransferase/hydroxymethyl-CoA reductase (mvaE) and hydroxymethylglutaryl-CoA synthase (mvaS) from Enterococcus faecalis, mevalonate kinase M. paludicola (MVK), linalool synthase from Actinidia arguta (LIS), and linalool synthase (LIS) fused with halophilic β-lactamase from Chromohalobacter sp. 560 with hexahistidine at the N-terminus, which increases the solubility of the enzyme in the host organism [9]. This strain in dual-phase fed-batch fermentation produced linalool at a concentration of 10.9 g/L. In a previous study, A. arguta LIS was found to support the highest of 16 screened enzymes for (S)-linalool production in P. ananatis [13]. However, P. ananatis is pathogenic on a broad range of plant hosts as well as humans and so is not suitable for industrial production [14].
Y. lipolytica is a non-conventional oleaginous yeast that is generally recognized as safe (GRAS) [15]. It can accumulate high levels of neutral lipids and therefore both efficiently synthesize a massive amount of acetyl-CoA as the precursor of the acyl group in lipids, and support strong regeneration of NADPH [16]. The mevalonate (MVA) pathway for the synthesis of terpenoid begins with the condensation of two molecules of acetyl-CoA [17] and so Y. lipolytica is considered an attractive host for terpenoid biosynthesis [18, 19]. Nevertheless, the highest described linalool titer in Y. lipolytica was only 5.34 mg/L in YPD medium and 6.96 mg/L in YP medium supplemented with citrate and pyruvate in shake flasks [12].
The standard approach in metabolic engineering for bioproduction is to increase the supply of product precursors. The reaction catalyzed by 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG1) is irreversible and HMG1 is considered to be the key rate-limiting enzyme in the MVA pathway [20]. In Y. lipolytica, almost all terpenoid production efforts have used HMG1 overexpression to improve terpenoid production [21]. Similarly, HMG1 overexpression showed a significant (4.7-fold) increase in linalool production by Y. lipolytica [12]. The overexpression of both HMG1 gene and the mevalonate kinase-encoding gene ERG12 showed a 7.6-fold increase in linalool titer. The five-carbon universal terpenoid building blocks, isopropyl diphosphate (IPP), and its allylic isomer dimethylallyl diphosphate (DMAPP) can be ligated in a head-to-tail manner to form geranyl diphosphate (GPP) via geranyl diphosphate synthase (GPPS) [21]. GPP must then be converted to linalool. However, the native Y. lipolytica enzyme ERG20 converts IPP and DMAP into both GPP and the side product FPP. Mutating ERG20 to ERG20F88W−N119W allows GPP synthase activity while removing this side activity [22].
In this study, (S)-linalool synthase from Actinida argute was expressed in Y. lipolitica. Markerless gene integration and deletion was performed using a CRISPR-Cas9-based toolkit [23]. DGK1 knockout, overexpression of GPPS from Catharanthus roseus (CrGPPS) by itself and in fusion with LIS in a Y. lipolytica strain with an optimized mevalonate pathway, resulted in the best titer yet reported (109.6 mg/L) for Y. lipolytica.
Materials and Methods
Strains and Culture Media
E. coli strain XL1-Blue (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB lacIqZΔM15 Tn10 (Tetr)]) was used for cloning and plasmid propagation. Cells were grown at 37 °C with constant shaking on 5 mL LB medium (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl), and ampicillin (100 μg/mL), chloramphenicol (120 μg/mL), or spectinomycin (50 μg/mL) were added for plasmid selection.
Y. lipolytica strains used in this study are derived from the wild-type Y. lipolytica W29 (ATCC 20460) strain. All the strains used in this study are listed in Supplementary Table S1. Rich media were based on YP containing 10 g/L yeast extract, 10 g/L bacto-peptone. YPsuc contained YP with 20 g/L sucrose. If necessary, nourseothricin (Nat, 250 μg/mL) was added to the media at a concentration of 250 μg/mL. Solid media for E. coli and Y. lipolytica were prepared by adding 20 g/L agar.
Construction of Backbone Vectors
The constructed plasmids and primers used in this study are presented in Supplementary Tables S2 and S3. All synthetic genes were codon-optimized for Y. lipolytica using the GenSmart™ Codon Optimization webtool (GenScript) and ordered from Twist Bioscience. The DNA sequences are presented in Supplementary Table S5. All described plasmids were assembled using standard cloning strategies [24], Gibson assembly [25], Golden Gate Assembly, and E. coli recombineering [23]. Enzymes for molecular biology were obtained from Thermo Fisher Scientific. The genes, homologous arms for chromosomal integration, promoters, and terminators were amplified using Phusion High-Fidelity DNA polymerase. All assembled products were verified by sequencing both strands.
The set of backbone plasmids for integration into IntA2, IntB1, IntD4, and IntF14 loci contains Y. lipolytica homology arms (IntUp and IntDn), promoters PTEFin and PEXP1, terminators TTEF and TXPR2, autonomously replicating sequence (ARS), and hygromycin (Hyg) resistance marker. The plasmids were assembled using Gibson assembly from seven fragments (Supplementary Table S3). All PCR-products were amplified from genomic DNA of W29 strain, except the chloramphenicol resistance gene (Cm), which was amplified from pMW-att-Cm [26] using primers Oligo21 and Oligo22, and the ERG20F88W−N119W gene, which was amplified from synthetic fragment ERG20F88W−N119W (Supplementary Table S5) using primers Oligo31 and Oligo32.
The set of backbone plasmids for integration into IntC2, IntC3, IntC13, IntC14, IntE12, and IntE16 loci contains Y. lipolytica homology arms (IntUp and IntDn) (Supplementary Table S5) and does not contain ARS or Hyg elements. Plasmids pT-flIntE12-LIS, pT-flIntE16-LIS, and pT-IntC14-LIS containing codon optimized synthetic gene encoding A. argute LIS (GenBank ID: GQ338153.1) and plasmid pT-IntC3-CrGPPS containing a truncated version (Met100–CrGPPS) of codon optimized CrGPPS gene encoding C. roseus CrGPPS (GenBank ID: JX417185.1) were synthesized by Twist Bioscience. The assembly of plasmids for integration into IntC2 and IntC13 was performed by Golden Gate assembly [27]. To obtain the pC13US1.1-PTEF-Rad52-TLIP expression plasmid harboring codon optimized ScRad52 gene encoding S. cerevisiae Rad52 (GenBank ID: CAA86623.1), Golden Gate assembly was used (Supplementary Table S3, S5).
Strain Construction
Y. lipolytica strains were engineered with a markerless CRISPR-Cas9 system. Guide sequences (sgRNA) and Cas9 proteins were expressed from an episomal pCasNA-series plasmid [23] (Supplementary Table S3) that was mixed with a repair cassette containing transcription unit or units flanked by arms homologous to the targeting chromosomal region. Summarized data about 10 integration loci, homology arms, and integration efficiency of the repair cassettes is presented in Supplementary Table S4. Yeast strains were transformed using a DTT/lithium acetate method [23]. All transformants were selected on agar YPsuc medium containing 250 μg/mL of nourseothricin. Six transformants per plate were tested by PCR for verification of transcription unit integration using primer sets (Int-chr-F and Int-chr-R) presented in Supplementary Table S2. Removal of an episomal pCasNA was performed by streaking the strain on agar YPsuc medium to get single colonies followed by the spot test on the media with and without nourseothricin. After the marker recovery procedure was completed, the marker-free integration cycle was restarted.
The 120 bp double-stranded oligos for DGK1 and for PAH1 genes knockout were obtained by annealing pairs of complementary single-stranded oligos Oligo35/Oligo36 and Oligo33/Oligo34 respectively, as described in oligonucleotide annealing protocol (https://www.sigmaaldrich.com/RU/en/technical-documents/protocol/genomics/pcr/annealing-oligos) (Supplementary Table S2). The resulting repair fragments for the DGK1 or PAH1 genes were co-transformed with helper plasmids pCNR-sgDGK1d/pCNR-sgDGK1f or pCNR-sgPAH1a/pCNR-sgPAH1b, respectively, into Y. lipolytica strains VKPM Y-3178, G0, G1, and G8 (Supplementary Table S1). Summarized data on efficiency of genes inactivation are presented in Table 1.
Culture Conditions
Strains were initially grown in 5 mL YPsuc medium in 50-mL test tubes and cultured overnight at 30 °C. For test tube cultivation, 10 mL of YP with sucrose 90 g/L was inoculated by the cell suspension to an OD600 of 0.2. The cultivation was run for 5 days at 28 °C and 275 rpm. One milliliter of isopropyl myristate (IPM) (ITW Reagents, Germany) was added to the test tubes at 48 h of cultivation. For shake-flask cultivation, 50 mL of YP with sucrose 90 g/L was inoculated to an OD600 of 0.2 and the cultured for 7 days at 28 °C and 275 rpm. Five milliliters of IPM was added to the test flasks at 48 h of cultivation.
Linalool Measurement
The culture broth was collected, and the cells were precipitated at 3841 × g for 5 min. The organic phase was taken for GC/MS analysis. The cell pellets were washed twice with ethanol and water (1:1 v/v) and centrifuged at 3841 × g for 5 min and lyophilized by a FreeZone 6 Plus system (Labconco, USA). Lyophilized cells were used for the dry-weight measurements (DCW). The concentrations of linalool in organic phase were determined using an GC/MS system on a “Maestro” (Interlab) (Agilent 7820A GC and 5977 MSD, Agilent Technologies). The carrier gas was helium. The oven temperature was maintained at 70 °C for 2 min after injection and then programmed at 15 °C/min to 260 °C, then at 30 °C/min to 320 °C and holding at 320 °C for 1 min. Sample injection was conducted with 1/5 split carrier gas. Registration of mass spectra was performed in full scanning mode of 35–300 AU masses. Evaporator temperature was 270 °C. Interface temperature kept 280 °C, quadrupole temperature was 150 °C, and source was 230 °C. Electron impact ionization, lionization energy was 70 eV. Injected sample volume was 1 μL. Twenty microliters of isopropyl myristate extract sample was added to 130 μL of hexane (dilution by a factor of 7.5).
Sugar Measurement
Supernatants obtained after removal of the cell pellets were used for analysis. The concentrations of glucose, sucrose, and fructose were determined using an HPLC system Dionex Ultimate 3000 (Thermo scientific) with a RID detector (temperature of 50 °C) and an YMC-Pack Polyamine II 250 × 4.6 mm I.D column (YMC, Japan). The mobile phase composition of ACN/H2O/EA (200:125:10, v/v) was used at a flow rate of 1.5 mL/min for 6–10 min at 50 °C.
Results and Discussion
Linalool Synthesis by Y. lipolytica
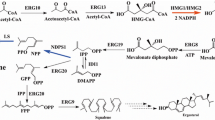
Y. lipolytica W29 (VKPM Y-4620), a lipid accumulating strain capable of growing on sucrose as the sole carbon source, was used as the platform strain for linalool production in this study. To increase the metabolic flux from MVA pathway, HMG1 and ERG12 genes were overexpressed (Fig. 1) [27]. Two copies of ERG20F88W−N119W gene encoding a mutated version of Y. lipolytica Erg20 with substitution mutations F88W and N119W, which shifts the activity of the enzyme to favor the generation of GPP, were introduced in GBK3 strain (Supplementary Table S1). Then, three individual copies each of LIS gene encoding linalool synthase from the plant Actinidia arguta were consecutively integrated into the genome of GBK3 strain resulted in G5, G6, and G7 strains, respectively (Supplementary Table S1, Fig. S1). Due to the fact that (S)-linalool is cytotoxic and semi-volatile [9], dual-phase flask cultivation in YP with 90 g/L sucrose and IPM was used. Enhancing the copy number of LIS gene led to increase linalool accumulation from 6.9 mg/L for one copy to 12.6 mg/L for three copies (Fig. 3).

Metabolic pathway involved in linalool biosynthesis. Enzymes expressed in this study are shown in green
Boosting GPP Synthesis by Expression of Exogenous GPPS
Low levels of monoterpene production are often associated with an insufficient precursor supply [28]. Therefore, to further increase linalool biosynthesis, an exogenous gene encoding a truncated version of geranyl diphosphate synthase, CrGPPS from C. roseus, was introduced in strain G7 (yielding strain G8, Supplementary Material Table S1). CrGPPS is a homomeric enzyme with two highly conserved aspartate-rich regions essential for catalytic function and substrate binding [29]. Unlike heterodimeric plant geranyl diphosphate synthases, CrGPPS lacks the conserved CxxxC motif critical for physical interaction between the two subunits forming a heterodimeric enzyme. In vitro analysis has shown that the single product of CrGPPS-catalyzed condensation of IPP and DMAPP was GPP, and only trace amount of GGPP was detected in the presence of either GPP or FPP along with IPP [29]. Indeed, G8 strain harboring CrGPPS gene had 2.5-fold increase in linalool titer (31.6 mg/L) compared to the parental G7 strain (Fig. 3).
Improving Linalool Production by Reducing Cytotoxicity
It has previously been shown that disruption of the phosphatidic acid phosphatase-encoding (PAH1) leads to enlargement of the endoplasmic reticulum (ER), which stimulates the production of recombinant triterpene biosynthesis enzymes and ultimately increases triterpenoid accumulation [30]. It was also noted that the cells lacking PAH1 are defective in cell wall integrity [31], which can increase permeability and so decrease the concentration of toxic linalool in cells. Previous reports have shown that deletions of DGK1 and PAH1 genes in Y. lipolytica also led to a distinctive elongated morphology of cell membranes, which in turn can cause terpene excretion and reduce cytotoxicity [28].
Y. lipolytica is characterized by a high efficiency of non-homologous end joining (NHEJ) for repair of DNA double-strand breaks (dsb) which hampers integration by homologous recombination (HR) [32]. Two strategies can be applied to improve the frequency and efficiency of homologous recombination (HR) in Y. lipolytica: disruption of the KU70 gene responsible for double stranded break repair in the NHEJ pathway [33] and heterologous expression of S. cerevisiae RAD52 gene, the key component of S. cerevisiae HR [34].
First, we tested the efficiency of oligo-mediated CRISPR-Cas9 gene inactivation in Y. lipolytica by disruption of DGK1 and PAH1 genes in three different strains: W29, W29 Δku70, and W29 Rad52. For DGK1 and PAH1 gene knockouts, synthetic 120 bp double-stranded oligos were used as repair templates together with two Cas9-helper episomal plasmids (Fig. 2) [23]. Gene disruption was confirmed by PCR from six isolated colonies for each variant (Supplementary Fig. S2).

Scheme of DGK1 gene knockout via oligo-mediated CRISPR-Cas9 homologous recombination. The DNA strand is indicated by the colorless rectangle. The DGK1 gene is indicated in blue. Two oligonucleotides, complementary to each other, were annealed to themselves to obtain an oligo duplex. The resulting DNA double strand, 120 base pairs long, was used as a patch for inactivation using CRISPR-Cas9. The double-stranded DNA breakage caused by the Cas9 protein are shown as scissors. The positions of primers annealing are depicted F and R
As expected, there was no gene disruption by HR in W29 strain (Table 1). The DGK1 gene knockout showed greater efficiency (100% in W29 Δku70 and 67% in W29 Rad52) than PAH1 (50% in W29 Δku70 and 17% in W29 Rad52). In both cases, better performance was observed in the Δku70 strain.
We then aimed to apply this technique to perform DGK1 and PAH1 knockouts in the G8 strain. PAH1 null mutant strain (G10) was characterized by reduced growth (9.1 g DCW/L biomass accumulated) as well as a 1.8-fold decrease in linalool titer (18.0 mg/L) (Fig. 3). On the contrary, DGK1 gene inactivation resulted in 1.2 higher linalool production (39.3 mg/L) compared to the parental G8 strain (Fig. 3).

Linalool production by the engineered Y. lipolytica strains during cultivation with sucrose as a carbon source with in situ extraction in organic solvent (IPM) in flask for 5 days
Constructing Fusion Proteins
One way to improve the utilization of substrates between proteins that catalyze sequential steps in a pathway is fusing two or more genes with a linker region [35]. In this study, LIS and CrGPPS genes were fused with a flexible GGGS linker and transformed into the best performing strain, G9 (Fig. 4). Both strains G11 (expressing a hybrid enzyme where CrGPPS fused through its C-terminus to LIS; CrGPPS-LIS) and G12 (expressing a hybrid enzyme where LIS fused through its C-terminus to CrGPPS; LIS-CrGPPS) demonstrated enhancing linalool production compared with the parental G9 strain (Fig. 4). The best performing strain, G12, accumulated 60.9 mg/L of linalool in flask culture at the 5th day of cultivation.

The linalool production of strains G11 and G12 expressing the fusion proteins CrGPPS-LIS or LIS-CrGPPS, respectively and G9 control parent strain
Time Course Flask Cultivation
Linalool production by the final engineered strain G12 was evaluated in flasks for 7 days of dual phase cultivation in rich YP medium with 90 g/L sucrose. IPM was added at the third day of cultivation. The biomass (DCW, g/L) accumulated linearly from the beginning of cultivation to 144 h (Fig. 5). Linalool production was evaluated starting from 72 h during “dual-phase” cultivation. After 7 days, the linalool titer of strain G12 reached 109.6 mg/L while biomass continued to increase linearly.

Time course flask cultivation of G12 strain. Concentrations of linalool (light green dot dash line), dry cell weight (DCW) (green two-dot dash line), sucrose (pink dashed lines), glucose (purple dotted line with circles), and fructose (blue line)
Sugar consumption during cultivation was also evaluated. Like all descendants of the parental VKPM Y-4620 strain, G12 can utilize sucrose as a single carbon source due to the overexpression of S. cerevisiae ScSUC2 and Y. lipolytica HXK1 genes [26]. Invertase (Suc2) is secreted to the culture medium where sucrose is hydrolyzed to glucose and fructose, which is then taken up by cells [36]. Hexokinase was also demonstrated to have an affinity to fructose, allowing its assimilation into glycolysis [37].
Sucrose was totally hydrolyzed by 48 h (Supplementary Fig. S3). Glucose concentration at 24 h was 9.7 g/L and was consumed totally by 72 h. However, fructose content remained at 16.8–36.7 g/L for 72 h and was consumed only by 168 h. We observed the same pattern of sugar consumption in the control strains G2 and G9 (Supplementary Fig. S4, S5) with a noticeable delay in fructose uptake. This may be due to delayed transport and the inefficient operation of hexokinase (HXK1) in Y. lipolytica [38].
Conclusions
Our linalool-producing strain was engineered by increasing the flux towards MVA precursors, expressing two types of GPP synthase, introducing three copies of a linalool synthase gene, and disrupting DGK1. The base strain with 2 copies of GPP synthase (ERG20F88W−N119W) and 3 copies of linalool synthases (LIS) accumulated 12.6 mg/L of linalool. Introduction of just one gene copy encoding C. roseus GPP synthase (CrGPPS) increased linalool production by 2.5 times resulting in 31.6 mg/L of linalool being produced. Disrupting DGK1 further increased accumulation of linalool. Finally, the fusion of two key enzymes (linalool synthase and geranyl diphosphate synthase) yielded a recombinant strain producing 109.6 mg/L of linalool at 168 h of cultivation, which exceeds previous results obtained in Y. lipolytica [12]. Thus, increasing the GPP precursor pool is critical for linalool synthesis.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Wojtunik-Kulesza, K. A., Kasprzak, K., Oniszczuk, T., & Oniszczuk, A. (2019). Natural monoterpenes: Much more than only a scent. Chemistry & Biodiversity, 16(12), e1900434. https://doi.org/10.1002/cbdv.201900434
Abd El-Baky, R., & Shawky, Z. (2016). Eugenol and linalool: Comparison of their antibacterial and antifungal activities. African Journal of Microbiology Research, 10(44), 1860–1872. https://doi.org/10.5897/AJMR2016.8283
Kamatou, G. P., & Viljoen, A. (2008). Linalool – A review of a biologically active compound of commercial importance. Natural Product Communications, 3, 1183–1192. https://doi.org/10.1177/1934578X0800300727
Stashenko, E. E., & Martinez, J. R. (2008). Sampling flower scent for chromatographic analysis. Journal of Separation Science, 31(11), 2022–2031. https://doi.org/10.1002/jssc.200800151
Aprotosoaie, A. C., Hancianu, M., Costache, I. I., & Miron, A. (2014). Linalool: A review on a key odorant molecule with valuable biological properties. Flavour and Fragrance Journal, 29(4), 193–219. https://doi.org/10.1002/ffj.3197
Pereira, I., Severino, P., Santos, A. C., Silva, A. M., & Souto, E. B. (2018). Linalool bioactive properties and potential applicability in drug delivery systems. Colloids and Surfaces. B, Biointerfaces, 171, 566–578. https://doi.org/10.1016/j.colsurfb.2018.08.001
Ozek, T., Tabanca, N., Demirci, F., Wedge, D., & Baser, K. H. C. (2010). Enantiomeric distribution of some linalool containing essential oils and their biological activities. Records of Natural Products, 4, 180–192.
Zhang, C., Chen, X., Lee, R. T. C., Rehka, T., Mauer-Stroh, S., & Rühl, M. (2021). Bioinformatics-aided identification, characterization of fungal linalool synthases and applications in linalool biosynthesis. Communications Biology, 4, 223. https://doi.org/10.21203/rs.3.rs-75943/v1
Nitta, N., Tajima, Y., Yamamoto, Y., Moriya, M., Matsudaira, A., Hoshino, Y., Nishio, Y., & Usuda, Y. (2021). Fermentative production of enantiopure (S)-linalool using a metabolically engineered Pantoea ananatis. Microbial Cell Factories, 20(1), 54. https://doi.org/10.1186/s12934-021-01543-0
Wang, X., Wu, J., Chen, J., Xiao, L., Zhang, Y., Wang, F., & Li, X. (2020). Efficient biosynthesis of R-(-)-linalool through adjusting the expression strategy and increasing GPP supply in Escherichia coli. Journal of Agriculture and Food Chemistry, 68(31), 8381–8390. https://doi.org/10.1021/acs.jafc.0c03664
Zhou, P., Du, Y., Fang, X., Xu, N., Yue, C., & Ye, L. (2021). Combinatorial modulation of linalool synthase and farnesyl diphosphate synthase for linalool overproduction in Saccharomyces cerevisiae. Journal of Agriculture and Food Chemistry, 69(3), 1003–1010. https://doi.org/10.1021/acs.jafc.0c06384
Cao, X., Wei, L. J., Lin, J. Y., & Hua, Q. (2017). Enhancing linalool production by engineering oleaginous yeast Yarrowia lipolytica. Bioresource Technology, 245(Pt B), 1641–1644. https://doi.org/10.1016/j.biortech.2017.06.105
Hoshino, Y., Moriya, M., Matsudaira, A., Katashkina, J. I., Nitta, N., Nishio, Y., & Usuda, Y. (2020). Stereospecific linalool production utilizing two-phase cultivation system in Pantoea ananatis. Journal of Biotechnology, 324, 21–27. https://doi.org/10.1016/j.jbiotec.2020.09.021
Coutinho, T. A., & Venter, S. N. (2009). Pantoea ananatis: An unconventional plant pathogen. Molecular Plant Pathology, 10(3), 325–335. https://doi.org/10.1111/j.1364-3703.2009.00542.x
Groenewald, M., Boekhout, T., Neuveglise, C., Gaillardin, C., van Dijck, P. W., & Wyss, M. (2014). Yarrowia lipolytica: Safety assessment of an oleaginous yeast with a great industrial potential. Critical Reviews in Microbiology, 40(3), 187–206. https://doi.org/10.3109/1040841x.2013.770386
Beopoulos, A., Nicaud, J. M., & Gaillardin, C. (2011). An overview of lipid metabolism in yeasts and its impact on biotechnological processes. Applied Microbiology and Biotechnology, 90(4), 1193–1206. https://doi.org/10.1007/s00253-011-3212-8
Zhang, Y., Nielsen, J., & Liu, Z. (2017). Engineering yeast metabolism for production of terpenoids for use as perfume ingredients, pharmaceuticals and biofuels. FEMS Yeast Research, 17(8). https://doi.org/10.1093/femsyr/fox080.
Zhang, G., Wang, H., Zhang, Z., Verstrepen, K. J., Wang, Q., & Dai, Z. (2021). Metabolic engineering of Yarrowia lipolytica for terpenoids production: Advances and perspectives. Critical Reviews in Biotechnology, 42(4), 618–633. https://doi.org/10.1080/07388551.2021.1947183
Miller, K. K., & Alper, H. S. (2019). Yarrowia lipolytica: More than an oleaginous workhorse. Applied Microbiology and Biotechnology, 103(23–24), 9251–9262. https://doi.org/10.1007/s00253-019-10200-x
Arnesen, J. A., Kildegaard, K. R., Cernuda Pastor, M., Jayachandran, S., Kristensen, M., & Borodina, I. (2020). Yarrowia lipolytica strains engineered for the production of terpenoids. Front Bioeng Biotechnol, 8, 945. https://doi.org/10.3389/fbioe.2020.00945
Li, Z. J., Wang, Y. Z., Wang, L. R., Shi, T. Q., Sun, X. M., & Huang, H. (2021). Advanced strategies for the synthesis of terpenoids in Yarrowia lipolytica. Journal of Agriculture and Food Chemistry, 69(8), 2367–2381. https://doi.org/10.1021/acs.jafc.1c00350
Ignea, C., Pontini, M., Maffei, M. E., Makris, A. M., & Kampranis, S. C. (2014). Engineering monoterpene production in yeast using a synthetic dominant negative geranyl diphosphate synthase. ACS Synthetic Biology, 3(5), 298–306. https://doi.org/10.1021/sb400115e
Yuzbashev, T., Yuzbasheva, E., Melkina, O., Patel, D., Bubnov, D., Dietz, H., & Ledesma-Amaro, R. (2022). A DNA assembly toolkit to unlock the CRISPR/Cas9 potential for metabolic engineering. https://doi.org/10.21203/rs.3.rs-1570357/v1.
Green, M., & Sambrook, J. (2012). Molecular cloning: A laboratory manual. 4th Edition, Vol. II, Cold Spring Harbor Laboratory Press, New York
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., 3rd., & Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods, 6(5), 343–345. https://doi.org/10.1038/nmeth.1318
Taratynova, M. O., Kosikhina, I. M., Vinogradova, E. B., Dementev, D. A., Korobov, V. S., Zolottsev, V. A., Yuzbashev, T. V., Yuzbasheva, E. Y., & Sineoky, S. P. (2021). Accumulation of neutral lipids and β-carotene by the Yarrowia lipolytica yeast on a medium with sucrose as a carbon source. Biotekhnologiya, 37(3), 29–41. https://doi.org/10.21519/0234-2758-2021-37-3-29-41
Yuzbasheva, E. Y., Taratynova, M. O., Fedyaeva, I. M., Dementev, D. A., Korobov, V. S., Fedorov, A. S., Vidal, L. S., Yuzbashev, T. V., Sineoky, S. P., & Mikheev, M. (2023). Large-scale bioproduction of natural astaxanthin in Yarrowia lipolytica. Bioresource Technology Reports, 21, 101289. https://doi.org/10.1016/j.biteb.2022.101289
Zhang, J. L., Bai, Q. Y., Peng, Y. Z., Fan, J., Jin, C. C., Cao, Y. X., & Yuan, Y. J. (2020). High production of triterpenoids in Yarrowia lipolytica through manipulation of lipid components. Biotechnology for Biofuels, 13, 133. https://doi.org/10.1186/s13068-020-01773-1
Rai, A., Smita, S. S., Singh, A. K., Shanker, K., & Nagegowda, D. A. (2013). Heteromeric and homomeric geranyl diphosphate synthases from Catharanthus roseus and their role in monoterpene indole alkaloid biosynthesis. Molecular Plant, 6(5), 1531–1549. https://doi.org/10.1093/mp/sst058
Arendt, P., Miettinen, K., Pollier, J., De Rycke, R., Callewaert, N., & Goossens, A. (2017). An endoplasmic reticulum-engineered yeast platform for overproduction of triterpenoids. Metabolic Engineering, 40, 165–175. https://doi.org/10.1016/j.ymben.2017.02.007
Park, Y., Han, G. S., Mileykovskaya, E., Garrett, T. A., & Carman, G. M. (2015). Altered lipid synthesis by lack of yeast Pah1 phosphatidate phosphatase reduces chronological life span. Journal of Biological Chemistry, 290(42), 25382–25394. https://doi.org/10.1074/jbc.M115.680314
Holkenbrink, C., Dam, M. I., Kildegaard, K. R., Beder, J., Dahlin, J., Domenech Belda, D., & Borodina, I. (2018). EasyCloneYALI: CRISPR/Cas9-based synthetic toolbox for engineering of the yeast Yarrowia lipolytica. Biotechnology journal, 13(9), e1700543. https://doi.org/10.1002/biot.201700543
Verbeke, J., Beopoulos, A., & Nicaud, J. M. (2013). Efficient homologous recombination with short length flanking fragments in Ku70 deficient Yarrowia lipolytica strains. Biotechnology Letters, 35(4), 571–576. https://doi.org/10.1007/s10529-012-1107-0
Ji, Q., Mai, J., Ding, Y., Wei, Y., Ledesma-Amaro, R., & Ji, X. J. (2020). Improving the homologous recombination efficiency of Yarrowia lipolytica by grafting heterologous component from Saccharomyces cerevisiae. Metabolic Engineering Communications, 11, e00152. https://doi.org/10.1016/j.mec.2020.e00152
Woolston, B. M., Edgar, S., & Stephanopoulos, G. (2013). Metabolic engineering: Past and future. Annual Review of Chemical and Biomolecular Engineering, 4, 259–288. https://doi.org/10.1146/annurev-chembioeng-061312-103312
Moeller, L., Zehnsdorf, A., Aurich, A., Bley, T., & Strehlitz, B. (2012). Substrate utilization by recombinant Yarrowia lipolytica growing on sucrose. Applied Microbiology and Biotechnology, 93(4), 1695–1702. https://doi.org/10.1007/s00253-011-3681-9
Lazar, Z., Rossignol, T., Verbeke, J., Crutz-Le Coq, A. M., Nicaud, J. M., & Robak, M. (2013). Optimized invertase expression and secretion cassette for improving Yarrowia lipolytica growth on sucrose for industrial applications. Journal of Industrial Microbiology and Biotechnology, 40(11), 1273–1283. https://doi.org/10.1007/s10295-013-1323-1
Hapeta, P., Szczepanska, P., Witkowski, T., Nicaud, J. M., Crutz-Le Coq, A. M., & Lazar, Z. (2021). The role of hexokinase and hexose transporters in preferential use of glucose over fructose and downstream metabolic pathways in the yeast Yarrowia lipolytica. International Journal of Molecular Sciences, 22(17), 9282. https://doi.org/10.3390/ijms22179282
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (grant number 075–15-2019–1659).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Maria O. Taratynova, Iuliia M. Fedyaeva, Dmitry A. Dementev, and Evgeniya Y. Yuzbasheva. The first draft of the manuscript was written by Maria O. Taratynova and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent to Publish
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Taratynova, M.O., Tikhonova, E.E., Fedyaeva, I.M. et al. Boosting Geranyl Diphosphate Synthesis for Linalool Production in Engineered Yarrowia lipolytica. Appl Biochem Biotechnol 196, 1304–1315 (2024). https://doi.org/10.1007/s12010-023-04581-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-023-04581-z