
Abstract
Granulocyte colony-stimulating factor (GCSF) stimulates the proliferation of neutrophils but it has low serum half-life. Therefore, the present study was done to investigate the effect of XTENylation on biological activity, pharmacokinetics, and pharmacodynamics of GCSF in a neutropenic rat model. XTEN tag was genetically fused to the N-terminal region of GCSF-encoding gene fragment and subcloned into pET28a expression vector. The cytoplasmic expressed recombinant protein was characterized through intrinsic fluorescence spectroscopy (IFS), dynamic light scattering (DLS), and size exclusion chromatography (SEC). In vitro biological activity of the XTEN-GCSF protein was evaluated on NFS60 cell line. Hematopoietic properties and pharmacokinetics were also investigated in a neutropenic rat model. An approximately 140 kDa recombinant protein was detected on SDS-PAGE. Dynamic light scattering and size exclusion chromatography confirmed the increase in hydrodynamic diameter of GCSF molecule after XTENylation. GCSF derivatives showed efficacy in proliferation of NFS60 cell line among which the XTEN-GCSF represented the lowest EC50 value (100.6 pg/ml). Pharmacokinetic studies on neutropenic rats revealed that XTEN polymer could significantly increase protein serum half-life in comparison with the commercially available GCSF molecules. PEGylated and XTENylated GCSF proteins were more effective in stimulation of neutrophils compared to the GCSF molecule alone. XTENylation of GCSF represented promising results in in vitro and in vivo studies. This approach can be a potential alternative to PEGylation strategies for increasing serum half-life of protein.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Several complications arise during chemotherapy regimens, among which neutropenia is one of the main consequences which can be treated by administration of granulocyte colony-stimulating factor (GCSF) in the myelo-suppressed patients [1, 2]. GCSF is produced by various cells including monocytes, fibroblasts, mesothelial, and endothelial cells [3] and its interaction with GCSF receptor induces production of granulocytes, proliferation, differentiation, and survival of neutrophils [4, 5]. Due to the small molecular size (19 kDa), GCSF can be eliminated quickly through kidney glomerular filtration [6]. This molecule has five cysteine residues and two disulfide bonds located at Cys36-Cys42 and Cys64-Cys74 which are essential for its activity and stability [7, 8], and the presence of O-linked carbohydrate chain on Thr133 has no effect on its activity [9]. Recombinant biosimilar GCSF molecules are available in the market in two main forms: Filgrastim and PEG-Filgrastim [10]. Although PEG-Filgrastim has longer half-life in comparison to the Filgrastim molecule (14 vs. 3.5 h) [11], its drawbacks include complicated manufacturing process, higher production costs, and development of host immune responses [12, 13]. Therefore, novel forms of GCSF with less disadvantages and extended half-life in comparison to the PEGylated form have attracted too much attention during the last decades. Several strategies have been developed to increase pharmaceutical half-life. For example, human serum albumin [7] and IgG1 Fc fragment [8] have been genetically fused to the GCSF and significantly increased circulation half-life of the attached GCSF protein. It seems that conjugation of chemical or biodegradable molecules such as PEG [9, 14], PAS motifs [15, 16], and acyl groups [17] could also increase hydrodynamic radius of the pharmaceutical products.
XTENylation is another approach to improve pharmacokinetics of biological components. This biodegradable polymer consists of six hydrophilic amino acids (Ala, Asp, Gly, Pro, Ser, and Thr) in non-repetitive state. The absence of hydrophobic amino acid residues can increase solubility of the conjugated protein and eventually decrease protein aggregations [18]. Monodispersity and low viscosity of the final products are the main advantages of these biomolecules in comparison to the PEGylated proteins [18,19,20,21]. During the last decade, XTENylation technology was applied for several medicines including glucagon [19, 22] and teduglutide [23]. Exenatide is eliminated from kidneys within 2-h post-administration, while its serum half-life significantly increased after genetic fusion to the XTEN moiety (VRS-859) (139 h) [21, 24]. The human growth hormone (hGH) has also been fused to the XTEN tag and represented desirable pharmacokinetic properties in comparison to the other fusion forms of the original protein [25].
The present study was performed to design and produce an XTENylated form of GCSF protein in order to improve its pharmacokinetic properties without affecting the pharmacodynamics of the protein compared to the commercially available GCSF molecules (Filgrastim and PEG-Filgrastim).
Materials and Methods
Design of the Expression Cassette
In this study, an XTEN576 sequence compromising of 576 amino acids was chosen from the published patents (EP 2755675B1, WO2016077505A2). GCSF amino acid sequence was obtained from drug bank (Accession no. DB00099) compromising of 174 amino acid residues. NcoI cleavage site and the nucleotide sequence encoding the histidine tag (His6) were inserted at 5′ end of the expression cassette, while TAA stop codon and HindIII cleavage site were designed at 3′ end. The designed cassette was synthesized and subcloned into pET28a expression vector (Novagen, USA). Identity of the expression vector was confirmed using restriction digestion and sequencing.
Expression and Purification of r-XTEN-GCSF
The recombinant vector was transformed into E. coli BL21 (DE3) expression host (Novagen, USA). Recombinant colonies were inoculated into 10 ml Luria–Bertani (LB) broth medium supplemented with 30 µg/ml kanamycin (Gibco, USA) and incubated at 30 °C shaker incubator until optical turbidity of the medium at 600 nm reached 0.4. Protein expression was induced by 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) as the inducer and incubation continued for further 24 h at 30 °C. Bacterial cells were collected through centrifugation at 9,000 rpm for 3 min. Bacterial lysates were run on 8% SDS-PAGE, and protein bands were visualized using Coomassie Brilliant Blue G250 dye (Sigma, USA).
To purify the recombinant protein, protein expression was done in 500 ml LB broth medium under the above mentioned conditions. To determine the solubility of the expressed protein, bacterial pellet was re-suspended in cold lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole; pH8.0) and disrupted by ultrasonic homogenizer (20 pulses with the same intervals). Bacterial lysate was centrifuged at 10,000 rpm for 30 min at 4 °C. The supernatant (soluble fraction) and the pellet (insoluble inclusion bodies) were analyzed on 8% SDS-PAGE.
The supernatant fraction of the sonicated bacterial lysate was filtered through 0.45-µm syringe filter and subjected to the Ni-agarose column (ABT-Agarose Bead Technologies, Spain). The column was washed with 30 volumes of washing buffer I (50 mM NaH2PO4, 300 mM NaCl, 30 mM imidazole, 0.1% Triton-X114; pH8.0) to remove weakly bounded proteins and E. coli lipopolysaccharides (LPS). The next washing step was performed using washing buffer II (50 mM NaH2PO4, 300 mM NaCl, 30 mM imidazole; pH8.0) in order to remove residual Triton-X114. The recombinant protein was eluted from the column using the elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole; pH8.0) at a flow rate of 1.5 ml/min.
The purified protein was dialyzed for 2 h against dialysis buffer (50 mM NaH2PO4, 300 mM NaCl; pH8.0) to remove excess imidazole molecules and for further 2 h against phosphate buffer (0.2 M NaHPO4, 0.2 M NaH2PO4; pH8.0). Protein concentration was performed using Centriprep-50 kDa (Amicon, USA). The purified concentrated protein was analyzed on 8% SDS-PAGE and protein concentration was measured by NanoDrop™ 3000 spectrophotometer.
Western Blotting
Recombinant XTEN-GCSF protein was transferred to nitrocellulose membrane (Amersham, UK) in a wet blotting system (Denagen Tajhiz, Iran) (70 V, 25 min). After overnight (o/n) blocking of the membrane with 2% w/v skim milk (Merck, Germany) in phosphate-buffered saline (PBS) at 4 °C, 1:1500 dilution of HRP conjugated rabbit anti-His antibody (Sigma, USA) was added to the membrane and incubated at room temperature (RT) for 2 h. 3,3′-diaminobenzidine (DAB) was used as the substrate for visualizing the corresponding protein bands.
Size Exclusion Chromatography (SEC)
To study the increased hydrodynamic radius of the recombinant XTENylated GCSF, size-exclusion chromatography (SEC) was performed according to the European Pharmacopoeia 9.1. Edition using TSKgel G3000SWxl column (Tosoh Bioscience, Japan) connected to a Shimadzu high-performance liquid chromatography (HPLC) system (Kyoto, Japan). For this purpose, samples were diluted to a final concentration of 0.2 µg/ml in 0.06 M sodium acetate (pH4.0). Eight micrograms of each protein was injected into the column and eluted by an isocratic mobile phase of 0.03 M di-ammonium hydrogen phosphate (pH7.0) with a flow rate of 0.5 ml/min at 30 °C. UV absorbance of the eluted protein was recorded at 215 nm. Molecular weight of the proteins was estimated according to the retention time of the gel filtration standard proteins (Bio-RAD, Cat No. 151–1901).
Fluorescence Measurement
Tertiary conformational changes of the recombinant protein were determined by Cary Eclipse Varian Spectrophotometer (Agilent Technologies, Germany). In brief, Filgrastim, PEG-Filgrastim (CinnaGen, Iran), and XTEN-GCSF were diluted in phosphate buffer (PB) to a final concentration of 300 µg/ml. Excitation of tryptophan residues was carried out at 295 nm (slit width of 5 nm), and emission was recorded at the range of 300–450 nm (slit width of 10 nm).
Dynamic Light Scattering
To determine the effect of XTEN polypeptide on hydrodynamic radius of the GCSF protein, dynamic light scattering (DLS) was performed. In brief, 0.5 mg/ml of each protein (Filgrastim, PEG-Filgrastim and XTEN-GCSF) was diluted in ddH2O and subjected to the Zetasizer ZEN3600 system (Malvern, Germany) at 25 °C. All results were reported as size by number.
In Vitro Cell Proliferation Study
Biological activity of the recombinant XTENylated GCSF was evaluated by MTT assay using GCSF-dependent M-NFS-60 cell line. The cells were seeded in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 0.025 mM 2-mercaptoethanol (2-ME), 0.025 mM sodium-pyruvate, and 33 IU/ml IL-3 (Sigma, USA) at a density of 5 × 103 cells/well in 96-well plates. Serially diluted proteins (Filgrastim, PEG-Filgrastim and XTEN-GCSF (0.0001, 0.001, 0.01, 0.1, 1, 10, 100, 1000, 10,000 ng/ml) were added to the wells and incubated for 3 days. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reagent was added to the wells, and the cells were incubated for further 4 h at 37 °C. Optical measurements were done using ELISA microplate reader (BioTeK, USA) at 550 nm after the addition of 1% SDS. Protein concentrations which induced halfway proliferation response after the specified treatment time were calculated using Prism software (v. 8.0) and expressed as EC50 values.
Pharmacodynamics
Animal studies were performed in agreement with the principles of the declaration of Helsinki approved by the Ethics Committee of Pasteur Institute of Iran (IR.PII.REC.1399.013). Normal Sprague Dawley male rats (6–7 weeks old; 200–250 g weight, 5 rats per each group) were randomly assigned to enter the study and were adopted for 1 week in appropriate conditions of light and humidity. All groups except group I received intraperitoneal (i.p) injection of 100 mg/kg of cyclophosphamide (CPA) on day 0 to induce neutropenia [14, 15]. On day 1, 100 µg/kg of Filgrastim, PEG-Filgrastim, or XTEN-GCSF was injected subcutaneously (s.c) in groups 2–4, respectively, while the 5th group received PBS alone as control. Blood samples were collected from tail vein in non-vacuumed K2EDTA Nex tubes (Nexamo Technoplast, India) on days 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, and 11 post-GCSF treatment. Neutrophils, lymphocytes, eosinophils, monocytes, and red and white blood cells were counted by hematology analyzer Celltac alpha (Nihon Kohden, Japan).
Pharmacokinetics
To investigate the comparative pharmacokinetic properties of Filgrastim, PEG-Filgrastim, and the recombinant XTEN-GCSF, blood sampling was scheduled for the assessment of GCSF in the above mentioned five groups at 0, 0.5, 1, 2, 4, 8, 10, 24, 48, 76, 92, 120, 144, 168, 192, and 240 h following subcutaneous drug administration. Plasma was immediately separated after 15 min of centrifugation at 3,000 rpm, and GCSF level was determined using Quantikine ELISA human GCSF kit (R&D systems, USA) according to the manufacturer’s instructions. Pharmacokinetic parameters including maximum plasma concentration (Cmax), the times to reach maximum concentration (Tmax), AUC(0-t), AUC(0-∞), T1/2, and MRT were calculated using non-compartmental analysis. Tmax and Cmax were directly determined from observational data. The area under the plasma concentration–time curve (AUC) and AUC up to the last measurable concentration (AUC(0-t)) were calculated using trapezoidal rule. The terminal rate constant (kel) was calculated using linear regression of terminal log-linear phase. The absorption rate constant (Ka) was analyzed using residual method.
Statistical Analysis
All data were expressed as mean ± SD. SPSS software (v. 21) was used for the statistical analysis. Mann–Whitney U test was used for comparing mean levels of protein expression between different groups. Differences between groups were considered to be significant if P values were below 0.05.
Results
Cloning, Expression, and Purification of XTEN-GCSF
The expression cassette subcloned into pET28a was verified through restriction digestion and sequencing (data not shown). After expression of XTEN-GCSF in LB medium at 30 °C for 24 h, Ni-agarose affinity chromatography effectively captured the soluble recombinant XTEN-GCSF through its histidine tag, and an approximately 140 kDa protein was observed on 8% SDS-PAGE which was confirmed by Western blotting with anti-His tag antibody on nitrocellulose membrane (Fig. S1).
Size Exclusion Chromatography
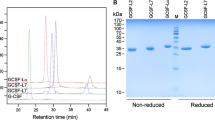
In the present study, on a calibrated SEC-HPLC column, monomers, dimers, and aggregated forms of the XTEN-GCSF were separated according to their relative molecular weight. As reported by SEC, XTENylation caused a very large increase in hydrodynamic volume of GCSF. As shown in Fig. 1, the main peak of monomer Filgrastim appeared in retention time of 19.6 min within the range of about 18 kDa, while the retention time of the main peak (monomer) of XTEN-GCSF was equal to 10.8 min, and it appeared in the area of about 670 kDa. According to the chromatograms, frequency of dimer formation in Filgrastim and XTEN-GCSF was equal to 3.3 and 3.8%, respectively. Lower amount of the aggregated protein was also observed in XTEN-GCSF in comparison to the Filgrastim (1.5 vs. 3.5%, respectively).

Size exclusion chromatograms of Filgrastim and XTEN-GCSF. Molecular weight marker includes bovine thyroglobulin (670 kDa), bovine c-globulin (158 kDa), chicken ovalbumin (44 kDa), horse myoglobin (17 kDa), and vitamin B12 (1.35 kDa). Last peak obtained at retention time of 22 min represents excipients from the dilution buffer
Intrinsic Fluorescence Measurements
To predict tertiary structural changes in GCSF molecule after fusion to the XTEN moiety, intrinsic fluorescence measurements were performed. Fluorescence intensity depends on aromatic amino acids within protein sequence. The effect of XTENylation on the measured fluorescence intensity of the two tryptophan residues (W59 and W119) within GCSF protein was followed by excitation and emission at 295 and 300–450 nm, respectively. The obtained spectra revealed that Filgrastim and PEG-Filgrastim had tryptophan maximum wavelength of 349 nm, while XTEN-GCSF showed a mild red shift (approximately 5 nm; λmax of 354 nm) (Fig. 2) which may be explained by the amino acid composition of XTEN moiety and their possible effects on the obtained fluorescence pattern through their hydroxyl groups.

Fluorescence spectra of Filgrastim, PEG-Filgrastim, and XTEN-GCSF
Dynamic Light Scattering
DLS analysis revealed that purified XTEN-GCSF protein sample was homogenous and non-aggregated (PDI: 0.448). It was also shown that fusion of XTEN moiety to N-terminal region of GCSF could increase the hydrodynamic radius of the protein in comparison with the standard GCSF molecules (Filgrastim and PEG-Filgrastim) (30.38 vs. 2.148 and 3.7 nm, respectively) (Figs. 3 and S2).

Hydrodynamic radius measurement. Asterisks represent significant differences between XTEN-GCSF and the two standard GCSF proteins (mean ± SD); (P ˂ 0.0001). Increased hydrodynamic radius of GCSF was found in the case of XTENylation
Biological Activity Assessment
Assessment of in vitro biological activity of GCSF derivatives was performed by the measurement of the proliferative effect on GCSF-dependent M-NFS-60 cell line. The cells were treated with serially diluted Filgrastim, PEG-Filgrastim, and XTEN-GCSF. Corresponding stimulus response graph (Fig. 4) showed that the number of cells has been sigmoidally increased as concentration of GCSF proteins raised. EC50 values (calculated potency index) of Filgrastim, PEG-Filgrastim, and XTEN-GCSF were measured as 44.62, 73.10, and 100.6 pg/ml, respectively, representing lower potency of XTENylated GCSF.

In vitro proliferation assay on GCSF dependent M-NFS-60 cell line. Cell density was measured at 550 nm after treatment with different GCSF derivatives. Data are represented as mean ± SD of triplicated experiments on a semi-log scale. Bars represent SD values
Pharmacodynamics
Pharmacodynamic activity of GCSF is usually described by its potency on proliferation and differentiation of neutrophils. The significant decrease of neutrophil count following the CPA injection began to rise after the injection of GCSF derivatives on day 2 among which PEG-Filgrastim and XTEN-GCSF significantly showed stronger effects in comparison with the group received Filgrastim (Fig. 5a). White blood cell (WBC) count was also increased gradually after receiving the GCSF injections from day 3, and all 3 GCSF derivatives represented the highest titers of WBC on day 9 (Fig. 5b), while other blood cells did not significantly changed (Fig. S3).

Neutrophil (a) and (b) white blood cells in neutropenic rats received single doses of GCSF derivatives. Data are means ± SD of 3 random rats/group on a linear scale. Bars represent the SD values
Pharmacokinetics
Plasma concentration time curves of Filgrastim, PEG-Filgrastim, and XTEN-GCSF have been presented in Fig. 6. All the calculated pharmacokinetic parameters using non-compartmental method are summarized in Table 1.

GCSF plasma concentration versus time after subcutaneous administrations of Filgrastim, PEG-Filgrastim, and XTEN-GCSF in rats. Data represents five individual rats in each group and is presented as means ± SD on a linear scale
According to the LOQ of Quantikine ELISA human GCSF kit, plasma concentrations were not accurately detectable 10 and 96 h after s.c. injection of Filgrastim, PEG-Filgrastim, and XTEN-GCSF, respectively. The peak plasma concentrations of about 552.35 ± 6.75, 481.75 ± 8.845, and 296.0 ± 6.0 ng/ml occurred 2.0 ± 0.0, 24.0 ± 0.0, and 10.0 ± 0.0 h after s.c. administration of the Filgrastim, PEG-Filgrastim, and XTEN-GCSF molecules, respectively.
The calculated AUC(0-∞) values showed much more drug exposure after administration of the PEGylated and XTENylated compounds (16,411.0 ± 2371.84 and 11,379.1 ± 632.8) vs. the Filgrastim received group (2315.3 ± 129.3 ng.h/ml) indicating the significant reduction in apparent total body clearance (CL/F) of the compounds (6.057 ± 0.83 and 8.8 ± 0.5 vs. 43.3 ± 2.3 ml/h/kg) for PEG-Filgrastim, XTEN-GCSF, and Filgrastim, respectively. The mean residence time (MRT) values were calculated about 17.92 ± 1.022 and 28.9 ± 2.1 h for PEG-Filgrastim and XTEN-GCSF, respectively compared to the 3.4 ± 0.31 h for Filgrastim showing a significant increase in terminal half-life of the PEGylated and XTENylated GCSF molecules (9.61 ± 0.52 and 16.48 ± 2.1 h vs. 1.847 ± 0.13 h for PEG-Filgrastim, XTEN-GCSF, and Filgrastim, respectively).
Discussion
Due to the high mortality rate in neutropenic patients, GCSF or granulocyte–macrophage colony-stimulating factor (GM-CSF) should be prescribed alongside broad-spectrum antibiotics to recover patients’ neutrophil count [26]. For increasing in vivo half-life of the GCSF molecule, several approaches have been developed [7, 8]. For example, half-life of the 3DHSA-GCSF compound has been reported to be 1.65 times higher than GCSF alone, or the fusion of the Fc domain to C-terminus of GCSF has led to its longer plasma circulation than GCSF molecule alone (> 48 vs. 24 h) [7, 8]. At the moment, the only long-acting commercially approved GCSF molecule is PEG-GCSF with several times higher half-life than GCSF [27]. In this study, the role of XTEN polymer in increasing half-life of the GCSF protein was investigated. It has been previously shown that biological tags could increase drugs’ half-life without any considerable side effects [18,19,20,21]. Due to the low complexity of the XTEN tag as well as GCSF molecule in terms of the number of disulfide bonds and tertiary structure, E. coli was chosen as the expression host cell. On the other hand, higher yield and lower cost of protein expression, and easy scale-up procedures have made E. coli as a suitable expression system [28, 29]. The recombinant XTEN-GCSF protein with an expected size of 72.6 kDa was observed as an approximately 140 kDa protein on SDS-PAGE which may be due to the lack of hydrophobic amino acids within this tag polymer resulting in slower movement of the protein within SDS-PAGE [18,19,20,21].
Being rich in negative charge and large hydrodynamic diameter of the proteins are two significant parameters influencing the renal infiltration and half-life of the proteins [16, 30]. The observed differences in size exclusion chromatograms may be explained by the ability of SEC in separation of proteins based on shape (hydrodynamic radius) as well as molecular weight [31, 32]. Previous studies have shown that XTENylation could enhance the solubility of the proteins [18] which has been confirmed by the lower amount of aggregated XTEN-GCSF forms in this study.
Tryptophan residues are very sensitive towards the local environment, and their changes in emission spectra usually happen in response to protein’s conformational alterations. Polarity of the solvent, the presence of hydrogen bonds, and other non-covalent interactions are the main factors influencing intensity and maximum wavelength of tryptophan residues in intrinsic fluorescence measurements [33]. The indole group of tryptophan residues plays a significant role in UV absorbance through intermolecular hydrogen binding in which conjugation of the fluorophore will be extended and a shift in higher wavelength will occur [34, 35]. Excitation of Filgrastim and its PEGylated form occurred at the maximum absorption wavelength of 350 nm, but XTEN-GCSF represented higher wavelength absorption (354 nm). The observed difference could be explained by the amino acid composition of XTEN polymer (A, E, G, P, S, and T) in which serine, threonine, and glutamic acid can form hydrogen bonds by the indole group and make mild changes in protein fluorescence pattern by transferring tryptophan residues from hydrophobic core of the protein to its surface. This finding is in accordance with the previous studies on GCSF molecules attached to the human serum albumin [36] or PEG molecule [37].
Aggregation of the proteins is a challenging issue in pharmaceutical industry, and experiments should be performed to ensure about the quality of the product. Dynamic light scattering (DLS) is one of the main techniques to study protein aggregation [38]. DLS data showed that the addition of XTEN polymer to the GCSF protein could increase the hydrodynamic radius of the protein without affecting its structure (PDI index < 0.5) which might represent its higher plasma half-life. Although in vitro proliferation assay showed that XTEN-GCSF could induce proliferation of NFS-60 cells, cellular response towards PEG-Filgrastim and XTEN-GCSF was lower than Filgrastim treated cells which can be explained by the inhibitory effect of the PEG and XTEN moieties on efficient binding of GCSF to its specific receptor [8, 39, 40].
Herein, a neutropenic rat model was developed to study in vivo biological activity of the XTEN-GCSF protein in comparison with the two commercially available proteins. Complete blood count (CBC) analysis showed that 3 tested proteins could maximize neutrophil count on day 2 compared to the control group receiving PBS among which the effect of XTEN and PEG-conjugated GCSF proteins was stronger than Filgrastim. This neutrophil-increasing effect and subsequent neutralization of CPA side effects maintained during the week post-injection of GCSF derivatives. This trend continued until the number of WBCs and neutrophils reached a balance. On the other hand, the XTEN-GCSF induced production of the highest count of WBC on day 9 which was quite similar to the other treatments (Filgrastim and PEG-Filgrastim). This finding reflects the appropriate distribution of XTENylated GCSF in comparison to the PEGylated form. In overall, any changes in pharmacodynamics curves could be explained by the potency of administered protein in reduction of adverse effects of chemotherapy agent and should be investigated alongside pharmacokinetic studies. GCSF enhances transformation of myoblasts to neutrophils which is different from the role of GM-CSF in development of different cell types (basophil, monocyte, neutrophil, and eosinophil) from the myoblasts [41]. The observed reduction in RBC count may be due to the frequent blood samplings at different time points.
After s.c. administration of PEG-Filgrastim and XTEN-GCSF, the peak of plasma concentration–time profile became broader in comparison with the group received Filgrastim, and therefore, the Tmax occurred later after dosing (from 2.0 ± 0.0 for Filgrastim to 24.0 ± 0.0 and 10.0 ± 0.0 h for PEG- and XTEN-GCSF, respectively) suggesting a negligible role of the neutrophil independent clearance pathways for both PEG-Filgrastim and XTEN-GCSF.
Although the maximum plasma concentration (Cmax) of PEG-Filgrastim was achieved much later than XTEN-GCSF (24.0 ± 0.0 vs. 10.0 ± 0.0 h), an obvious delay was shown in absorption process of XTEN-GCSF according to the plasma concentration–time curves reflecting absorption rate constants of XTEN-GCSF and PEG-Filgrastim (0.07 ± 0.001 vs. 0.1 ± 0.001 h−1, respectively). This observation could be explained by respective molecular size (30.4 vs. 3.7 nm) playing a predominant role in restriction of lymphatic absorption pathway.
Cmax values of the subcutaneously administered PEGylated and XTENylated forms were approximately 88 and 54% of the values obtained after Filgrastim administration (481.75 ± 8.8 and 296.0 ± 6.0 vs. 552.35 ± 6.75 ng/ml, respectively), which could also be described by the reduced absorption rates after PEGylation and XTENylation processes.
In the present study, both PEGylation and XTEN tagging approaches resulted in reduced renal clearance of GCSF which has been reflected in the calculated apparent total clearance of PEG-Filgrastim and XTEN-GCSF compared to the Filgrastim (6.1 ± 0.1 and 8.8 ± 0.5 vs. 43.3 ± 2.3 ml/h.kg, respectively). Therefore, a significant increase in AUC(0-∞) values was also observed (16,411.0 ± 2371.84 and 11,379.1 ± 632.8 ng.h/ml for PEG-Filgrastim and XTEN-GCSF vs. 2315.3 ± 129.3 ng.h/ml for Filgrastim, respectively).
Although the calculated AUC(0-∞) of XTENylated form was lower than PEGylated Filgrastim, the average time of protein remaining within the rats’ blood circulation (MRT) was still considerably high due to the significant decrease in elimination rate constant (0.07 ± 0.01 and 0.04 ± 0.01 h−1 for PEG-Filgrastim and XTEN-GCSF vs. 0.38 ± 0.05 h−1 for Filgrastim, respectively). On the other hand, the results of pharmacokinetic study indicated that tagging GCSF molecule with XTEN polymer could retain the protein for a longer time which is in accordance with the previous studies representing higher protein’s half-life for Exenatide [25] GLP-2 [23] and enfuvirtide [18] when genetically fused to the XTEN.
Conclusions
Our findings showed that XTENylation could enhance serum circulation of GCSF by increasing hydrodynamic diameter of the protein and improve its pharmacodynamic properties without having significant effects on higher structures of the GCSF. Based on the repeatedly reported drawbacks of PEGylation including complicated production, multistep purification process, high cost of manufacturing, polydispersity of the final product, and the increased risk of renal tubular vacuolation, it seems that using biopolymers, such as XTEN, may be beneficial for development of the next generation of pharmaceutics.
Data Availability
Not applicable.
References
Lyman, G. H., Abella, E., & Pettengell, R. (2014). Risk factors for febrile neutropenia among patients with cancer receiving chemotherapy: A systematic review. Critical Reviews in Oncology/Hematology, 90, 190–199.
Newburger, P. E., & Dale, D. C. (2013). Evaluation and management of patients with isolated neutropenia. Seminars in Hematology, 50, 198–206.
Schneider, A., Kuhn, H. G., & Schabitz, W. R. (2005). A role for G-CSF (granulocyte-colony stimulating factor) in the central nervous system. Cell Cycle, 4, 1753–1757.
Dwivedi, P., & Greis, K. D. (2017). Granulocyte colony-stimulating factor receptor signaling in severe congenital neutropenia, chronic neutrophilic leukemia, and related malignancies. Experimental Hematolology, 46, 9–20.
Pollmächer, T., Korth, C., Schreiber, W., Hermann, D., & Mullington, J. (1996). Effects of repeated administration of granulocyte colony-stimulating factor (G-CSF) on neutrophil counts, plasma cytokine, and cytokine receptor levels. Cytokine, 8, 799–803.
Kumari, M., Sahni, G., & Datta, S. (2020). Development of site-specific PEGylated granulocyte colony stimulating factor with prolonged biological activity. Frontiers in Bioengineering and Biotechnology, 8, 572077.
Zhao, S., Zhang, Y., Tian, H., Chen, X., Cai, D., Yao, W., & Gao, X. (2013). Extending the serum half-life of G-CSF via fusion with the domain III of human serum albumin. BioMed Research International, 2013, 107238.
Do, B. H., Kang, H. J., Song, J-A., Nguyen, M. T., Park, S., Yoo, J., Nguyen, A. N., Kwon, G. G., Jang, J., Jang, J., Lee, S., So, S., Sim, S., Lee, K. J., Osborn, M. J., & Choe, H. (2017). Granulocyte colony-stimulating factor (GCSF) fused with Fc domain produced from E. coli is less effective than polyethylene glycol-conjugated GCSF. Scientific Reports, 7, 6480.
Tanaka, H., Satake-Ishikawa, R., Ishikawa, M., Matsuki, S., & Asano, K. (1991). Pharmacokinetics of recombinant human granulocyte colony-stimulating factor conjugated to polyethylene glycol in rats. Cancer Research, 51, 3710–3714.
Halpern, W., Riccobene, T. A., Agostini, H., Baker, K., Stolow, D., Gu, M. L., Hirsch, J., Mahoney, A., Carrell, J., Boyd, E., & Grzegorzewski, K. J. (2002). Albugranin™, a recombinant human granulocyte colony stimulating factor (G-CSF) genetically fused to recombinant human albumin induces prolonged myelopoietic effects in mice and monkeys. Pharmaceutical Research, 19, 1720–1729.
van der Auwera, P., Platzer, E., Xu, Z. X., Schulz, R., Feugeas, O., Capdeville, R., & Edwards, D. J. (2001). Pharmacodynamics and pharmacokinetics of single doses of subcutaneous pegylated human G-CSF mutant (Ro 25–8315) in healthy volunteers: Comparison with single and multiple daily doses of filgrastim. American Journal of Hematolology., 66, 245–251.
Strohl, W. R. (2015). Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs. 29, 215–239.
Bendele, A., Seely, J., Richey, C., Sennello, G., & Shopp, G. (1998). Short communication: Renal tubular vacuolation in animals treated with polyethylene-glycol-conjugated proteins. Toxicological Sciences, 42, 152–157.
He, X., Yin, H. L., Wu, J., Zhang, K., Liu, Y., Yuan, T., Rao, H. L., Li, L., Yang, G., & Zhang, X. M. (2011). A multiple-dose pharmacokinetics of polyethylene glycol recombinant human interleukin-6 (PEG-rhIL-6) in rats. Journal of Zhejiang University Scieince B, 12, 32–39.
Schlapschy, M., Binder, U., Borger, C., Theobald, I., Wachinger, K., Kisling, S., Haller, D., & Skerra, A. (2013). PASylation: A biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Engineering Design and Selection, 26, 489–501.
Mazaheri, S., Talebkhan, Y., Mahboudi, F., Nematollahi, L., Cohan, R. A., MirabzadehArdakani, E., Bayat, E., Sabzalinejad, M., Sardari, S., & Torkashvand, F. (2020). Improvement of certolizumab fab′ properties by PASylation technology. Scientific Reports, 10, 18464.
Bech, E. M., Pedersen, S. L., & Jensen, K. J. (2018). Chemical strategies for half-life extension of biopharmaceuticals: Lipidation and its alternatives. ACS Medicinal Chemistry Letters, 9, 577–580.
Ding, S., Song, M., Sim, B.-C., Gu, C., Podust, V. N., Wang, C.-W., McLaughlin, B., Shah, T. P., Lax, R., Gast, R., Sharan, R., Vasek, A., Hartman, M. A., Deniston, C., Srinivas, P., & Schellenberger, V. (2014). Multivalent antiviral XTEN–Peptide conjugates with long in vivo half-life and enhanced solubility. Bioconjugate Chemistry, 25, 1351–1359.
Geething, N. C., To, W., Spink, B. J., Scholle, M. D., Wang, C., Yin, Y., Yao, Y., Schellenberger, V., Cleland, J. L., Stemmer, W. P. C., & Silverman, J. (2010). Gcg-XTEN: An improved glucagon capable of preventing hypoglycemia without increasing baseline blood glucose. PLoS One, 5,
Podust, V. N., Balan, S., Sim, B.-C., Coyle, M. P., Ernst, U., Peters, R. T., & Schellenberger, V. (2016). Extension of in vivo half-life of biologically active molecules by XTEN protein polymers. Journal of Controlled Release, 240, 52–66.
Schellenberger, V., Wang, C., Geething, N. C., Spink, B. J., Campbell, A., To, W., Scholle, M. D., Yin, Y., Yao, Y., Bogin, O., Cleland, L., Silverman, J., & Stemmer, W. P. C. (2009). A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nature Biotechnology, 27, 1186–1190.
Varanko, A., Saha, S., & Chilkoti, A. (2020). Recent trends in protein and peptide-based biomaterials for advanced drug delivery. Advanced Drug Delivery Reviews, 156, 133–187.
Alters, S. E., McLaughlin, B., Spink, B., Lachinyan, T., Wang, C., Podust, V., Schellenberger, V., & Stemmer, W. P. C. (2012). GLP2-2G-XTEN: A pharmaceutical protein with improved serum half-life and efficacy in a rat Crohn’s disease model. PLoS One, 7,
Amatya, R., Park, T., Hwang, S., Yang, J., Lee, Y., Cheong, H., Moon, C., Kwak, H. D., Min, K. A., & Shin, M. C. (2020). Drug delivery strategies for enhancing the therapeutic efficacy of toxin-derived anti-diabetic peptides. Toxins, 12, 313.
Moore, W. V., Nguyen, H. J., Kletter, G. B., Miller, B. S., Rogers, D., Ng, D., Moore, J. A., Humphriss, E., Cleland, J. L., & Bright, G. M. (2016). A randomized safety and efficacy study of somavaratan (VRS-317), a long-acting rhGH, in pediatric growth hormone deficiency. The Journal of Clinical Endocrinology and Metabolism, 101, 1091–1097.
NICE. Neutropenic sepsis : Prevention and management in people with cancer. NICE Guidel. 2012;1–31.
Zamboni, W. C. (2003). Pharmacokinetics of pegfilgrastim. Pharmacotherapy, 23, 9S-14S.
Rosano, G. L., & Ceccarelli, E. A. (2014). Recombinant protein expression in Escherichia coli: Advances and challenges. Frontiers in Microbiology, 5, 172.
Baeshen, M. N., Al-Hejin, A. M., Bora, R. S., Ahmed, M. M. M., Ramadan, H. A. I., Saini, K. S., Baeshen, N. A., & Redwan, E. M. (2015). Production of biopharmaceuticals in E. coli: Current scenario and future perspectives. Journal of Microbiology and Biotechnology, 25, 953–962.
Tryggvason, K., & Wartiovaara, J. (2005). How does the kidney filter plasma? Physiology, 20, 96–101.
Hagel, L. (1998). in Current protocols in molecular biology: Gel-filtration chromatography. Wiley, New York, pp. 10.9.1–10.9.2.
Mickiene, G., Dalgediene, I., Dapkunas, Z., Zvirblis, G., Pesliakas, H., Kaupinis, A., Valius, M., Mistiniene, E., & Pleckaityte, M. (2017). Construction, purification, and characterization of a homodimeric granulocyte colony-stimulating factor. Molecular Biotechnology, 59, 374–384.
Ghisaidoobe, A., & Chung, S. J. (2014). Intrinsic tryptophan fluorescence in the detection and analysis of proteins: A focus on Förster resonance energy transfer techniques. International Journal of Molecular Sciences, 15, 22518–22538.
Lakowicz, J. R. (2006). in Principles of fluorescence spectroscopy (pp. 529–579). Springer.
Koenig, S., Müller, L., & Smith, D. K. (2001). Dendritic biomimicry: Microenvironmental hydrogen-bonding effects on tryptophan fluorescence. Chemistry, 7, 979–986.
Huang, Y.-S., Wen, X.-F., Yang, Z.-Y., Wu, Y.-L., Lu, Y., & Zhou, L.-F. (2014). Development and characterization of a novel fusion protein of a mutated granulocyte colony-stimulating factor and human serum albumin in Pichia pastoris. PLoS One, 9,
Mero, A., Grigoletto, A., Maso, K., Yoshioka, H., Rosato, A., & Pasut, G. (2016). Site-selective enzymatic chemistry for polymer conjugation to protein lysine residues: PEGylation of G-CSF at lysine-41. Polymer Chemistry, 7, 6545–6553.
Stetefeld, J., McKenna, S. A., & Patel, T. R. (2016). Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophysical Reviews, 8, 409–427.
Wadhwa, M., Bird, C., Dougall, T., Rigsby, P., Bristow, A., Thorpe, R., & participants of the study. (2015). Establishment of the first international standard for PEGylated granulocyte colony stimulating factor (PEG-G-CSF): Report of an international collaborative study. Journal of Immunological Methods, 416, 17–28.
Kang, J. S., & Lee, K. C. (2013). In vivo pharmacokinetics and pharmacodynamics of positional isomers of mono-PEGylated recombinant human granulocyte colony stimulating factor in rats. Biological & Pharmaceutical Bulletin, 36, 1146–1151.
Mehta, H. M., Malandra, M., & Corey, S. J. (2015). G-CSF and GM-CSF in neutropenia. The Journal of Immunology, 195, 1341–1349.
Acknowledgements
The authors wish to express their deep gratitude to all who provided deep technical supports.
Funding
This project was supported by Pasteur Institute of Iran and the Biotechnology Development Headquarter of the Vice-Presidency for Science & Technology, Tehran, Iran.
Author information
Authors and Affiliations
Contributions
YT, LN, and YHA: Design the study and revise of the manuscript. FYN: Major experimental work, data analysis, and contribution in writing the manuscript. PGh, EB, ShD, and HAMA: Contribution in experimental work. EM: Contribution in animal studies.
Corresponding authors
Ethics declarations
Ethics Approval
All experiments and procedures were approved by the Ethics Committee of Pasteur Institute of Iran (IR.PII.REC.1399.013) and performed in accordance with the approved guidelines and regulations.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.

Fig. S1
Western blotting using HRP conjugated rabbit anti-His antibody. #1, 2: Recombinant E. coli cell lysates before and after induction; #3, 4: Purified XTEN-GCSF samples; M: Pre-stained protein molecular weight marker. (PNG 88 kb)
Fig. S2
Dynamic light scattering histogram. Assessment of the size of GCSF derivatives. (PNG 760 kb)

Fig. S3
Blood counting of rats received single doses of GCSF derivatives. (a) Filgrastim; (b) PEG-Filgrastim; (c) XTEN-GCSF. Data are means ± SE SD of 3 random rats/group on a linear scale. Bars represent the SD values. (PNG 17 kb)

Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Nikravesh, F.Y., Gholami, P., Bayat, E. et al. Expression, Purification, and Biological Evaluation of XTEN-GCSF in a Neutropenic Rat Model. Appl Biochem Biotechnol 196, 804–820 (2024). https://doi.org/10.1007/s12010-023-04522-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-023-04522-w